
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient synthesis of ether phosphonates using trichloroacetimidate and acetate coupling methods
Walid. Fathalla *a,
Ibrahim. A. I. Alib and
Pavel. Pazderac
*a,
Ibrahim. A. I. Alib and
Pavel. Pazderac
aPhysics and Math. Engineering Dept., Faculty of Engineering, Port-Said University, Port Said, Egypt. E-mail: walid3369@yahoo.com
bDepartment of Chemistry, Faculty of Science, Suez Canal University, Ismailia, Egypt
cCentre for Syntheses at Sustainable Conditions and Their Management, Chemistry Dept., Faculty of Science, Masaryk University, Brno, Czech Republic
First published on 15th February 2018
Abstract
A series of ether phosphonates have been prepared by trichloroacetimidate and acetate coupling methods. Trichloroacetimidates or acetates were treated with primary and secondary alcohols as O-nucleophiles in the presence of catalytic TMSOTf to afford 21 examples of diethyl alkyloxy(substitutedphenyl)methyl phosphonates via C–O bond formation in 55–90% yields and short reaction time.
Introduction
Phosphonates are an important class of organic compounds showing remarkable applications as isosteric analogs of natural phosphate esters.1,2 Phosphonates are key intermediates in a variety of synthetically important reactions. The reaction of stabilized phosphonate carbanions (ylides) with aldehydes or ketones to afford E-selective alkenes is known as Horner–Wadsworth–Emmons reaction (a modified Wittig reaction).3–5 A number of methods have been reported for the preparation of phosphonates. The Michaelis–Arbuzov reaction6,7 is an abundant method for the preparation of phosphonates via P–C bond formation. It involves the reaction of an aryl/alkyl halide with trialkyl phosphite to give alkyl phosphonate. Hirao method8,9 shows the palladium-catalyzed cross-coupling of dialkylphosphite with aromatic electrophiles for the synthesis of arylphosphonates via aryl C–P bond formation. Recently, α-functional phosphonates, α-aminophosphonates,10 α-acetoxyphosphonates11 and α-hydroxyphosphonates12–15 showed interesting medicinal applications as antiviral agents and attracted the attention of many research groups. Abramov and Pudovik method16,17 described the reaction of aldehydes with di and trialkyl phosphite in the presence of a base to afford α-hydroxyphosphonates. An efficient use of α-hydroxy phosphonates enables a mild process for the preparation of α-substituted methyl phosphonates with promising biological activities.The use of trichloroacetimidate and acetate methods are well recognized in organic and carbohydrate synthesis via C–O or C–C bond formations.18–21 Earlier we described the reaction of O-phthalimidomethyl trichloroacetimidate and O-diphenylmethyl trichloroacetimidate with C-nucleophiles in the presence of TMSOTf to afford a series of N-substituted phthalimides,20 and benzhydryl derivatives.21 Also, we showed the preparation of a number of N-protected non proteinogenic α-amino acid esters using trichloroacetimidate or acetate coupling methods via C–C bond formation from the corresponding methyl 2-benzamido-2-hydroxyacetate and benzyl(methoxycarbonyl)(hydroxy)methylcarbamate substrates and C-nucleophiles.22
The strategies of both trichloroacetimidate and acetate methods involve the transformation of the substrate hydroxyl group into an excellent leaving group, such as trichloroacetimidate or acetate by the reaction with trichloroacetonitrile or acetic anhydride, respectively. The successive addition of poor nucleophiles (alcohols) to these active intermediates in the presence of Lewis acid give interesting products via C–O bond formations, Fig. 1.
 | ||
| Fig. 1 The trichloroacetimidate and acetate coupling methods from functionalized alcohol substrates. | ||
These findings motivated the development of a series of diethyl alkyloxy(substitutedphenyl)methyl phosphonate 6–8(a–f), 10 and 12 using trichloroacetimidate and acetate coupling methods via C–O bond formation.
Results and discussion
Nucleophilic addition of diethyl phosphite to benzaldehydes 1a–d in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio using triethylamine as a base gave the diethyl(hydroxy)arylmethyl phosphonates 2a–d in good yields (Scheme 1). The base-catalyzed addition reaction of trichloroacetonitrile to α-hydroxyphosphonates 2a–d hydroxyl group in the presence of DBU gave the desired diethyl 2-(2,2,2-trichloro-1-iminoethoxy)(3,4-dimethoxyphenyl)methylphosphonates 3a–d, in 80–94% yield, Scheme 1. The reactions of α-hydroxyphosphonates 2b–d with acetic anhydride in the presence of DMAP (N,N-dimethyl aminopyridine) at room temperature afforded diethylmethylcarbonyloxy(4-methoxyphenyl)methylphosphonates 4b–d, Scheme 1.
:1 molar ratio using triethylamine as a base gave the diethyl(hydroxy)arylmethyl phosphonates 2a–d in good yields (Scheme 1). The base-catalyzed addition reaction of trichloroacetonitrile to α-hydroxyphosphonates 2a–d hydroxyl group in the presence of DBU gave the desired diethyl 2-(2,2,2-trichloro-1-iminoethoxy)(3,4-dimethoxyphenyl)methylphosphonates 3a–d, in 80–94% yield, Scheme 1. The reactions of α-hydroxyphosphonates 2b–d with acetic anhydride in the presence of DMAP (N,N-dimethyl aminopyridine) at room temperature afforded diethylmethylcarbonyloxy(4-methoxyphenyl)methylphosphonates 4b–d, Scheme 1.
 | ||
| Scheme 1 Synthetic pathway of phosphonates trichloroacetimidates and acetates 3a–d and 4b–d. | ||
The structure assignment of α-hydroxyphosphonates 2a–d, active intermediates trichloroacetimidate derivatives 3a–d and acetate derivatives 4b–d were based on 1H, 13C and 31P NMR spectroscopic and elemental analysis are shown in the experimental part.
Both active intermediates trichloroacetimidates 3a–d and acetates 4b–d are excellent precursors for the structure modification of the diethyl phosphonate residue via trichloroacetimidate and acetate coupling methods. However, our early results concerning this synthetic method applying diethyl 2-(2,2,2-trichloro-1-iminoethoxy)(phenyl)methyl phosphonate (3a) failed to give us the desired products. Thus the reaction of trichloroacetimidate 3a with isobutyl alcohol in the presence of a catalytic amount of TMSOTf at room temperature did not give diethyl 2-methylpropyloxy(phenyl)methyl phosphonate (5a) and instead decomposition was observed and a complex mixture was obtained, Scheme 2. On the other hand, trichloroacetimidates 3b–d reacted with a variety of O-nucleophile acceptors (alcohols) in the presence of catalytic amount of TMSOTf at room temperature and readily gave a series of functionalized ether phosphonates 6–8(a–f), Scheme 2, Table 1. Similarly, the acetate derivatives 4b–d reacted with a variety of O-nucleophile acceptors (alcohols) in the presence of a catalytic amount of TMSOTf at room temperature in dichloromethane and readily gave a series of functionalized ether phosphonates 6–8(a–f), Scheme 2, Table 1. A comparative study of both trichloroacetimidate and acetate methodologies was carried out to examine the efficiency of reaction time and % of yield in both methods. The results showed that, although all compounds were prepared in good yields, there was a slight improvement in the % of yield and in the reaction time (monitored by TLC) using the trichloroacetimidate method, see Experimental section.
 | ||
| Scheme 2 Synthetic pathway of phosphonates 6–8(a–f) via C–O bond formation. | ||
| No. | Structure | TCI and Ac yield (time) | No. | Structure | TCI and Ac yield (time) |
|---|---|---|---|---|---|
| a Abbrev. TCI: trichloroacetimidate method, Ac: acetate method. | |||||
| 6a |  |
81% (2 h), 76% (4 h) | 7d |  |
78% (3 h), 59% (4 h) |
| 6b |  |
72% (2 h), 64% (5 h) | 7e | 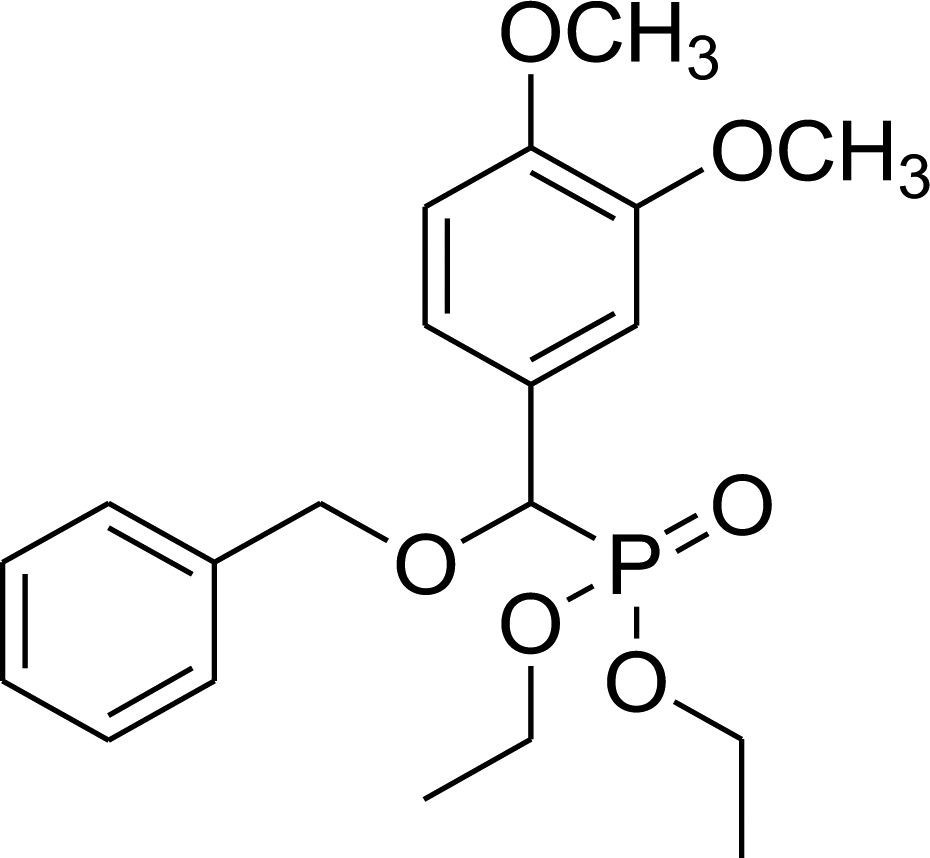 |
79% (1 h), 73% (5 h) |
| 6c |  |
75% (3 h), 71% (5 h) | 7f |  |
86% (2 h), 79% (4 h) |
| 6d |  |
82% (2 h), 76% (5 h) | 8a |  |
79% (2 h), 75% (2 h) |
| 6e |  |
67% (2 h), 71% (3 h) | 8b |  |
74% (1 h), 66% (3 h) |
| 6f |  |
71% (3 h), 56% (4 h) | 8c |  |
80% (2 h), 78% (4 h) |
| 7a |  |
86% (1 h), 77% (4 h) | 8d |  |
81% (3 h), 72% (3 h) |
| 7b |  |
78% (1 h), 68% (4 h) | 8e |  |
71% (2 h), 63% (3 h) |
| 7c |  |
86% (2 h), 74% (4 h) | 8f |  |
79% (2 h), 69% (4 h) |
According to the presumed mechanism, the TMSOTf is used to eliminate the trichloroacetimidate and acetate leaving groups with the subsequent formation of carbocation. This carbocation could be stabilized by an efficient electron donor and a phenyl group but it is insufficient. In contrast there was good stabilization in the case of a phenyl residue bearing an electron-donating –OR group at the 4-position. The final step is the nucleophilic attack of the weak alcohol nucleophiles at the stabilized carbocation to form the desired products 6–8(a–f) via an overall SN1 mechanism. The attempted reaction of diethyl 2-methylpropyloxy(4-methoxyphenyl)methyl phosphonate (6a) with isopropyl alcohol under the same reaction conditions, in the presence of a catalytic amount of TMSOTf at room temperature in dichloromethane, failed to give the corresponding isopropyl ether phosphonate derivative 6b via transetherification, Scheme 2.
Structure assignment of ether derivatives 6–8(a–f) were based on 1H, 13C and 31P-NMR spectral and physicochemical analysis. The 1H-NMR spectrum of 6a shows an interesting doublet signal at δ = 4.52 ppm with coupling constant 15.0 Hz typically associated with CHP. This chemical shift is common for all isolated phosphonate derivatives 6–8(a–f) with chemical shifts ranging from 4.37–4.93 ppm and with coupling constant ranging from 12–18 Hz. The 1H-NMR spectrum exhibit signals at δ 0.90, 0.93, 1.84–1.97 and 3.16–3.28 ppm due to two CH3, CH and CH2 groups, respectively of the isobutyl-residue and signals at δ 1.24–1.28 and 4.02–4.12 ppm due to two OCH2CH3 residue. The 13C NMR spectrum of 6a shows interesting doublet signals at δ 16.3, 16.4, 62.7, 63.0, 76.9 and 77.5 ppm having coupling constants JCP 6.0, 6.0, 6.8, 6.8, 32.4 and 169.6 Hz corresponding to CH3, CH3, OCH2P, OCH2P, CH2OCHP and CHP groups, respectively. These signals are common for all isolated products, which confirm the C–O bond formation. The previously mentioned signals are good evidences for either the C–P coupling, the diastereotopic nature or both for these individual groups. The 31P-NMR spectrum of 6a showed a single signal at δ = 19.5 ppm corresponding to phosphonate phosphorus and was used as an indicator for the purity of the products.
The trichloroacetimidate method proved to be an excellent method for the structure modification of diethyl hydroxy(substituted phenyl)methylphosphonate 2a–d. Next, the use of an interesting derivatized alcohols was explored: (5-methyl-2-phenyl-1,3-dioxan-5-yl)methanol 9 and 2-(N-methyl-N-phenylamino)ethanol 11. The dioxane derivative 9 was prepared by heating 1,1,1-tris(hydroxymethyl)ethane with benzaldehyde in the presence of toluene-p-sulfonic acid in benzene and this compound is known to possess potential medicinal chemistry relevance.23 Thus the reaction of trichloroacetimidate 3a–b with dioxane derivative 9 in the presence of a catalytic amount of TMSOTf at room temperature in dichloromethane and gave diethyl((5-methyl-2-phenyl-1,3-dioxan-5-yl)methoxy)(substitutedphenyl)methyl phosphonate 10a–b in 62–70% yields, Scheme 3. Also trichloroacetimidate 3a reacted with 2-(N-methyl-N-phenylamino)ethanol (11) in the presence of a catalytic amount of TMSOTf at room temperature in dichloromethane and gave diethyl 2-(N-methyl-N-phenylamino)ethyloxy(4-methoxyphenyl)methyl phosphonate 12 in 29% yield, Scheme 3.
 | ||
| Scheme 3 Synthesis of phosphonates 10a–b and 12 via C–O bond formation. | ||
Conclusion
In conclusion, an efficient and very simple method for the synthesis of various ether phosphonates by trichloroacetimidate and acetate coupling methods is described. Trichloroacetimidates 3a–d or acetates 4b–d were treated with Lewis acid followed by reaction with O-nucleophiles to afford the desired products via C–O bond formation. Both methods gave good yields of desired products in short reaction times, and generally slightly better results were obtained using the trichloroacetimidate method. Applications of this methodology to synthesize various phosphonate analogues of alcohols containing primary and secondary hydroxy groups as O-nucleophiles are under progress in our laboratory.Experimental
Solvents were purified and dried in the usual way. The boiling range of the petroleum ether used was 40–60 °C. Thin-layer chromatography (TLC) utilized silica gel 60 F254 plastic plates (E. Merck, layer thickness 0.2 mm). Detection was by UV absorption. Melting points were determined on a Buchi 510 melting point apparatus. The 1H, 13C and 31P NMR spectra were recorded at 300 MHz, 75.5 and 194.4 MHz, respectively (Bruker AC 300) in CDCl3 with tetramethylsilane as an internal standard. The NMR analysis were performed at Organic Chemistry Department Masaryk University, Brno, Czech Republic. Elemental analyses were performed on a Flash EA-1112 instrument at the Microanalytical Laboratory, Faculty of Science, Suez Canal University, Ismailia, Egypt. 3,4-Dimethoxybenzaldehyde 1c and 3-methoxy-4-(prop-2-ynyloxy)benzaldehyde 1d was prepared by alkylation of vaniline as described in the literature.24Diethyl hydroxy(substitutedphenyl)methyl phosphonate 2a–d25
To a mixture of (20.7 g, 0.15 mol) of diethyl phosphonate and 1.09 g (0.011 mol) of triethylamine was added (0.15 mol) of aldehydes 1a–d portion wise during a period of 1 h at 50–70 °C. The reaction mixture was subsequently stirred at 70 °C for 2 h. After cooling it was left over night to give white crystals, filter and wash with cold toluene and then recrystallized from toluene to give pure crystals.Diethyl hydroxy(phenyl)methyl phosphonate (2a)
27.5 g, yield 76% white crystals. Mp 80–81 °C. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.19–1.34 (m, 6H, 2CH3), 3.82 (s, 3H, OCH3), 3.91–4.10 (m, 4H, 2CH2), 3.54 (bs, 1H, OH), 5.00 (d, JHP = 8.0 Hz, 1H, CHP), 7.24–7.46 (m, 5H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 4.0, CH3), 16.4 (d, JCP = 4.0, CH3), 55.3 (OCH3), 62.7 (d, JCP = 7.6, CH2OP), 63.4 (d, JCP = 7.6, CH2OP), 70.2 (d, JCP = 158.4, CHP), 127.1, 127.2, 128.1, 128.3, 136.8 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 21.6 (P). Found, %: C, 53.89; H, 6.94. For C11H17O4P (244.1). Calculated, %: C, 54.10; H, 7.02.Diethyl hydroxy(4-methoxyphenyl)methyl phosphonate (2b)
34.4 g, yield 84% white crystals. Mp 90–91 °C. 1H-NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.19–1.29 (m, 6H, 2CH3), 3.80 (s, 3H, OCH3), 3.89–4.10 (m, 4H, 2CH2), 4.33 (bs, 1H, OH), 4.95 (d, JHP = 9.0 Hz, 1H, CHP), 6.87 (d, J = 9.0 Hz, 2H, H-Ar), 7.39–7.43 (m, 2H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 4.0, CH3), 16.4 (d, JCP = 4.0, CH3), 55.2 (OCH3), 62.9 (d, JCP = 7.6, CH2OP), 63.2 (d, JCP = 7.6, CH2OP), 70.3 (d, JCP = 161.6, CHP), 113.0, 113.7, 128.4, 128.5, 128.8, 159.5 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 21.7 (P). Found, %: C, 52.39; H, 6.81. For C12H19O5P (274.1). Calculated, %: C, 52.55; H, 6.98.Diethyl hydroxy(3,4-dimethoxyphenyl)methyl phosphonate (2c)
40.6 g, yield 89% white crystals. Mp 101–103 °C. 1H-NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.16–1.26 (m, 6H, 2CH3), 3.83 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 3.92–4.07 (m, 4H, 2CH2), 4.62 (bs, 1H, OH), 4.89 (d, JHP = 9.0 Hz, 1H, CHP), 6.81 (d, J = 9.0 Hz, 1H, H-Ar), 6.95–7.07 (m, 1H, H-Ar), 7.08 (s, 1H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 4.0, CH3), 16.4 (d, JCP = 4.0, CH3), 55.8 (OCH3), 55.9 (OCH3), 62.9 (d, JCP = 7.6, CH2OP), 63.2 (d, JCP = 7.6, CH2OP), 70.4 (d, JCP = 161.6, CHP), 110.5, 110.8, 110.9, 129.3, 148.7, 148.8 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 21.7 (P). Found, %: C, 51.12; H, 6.89. For C13H21O6P (304.1). Calculated, %: C, 51.31; H, 6.96.Diethyl hydroxy(3-methoxy-4-(prop-2-yn-1-yloxy)phenyl)methyl phosphonate (2d)
36.9 g, yield 75% white crystals. Mp 87–88 °C. 1H-NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.19–1.37 (m, 6H, 2CH3), 2.50 (t, J = 3.0 Hz, 1H, CH), 3.86 (s, 3H, OCH3), 3.92–4.17 (m, 4H, 2CH2), 4.39 (bs, 1H, OH), 4.74 (d, J = 3.0 Hz, 2H, OCH2), 4.93 (d, JHP = 9.0 Hz, 1H, CHP), 6.99–7.12 (m, 3H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 4.0, CH3), 16.4 (d, JCP = 4.0, CH3), 55.9 (OCH3), 56.8 (OCH2), 63.0 (d, JCP = 7.6, CH2OP), 63.2 (d, JCP = 7.6, CH2OP), 70.5 (d, JCP = 160.8, CHP), 75.8 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 78.5 (CCH), 110.9, 114.0, 119.5, 130.7, 146.6, 149.6 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 21.5 (P). Found, %: C, 54.68; H, 6.43. For C15H21O6P (328.1). Calculated, %: C, 54.88; H, 6.45.
CH), 78.5 (CCH), 110.9, 114.0, 119.5, 130.7, 146.6, 149.6 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 21.5 (P). Found, %: C, 54.68; H, 6.43. For C15H21O6P (328.1). Calculated, %: C, 54.88; H, 6.45.
Preparation of diethyl 2-(2,2,2-trichloro-1-iminoethoxy)(substitutedphenyl) methyl phosphonate 3a–d
A stirred solution of diethyl hydroxy(substitutedphenyl)methylphosphonate 2a–d (10.0 mmol) in dry dichloromethane (30 mL) was treated with trichloroacetonitrile (2.9 mL, 20 mmol) and DBU (0.8 mL, 5.0 mmol). The reaction mixture was stirred at room temperature for 2 h. The solvent was evaporated under reduced pressure and the product was purified by column chromatography 5% triethylamine in dichloromethane, to give 3a–d as whitish oil.Diethyl 2-(2,2,2-trichloro-1-iminoethoxy)(phenyl)methyl phosphonate (3a)
1.92 g, yield 91% brownish oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.16–1.29 (m, 6H, 2CH3), 3.78 (s, 3H, OCH3), 3.94–4.17 (m, 4H, 2CH2), 6.22 (d, JHP = 15.0 Hz, 1H, CHP), 6.95–7.53 (m, 5H, H-Ar), 8.55 (brs, 1H, NH). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 16.4 (P). Found, %: C, 40.04; H, 4.32; N, 3.48. For C13H17Cl3NO4P (387.1). Calculated, %: C, 40.18; H, 4.41; N, 3.60.Diethyl(2,2,2-trichloro-1-iminoethoxy)(4-methoxyphenyl)methyl phosphonate (3b)
3.6 g, yield 87% brownish oil. 1H-NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.20–1.27 (m, 6H, 2CH3), 3.76 (s, 3H, OCH3), 3.97–4.16 (m, 4H, 2CH2), 6.20 (d, JHP = 15.0 Hz, 1H, CHP), 6.86 (d, J = 9.0 Hz, 2H, H-Ar), 7.44 (d, J = 6.0 Hz, 2H, H-Ar), 8.51 (brs, 1H, NH). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 55.2 (OCH3), 63.3 (d, JCP = 6.0, CH2OP), 63.5 (d, JCP = 6.0, CH2OP), 74.8 (d, JCP = 172.1, CHP), 90.9 (CCl3), 113.8, 113.9, 124.7, 124.7, 129.2, 129.3 (C-Ar), 161.1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 16.5 (P). Found, %: C, 39.96; H, 4.42; N, 3.29. For C14H19Cl3NO5P (417.0). Calculated, %: C, 40.17; H, 4.57; N, 3.35.
N). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 16.5 (P). Found, %: C, 39.96; H, 4.42; N, 3.29. For C14H19Cl3NO5P (417.0). Calculated, %: C, 40.17; H, 4.57; N, 3.35.
Diethyl(2,2,2-trichloro-1-iminoethoxy)(3,4-dimethoxyphenyl)methyl phosphonate (3c)
4.2 g, yield 94% brownish oil. 1H-NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.25–1.32 (m, 6H, 2CH3), 3.89 (s, 6H, 2 OCH3), 4.02–4.23 (m, 4H, 2CH2), 6.23 (d, JHP = 12.0 Hz, 1H, CHP), 6.86 (d, J = 9.0 Hz, 1H, H-Ar), 7.09 (d, J = 9.0 Hz, 2H, H-Ar), 8.53 (brs, 1H, NH). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.4 (d, JCP = 6.0, CH3), 16.5 (d, JCP = 6.0, CH3), 55.8 (OCH3), 63.3 (d, JCP = 6.0, CH2OP), 63.7 (d, JCP = 6.0, CH2OP), 74.9 (d, JCP = 172.1, CHP), 91.0 (CCl3), 110.8, 120.6, 120.7, 125.2, 148.9, 149.5 (C-Ar), 161.2 (CN). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 16.5 (P). Found, %: 40.02; H, 4.67; N, 3.05. For C15H21Cl3NO6P (447.0). Calculated, %: C, 40.15; H, 4.72; N, 3.12.
Diethyl(2,2,2-trichloro-1-iminoethoxy)(3-methoxy-4-(prop-2-yn-1-yloxy)phenyl)methyl phosphonate (3d)
3.8 g, yield 80% brownish oil. 1H-NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.22 (m, 6H, 2CH3), 2.50 (s, 1H, CH), 3.85 (s, 3H, OCH3), 3.99–4.16 (m, 4H, CH, 2CH2), 4.73 (d, J = 3.0 Hz, 2H, OCH2), 6.20 (d, JHP = 12.0 Hz, 1H, CHP), 6.99–7.28 (m, 3H, H-Ar), 8.53 (brs, 1H, NH). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.4 (d, JCP = 6.0, CH3), 16.5 (d, JCP = 6.0, CH3), 55.9 (OCH3), 56.7 (OCH2), 63.3 (d, JCP = 6.8, CH2OP), 63.6 (d, JCP = 6.8, CH2OP), 74.1 (d, JCP = 171.4, CHP), 75.9 (CCH), 78.4 (CCH), 90.9 (CCl3), 111.2, 113.9, 120.3, 126.6, 147.3, 149.6 (C-Ar), 161.1 (CN). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 16.3 (P). Found, %: C, 43.11; H, 4.36; N, 2.82. For C17H21Cl3NO6P (471.0). Calculated, %: C, 43.20; H, 4.48; N, 2.96.
Preparation of diethyl methylcarbonyloxy(substitutedphenyl)methyl phosphonate 4b–d
A solution of diethyl hydroxy(substitutedphenyl)methylphosphonate 2a–c (10.0 mmol) in acetic anhydride (1.2 mL, 12.0 mmol) and DMAP (0.61 g, 5.0 mmol) and NEt3 (1.1 mL, 12.0 mmol) was stirred at room temperature for 4 h. The reaction mixture was evaporated under reduced pressure. The residue was purified by column chromatography 4:1 pet. ether/ethyl-acetate, to give 4a–c as white oil.
Diethyl methylcarbonyloxy(4-methoxyphenyl)methyl phosphonate (4b)
2.94 g, yield 93% whitish oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.19 (t, J = 6.0 Hz, 3H, CH3), 1.28 (t, J = 6.0 Hz, 3H, CH3), 2.13 (s, 3H, CH3), 3.79 (s, 3H, OCH3), 3.87–4.13 (m, 4H, 2CH2), 6.07 (d, JHP = 12.0 Hz, 1H, CHP), 6.87 (d, J = 6.0 Hz, 2H, H-Ar), 7.40–7.44 (m, 2H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.2 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 20.9 (CH3), 55.2 (OCH3), 63.1 (d, JCP = 6.8, CH2OP), 63.6 (d, JCP = 6.8, CH2OP), 70.1 (d, JCP = 172.9, CHP), 113.9, 125.5, 129.5, 129.6, 159.9 (C-Ar), 169.3 (CO). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 18.0 (P). Found, %: C, 53.13; H, 6.57. For C14H21O6P (316.1). Calculated, %: C, 53.16; H, 6.69.Diethyl methylcarbonyloxy(3,4-dimethoxyphenyl)methyl phosphonate (4c)
2.94 g, yield 85% whitish oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.15 (t, J = 6 Hz, 3H, CH3), 1.24 (t, J = 6.0 Hz, 3H, CH3), 2.10 (s, 3H, CH3), 3.82 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 3.91–4.11 (m, 4H, 2CH2), 6.02 (d, JHP = 12.0 Hz, 1H, CHP), 6.79 (d, J = 6.0 Hz, 1H, H-Ar), 6.98–7.02 (m, 2H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 20.8 (CH3), 55.8 (CH3), 55.9 (CH3), 63.1 (d, JCP = 6.8, CH2OP), 63.7 (d, JCP = 6.8, CH2OP), 70.3 (d, JCP = 172.9, CHP), 110.9, 120.9, 121.0, 125.7, 148.9, 149.4 (C-Ar), 169.2 (CO). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 17.9 (P). Found, %: C, 51.85; H, 6.54. For C15H23O7P (346.1). Calculated, %: C, 52.02; H, 6.69.Diethyl methylcarbonyloxy(3-methoxy-4-propargyloxyphenyl)methyl phosphonate (4d)
2.44 g, yield 66% whitish oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.13 (t, J = 6.0 Hz, 3H, CH3), 1.31 (t, J = 6.0 Hz, 3H, CH3), 2.21 (s, 3H, COCH3), 2.51 (s, 1H, CH), 3.87 (s, 3H, OCH3), 3.92–4.13 (m, 4H, CH, 2CH2), 4.74 (d, J = 3.0 Hz, 2H, CH2), 6.07 (d, JHP = 12.0 Hz, 1H, CHP), 6.98–7.08 (m, 3H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 20.9 (CH3), 56.0 (OCH3), 56.7 (OCH2), 63.2 (d, JCP = 6.0, CH2OP), 63.3 (d, JCP = 6.0, CH2OP), 70.1 (d, JCP = 172.9, CHP), 75.9 (CCH), 78.4 (CCH), 111.9, 114.0, 120.7, 127.2, 147.3, 149.6 (C-Ar), 169.2 (CO). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 17.9 (P). Found, %: C, 55.04; H, 6.19. For C17H23O7P (370.1). Calculated, %: C, 55.13; H, 6.26.
General procedure for the preparation of diethyl alkyloxy(substitutedphenyl)methyl phosphonate 6–8(a–f), 10 and 12
A solution of trichloroacetimidate 3a–d or acetate 4b–d (1.0 mmol) in dichloromethane (40 mL) was added O-nucleophiles (alcohols) (1.0 mmol) and TMSOTf (0.1 mmol). The reaction mixture was stirred at room temperature till completion of the reaction (TLC monitored and the time was recorded). The reaction mixture was neutralized with solid sodium bicarbonate, filtered and concentrated in vacuo. The residue was purified by flash chromatography 1:2 pet. ether/ethyl-acetate to give the pure alkyloxy phosphonates 6–8(a–f), 10 and 12 as colorless oil.
Diethyl 2-methylpropyloxy(4-methoxyphenyl)methyl phosphonate (6a)
0.26 g, yield 78% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 0.90 (d, J = 3.0 Hz, 3H, CH3), 0.93 (d, J = 3.0 Hz, 3H, CH3), 1.24–1.28 (m, 6H, 2CH3), 1.84–1.97 (m, 1H, CH), 3.16–3.28 (m, 2H, CH2), 3.82 (s, 3H, OCH3), 4.02–4.12 (m, 4H, 2CH2), 4.52 (d, JHP = 15.0 Hz, 1H, CHP), 6.90 (d, J = 6.0 Hz, 2H, H-Ar), 7.28–7.39 (m, 2H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 19.2 (CH3), 19.3 (CH3), 28.4 (CH), 55.2 (OCH3), 62.7 (d, JCP = 6.8, CH2OP), 63.0 (d, JCP = 6.8, CH2OP), 76.9 (d, JCP = 32.4, CH2OCHP), 77.5 (d, JCP = 169.6, CHP), 113.7, 113.8, 127.0, 129.2, 129.3, 159.7 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.5 (P). Found, %: C, 58.04; H, 8.16. For C16H27O5P (330.2). Calculated, %: C, 58.17; H, 8.24.Diethyl 1-methylethyloxy(4-methoxyphenyl)methyl phosphonate (6b)
0.26 g, yield 83% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.08 (d, J = 6.0 Hz, 3H, CH3), 1.14 (d, J = 6.0 Hz, 3H, CH3), 1.19–1.25 (m, 6H, 2CH3), 3.57–3.76 (m, 1H, CH), 3.87 (s, 3H, OCH3), 3.92–4.09 (m, 4H, 2CH2), 4.64 (d, JHP = 18.0 Hz, 1H, CHP), 6.84 (d, J = 9.0 Hz, 2H, H-Ar), 7.33–7.36 (m, 2H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 20.9 (CH3), 23.0 (CH3), 55.1 (OCH3), 62.7 (d, JCP = 8.0, CH2OP), 63.1 (d, JCP = 8.0, CH2OP), 71.3 (d, JCP = 13.6, CHOCHP), 75.5 (d, JCP = 171.4, CHP), 113.6, 113.7, 117.6, 129.3, 159.5, 159.6 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 20.0 (P). Found, %: C, 56.78; H, 7.83. For C15H25O5P (316.1). Calculated, %: C, 56.95; H, 7.97.Diethyl propargyloxy(4-methoxyphenyl)methyl phosphonate (6c)
0.19 g, yield 62% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.21 (t, J = 6.0 Hz, 3H, CH3), 1.30 (t, J = 6.0 Hz, 3H, CH3), 2.46 (s, 1H, CH), 3.81 (s, 3H, OCH3), 3.89–3.95 (m, 2H, CH2), 4.01–4.08 (m, 4H, 2CH2), 4.93 (d, JHP = 15.0 Hz, 1H, CHP), 6.70–6.91 (m, 4H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 55.2 (OCH3), 56.4 (d, JCP = 15.9, CH2OCHP), 62.8 (d, JCP = 7.6, CH2OP), 63.1 (d, JCP = 7.6, CH2OP), 75.5 (CCH), 75.6 (d, JCP = 172.1, CHP), 78.6 (CCH), 113.9, 114.0, 125.2, 129.8, 129.9, 160.0 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.2 (P). Found, %: C, 57.57; H, 6.63. For C15H21O5P (312.1). Calculated, %: C, 57.69; H, 6.78.
Diethyl 4-chlorobutyloxy(4-methoxyphenyl) methyl phosphonate (6d)
0.17 g, yield 46% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.17 (t, J = 6.0 Hz, 3H, CH3), 1.28 (t, J = 6.0 Hz, 3H, CH3), 1.56–1.92 (m, 4H, 2CH2), 3.38–3.64 (m, 4H, 2CH2), 3.83 (s, 3H, OCH3), 3.98–4.08 (m, 4H, 2CH2), 4.56 (d, JHP = 12.0 Hz, 1H, CHP), 6.84 (d, J = 9.0 Hz, 2H, H-Ar), 7.33–7.23 (m, 2H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 26.7 (CH2), 29.3 (CH2), 44.9 (ClCH2), 55.6 (OCH3), 62.6 (d, JCP = 8.0, CH2OP), 63.1 (d, JCP = 8.0, CH2OP), 71.6 (d, JCP = 13.6, CH2OCHP), 77.5 (d, JCP = 172.1, CHP), 113.6, 114.0, 125.1, 129.7, 129.9, 159.8 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.1 (P). Found, %: C, 52.56; H, 7.03. For C16H26ClO5P (364.1). Calculated, %: C, 52.68; H, 7.18.Diethyl benzyloxy(4-methoxyphenyl)methylphosphonate (6e)
0.33 g, yield 90% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.20 (t, J = 6.0 Hz, 3H, CH3), 1.27 (t, J = 6.0 Hz, 3H, CH3), 3.82 (s, 3H, OCH3), 3.92–4.13 (m, 4H, 2CH2), 4.37 (d, JHP = 12.0 Hz, 1H, CHP), 4.60–4.71 (m, 2H, CH2), 6.92 (d, J = 6.0 Hz, 2H, H-Ar), 7.31–40 (m, 7H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 55.3 (OCH3), 62.9 (d, JCP = 6.0, CH2OP), 63.2 (d, JCP = 6.0, CH2OP), 71.4 (d, JCP = 15.1, CH2OCHP), 76.4 (d, JCP = 172.9, CHP), 113.9, 114.0, 126.2, 128.0, 128.2, 128.4, 129.6, 129.7, 137.1, 159.9 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.6 (P). Found, %: C, 62.48; H, 6.76. For C19H25O5P (364.1). Calculated, %: C, 62.63; H, 6.92.Diethyl 2-phenylethyloxy(4-methoxyphenyl)methyl phosphonate (6f)
0.15 g, yield 39% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.13–1.21 (m, 6H, 2CH3), 2.84–2.89 (m, 2H, CH2), 3.59–3.69 (m, 2H, CH2), 3.73 (s, 3H, OCH3), 3.89–4.01 (m, 4H, 2CH2), 4.56 (d, JHP = 15.0 Hz, 1H, CHP), 6.83 (d, J = 9.0 Hz, 2H, H-Ar), 7.14–7.28 (m, 7H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 36.2 (CH2), 55.7 (OCH3), 62.8 (d, JCP = 8.0, CH2OP), 63.2 (d, JCP = 8.0, CH2OP), 71.3 (CH2) (d, JCP = 13.6, CH2OCHP), 78.4 (d, JCP = 170.6, CHP), 113.7, 113.8, 126.2, 126.5, 128.2, 128.9, 129.3, 129.4, 138.6, 159.7, 159.8, 163.6 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.1 (P). Found, %: C, 63.26; H, 7.13. For C20H27O5P (378.2). Calculated, %: C, 63.48; H, 7.19.Diethyl 2-methylpropyloxy(3,4-dimethoxyphenyl)methyl phosphonate (7a)
0.23 g, yield 65% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 0.86 (d, J = 3.0 Hz, 3H, CH3), 0.89 (d, J = 3.0 Hz, 3H, CH3), 1.19–1.23 (m, 6H, 2CH3), 1.80–1.91 (m, 1H, CH), 3.12–3.24 (m, 2H, CH2), 3.83 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 4.01–4.07 (m, 4H, 2CH2), 4.48 (d, JHP = 18.0 Hz, 1H, CHP), 6.79–6.92 (m, 2H, H-Ar), 7.00 (s, 1H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 5.3, CH3), 16.4 (d, JCP = 5.3, CH3), 19.1 (CH3), 19.2 (CH3), 28.4 (CH), 55.8 (2 OCH3), 62.7 (d, JCP = 6.8, CH2OP), 63.0 (d, JCP = 6.8, CH2OP), 78.7 (d, JCP = 160.0, CHP), 76.9 (d, JCP = 32.5, CH2OCHP), 110.7, 110.8, 111.0, 120.6, 127.4, 149.0 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.4 (P). Found, %: C, 56.64; H, 8.07. For C17H29O6P (360.2). Calculated, %: C, 56.66; H, 8.11.Diethyl 1-methylethyloxy(3,4-dimethoxyphenyl)methyl phosphonate (7b)
0.26 g, yield 74% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.08 (d, J = 6.0 Hz, 6H, 2CH3), 1.11–1.22 (m, 6H, 2CH3), 3.54–3.62 (m, 1H, CH), 3.80 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 3.92–4.06 (m, 4H, 2CH2), 4.59 (d, JHP = 18.0 Hz, 1H, CHP), 6.76–6.91 (m, 2H, H-Ar), 6.99 (s, 1H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 5.3, CH3), 16.4 (d, JCP = 5.3, CH3), 21.0 (CH3), 23.0 (CH3), 55.7 (OCH3), 55.8 (OCH3), 62.8 (d, JCP = 6.8, CH2OP), 63.1 (d, JCP = 6.8, CH2OP), 71.5 (OCH) (d, JCP = 13.6, CHOCHP), 75.6 (d, JCP = 167.6, CHP), 110.7, 110.8, 111.0, 120.4, 128.1, 148.8 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.8 (P). Found, %: C, 55.39; H, 7.67. For C16H27O6P (346.2). Calculated, %: C, 55.48; H, 7.86.Diethyl propargyloxy(3,4-dimethoxyphenyl)methyl phosphonate (7c)
0.19 g, yield 56% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.05 (t, J = 6.0 Hz, 3H, CH3), 1.15 (t, J = 6.0 Hz, 3H, CH3), 2.39 (s, 1H, CH), 3.72 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 3.87–3.94 (m, 2H, CH2), 3.99–4.17 (m, 4H, 2CH2), 4.75 (d, JHP = 15.0 Hz, 1H, CHP), 6.70–6.91 (m, 3H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.2 (d, JCP = 6.0, CH3), 16.3 (d, JCP = 6.0, CH3), 55.7 (OCH3), 55.8 (OCH3), 56.3 (d, JCP = 15.9, CH2OCHP), 62.8 (d, JCP = 6.8, CH2OP), 63.2 (d, JCP = 6.8, CH2OP), 75.6 (CCH), 75.7 (d, JCP = 172.1, CHP), 78.4 (CCH), 110.8, 111.2, 121.3, 125.6, 148.9, 149.4 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.0 (P). Found, %: C, 55.95; H, 6.60. For C16H23O6P (342.1). Calculated, %: C, 56.14; H, 6.77.
Diethyl 4-chlorobutyloxy(3,4-dimethoxyphenyl)methyl phosphonate (7d)
0.27 g, yield 69% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.19 (t, J = 6.0 Hz, 3H, CH3), 1.34 (t, J = 6.0 Hz, 3H, CH3), 1.50–1.72 (m, 4H, 2CH2), 3.51–3.70 (m, 2H, CH2), 3.88 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 4.02–4.10 (m, 2H, CH2), 4.53 (d, JHP = 15.0 Hz, 1H, CHP), 6.71–6.87 (m, 2H, H-Ar), 7.63 (s, 1H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 27.3 (CH2), 29.3 (CH2), 44.7 (ClCH2), 55.4 (OCH3), 56.9 (OCH3), 62.6 (d, JCP = 6.8, CH2OP), 63.5 (d, JCP = 6.8, CH2OP), 71.4 (d, JCP = 13.6, CH2OCHP), 78.1 (d, JCP = 170.6, CHP), 110.5, 112.0, 121.8, 126.4, 149.0, 149.7 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.2 (P). Found, %: C, 51.56; H, 7.01. For C17H28ClO6P (394.1). Calculated, %: C, 51.71; H, 7.15.Diethyl benzyloxy(3,4-dimethoxyphenyl)methyl phosphonate (7e)
0.32 g, yield 81% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.17 (t, J = 6.0 Hz, 3H, CH3), 1.25 (t, J = 6.0 Hz, 3H, CH3), 3.86 (s, 3H, OCH3), 3.99 (s, 3H, OCH3), 4.02–4.12 (m, 4H, 2CH2), 4.37 (d, JHP = 12.0 Hz, 1H, CHP), 4.61 (dd, J = 9.0, 12.0 Hz, 2H, CH2), 6.83 (d, J = 9.0 Hz, 1H, H-Ar), 6.94–6.96 (m, 1H, H-Ar), 7.05 (s, 1H, H-Ar), 7.29 (s, 5H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 55.8 (OCH3), 55.9 (OCH3), 62.7 (d, JCP = 6.8, CH2OP), 63.2 (d, JCP = 6.8, CH2OP), 71.4 (d, JCP = 14.3, CH2OCHP), 76.6 (d, JCP = 171.4, CHP), 110.8, 110.9, 111.2, 111.3, 121.1, 121.2, 126.7, 127.9, 128.2, 128.3, 137.1, 149.1 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.5 (P). Found, %: C, 60.87; H, 6.74. For C20H27O6P (394.2). Calculated, %: C, 60.91; H, 6.90.Diethyl 2-phenylethyloxy(3,4-dimethoxyphenyl)methyl phosphonate (7f)
0.28 g, yield 69% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.21 (t, J = 6.0 Hz, 3H, CH3), 1.26 (t, J = 6.0 Hz, 3H, CH3), 2.90 (t, J = 9.0, 6.0 Hz, 2H, CH2), 3.63–3.79 (m, 2H, CH2), 3.85 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 3.97–4.04 (s, 4H, 2CH2), 4.55 (d, JHP = 15.0 Hz, 1H, CHP), 6.79–6.93 (m, 3H, H-Ar), 7.14–7.28 (m, 5H, H-Ar). 13C-NMR (75.0 MHz, CDCl3) δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 36.2 (CH2), 55.8 (OCH3), 55.9 (OCH3), 62.8 (d, JCP = 6.8, CH2OP), 63.2 (d, JCP = 6.8, CH2OP), 71.3 (d, JCP = 14.3, CH2OCHP), 78.6 (d, JCP = 169.9, CHP), 110.7, 110.8, 110.9, 120.7, 120.8, 126.2, 127.1, 128.2, 128.9, 138.7, 149.0, 149.2 (C-Ar). 31P-NMR (194.4 MHz, CDCl3) δ, ppm: 19.1 (P). Found, %: C, 61.52; H, 7.08. For C21H29O6P (408.2). Calculated, %: C, 61.76; H, 7.16.Diethyl 2-methylpropyloxy(3-methoxy-4-propargyloxyphenyl)methyl phosphonate (8a)
0.25 g, yield 66% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 0.90 (t, J = 3.0 Hz, 6H, 2CH3), 1.21–1.27 (m, 6H, 2CH3), 1.82–1.95 (m, 1H, CH), 2.50 (d, J = 3.0 Hz, 1H, CH), 3.15–3.27 (m, 2H, CH2), 3.86 (s, 3H, OCH3), 3.98–4.09 (m, 4H, 2CH2), 4.51 (d, JHP = 15.0 Hz, 1H, CHP), 4.73 (d, J = 3.0 Hz, 2H, CH2), 6.90–7.04 (m, 3H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 5.3, CH3), 16.4 (d, JCP = 5.3, CH3), 19.2 (CH3), 19.3 (CH3), 28.4 (CH), 55.8 (OCH3), 56.8 (OCH2), 62.8 (d, JCP = 6.8, CH2OP), 63.1 (d, JCP = 6.8, CH2OP), 75.8 (CCH), 76.9 (d, JCP = 31.7, CH2OCHP), 77.7 (CCH), 78.6 (d, JCP = 160.6, CHP), 111.3, 113.9, 120.5, 128.9, 146.8, 149.6 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.3 (P). Found, %: C, 59.23; H, 7.46. For C19H29O6P (384.2). Calculated, %: C, 59.37; H, 7.60.
Diethyl 1-methylethyloxy(3-methoxy-4-propargyloxyphenyl)methyl phosphonate (8b)
0.34 g, yield 89% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.17–1.25 (m, 12H, 4CH3), 2.49 (d, J = 3.0 Hz, 1H, CH), 3.58–3.66 (m, 1H, CH), 3.84 (s, 3H, OCH3), 3.93–4.09 (m, 4H, 2CH2), 4.63 (d, JHP = 18.0 Hz, 1H, CHP), 4.70 (d, J = 3.0 Hz, 2H, CH2), 6.89–7.04 (m, 3H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 21.0 (CH3), 23.1 (CH3), 55.9 (OCH3), 56.8 (OCH2), 62.8 (d, JCP = 7.8, CH2OP), 63.3 (d, JCP = 7.8, CH2OP), 71.6 (d, JCP = 13.6, CHOCHP), 75.8 (CCH), 75.6 (d, JCP = 171.4, CHP), 78.5 (CCH), 111.3, 111.5, 113.9, 120.4, 129.6, 146.8 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.8 (P). Found, %: C, 59.22; H, 7.46. For C19H29O6P (384.2). Calculated, %: C, 59.37; H, 7.60.
Diethyl propargyloxy(3-methoxy-4-propargyloxyphenyl)methyl phosphonate (8c)
0.27 g, yield 74% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.11 (t, J = 6.0 Hz, 3H, CH3), 1.20 (t, J = 6.0 Hz, 3H, CH3), 2.42 (t, J = 3.0 Hz, 1H, CH), 2.47 (t, J = 3.0 Hz, 1H, CH), 3.78 (s, 3H, OCH3), 3.82–3.94 (m, 2H, CH2), 3.98–4.08 (m, 4H, 2CH2), 4.65 (d, J = 3.0 Hz, 2H, OCH2), 4.81 (d, JHP = 15.0 Hz, 1H, CHP), 6.86–6.98 (m, 3H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.2 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 55.9 (OCH3), 56.5 (d, JCP = 15.9, CH2OCHP), 56.9 (OCH2), 62.9 (d, JCP = 6.8, CH2OP), 63.2 (d, JCP = 6.8, CH2OP), 75.7 (d, JCP = 169.1, CHP), 75.7 (CCH), 76.0 (CCH), 78.3 (CCH), 78.5 (CCH), 111.6, 113.9, 121.1, 127.1, 147.2, 149.7 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 18.9 (P). Found, %: C, 58.92; H, 6.29. For C18H23O6P (366.1). Calculated, %: C, 59.01; H, 6.33.
Diethyl 4-chlorobutyloxy(3-methoxy-4-propargyloxyphenyl)methyl phosphonate (8d)
0.26 g, yield 62% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.15–1.48 (m, 6H, 2CH3), 1.58–1.89 (m, 4H, 2CH2), 2.51 (t, J = 3.0 Hz, 1H, CH), 3.41–3.61 (m, 4H, 2CH2), 3.84 (s, 3H, OCH3), 3.99–4.06 (m, 4H, 2CH2), 4.48 (d, JHP = 12.0 Hz, 1H, CHP), 4.71 (d, J = 3.0 Hz, 2H, OCH2), 6.89–7.13 (m, 3H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 26.9 (CH2), 29.3 (CH2), 44.7 (CH2), 55.9 (OCH3), 56.0 (OCH3), 56.7 (OCH2), 62.9 (d, JCP = 6.8, CH2OP), 63.1 (d, JCP = 6.8, CH2OP), 69.9 (d, JCP = 13.6, CH2OCHP), 75.9 (CCH), 78.4 (d, JCP = 170.6, CHP), 78.6 (CCH), 110.8, 112.0, 121.8, 126.4, 149.0, 149.7 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.2 (P). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.2 (P). Found, %: C, 54.34; H, 6.65. For C19H28ClO6P (418.1). Calculated, %: C, 54.48; H, 6.74.
Diethyl benzyloxy(3-methoxy-4-propargyloxyphenyl)methyl phosphonate (8e)
0.36 g, yield 87% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.19 (d, J = 6.0 Hz, 3H, CH3), 1.24 (d, J = 6.0 Hz, 3H, CH3), 2.52 (d, J = 3.0 Hz, 1H, CH), 3.87 (s, 3H, OCH3), 3.90–4.13 (m, 4H, 2CH2), 4.38 (d, J = 12.0 Hz, 1H, CH), 4.40 (d, JHP = 12.0 Hz, 1H, CHP), 4.67 (d, J = 12.0 Hz, 1H, CH), 4.77 (d, J = 3.0 Hz, 2H, CH2), 6.94–6.98 (m, 3H, H-Ar), 7.06–7.32 (m, 5H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.5 (d, JCP = 6.0, CH3), 55.9 (OCH3), 56.8 (OCH2), 62.8 (d, JCP = 6.8, CH2OP), 63.1 (d, JCP = 6.8, CH2OP), 71.5 (d, JCP = 14.3, CH2OCHP), 75.9 (CCH), 76.6 (d, JCP = 170.6, CHP), 78.5 (CCH), 111.6, 111.7, 113.9, 120.8, 120.9, 127.9, 128.2, 128.4, 137.1, 147.1, 149.8 (C-Ar), 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.4 (P). Found, %: C, 63.03; H, 6.48. For C22H27O6P (418.2). Calculated, %: C, 63.15; H, 6.50.
Diethyl 2-phenylethyloxy(3-methoxy-4-propargyloxyphenyl)methyl phosphonate (8f)
0.35 g, yield 82% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.16–1.24 (m, 6H, 2CH3), 2.51 (s, 1H, CH), 2.88–2.93 (m, 2H, CH2), 3.67–3.71 (m, 2H, CH2), 3.79 (s, 3H, OCH3), 3.92–4.04 (m, 4H, 2CH2), 4.56 (d, JHP = 15.0 Hz, 1H, CHP), 4.73 (d, J = 3.0 Hz, 2H, CH2), 6.86–6.98 (m, 3H, H-Ar), 7.17–7.38 (m, 5H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 36.2 (CH2), 55.8 (OCH3), 56.8 (OCH2), 62.9 (d, JCP = 6.8, CH2OP), 63.2 (d, JCP = 6.8, CH2OP), 71.5 (d, JCP = 13.6, CH2OCHP), 75.8 (CCH), 78.5 (CCH), 78.6 (d, JCP = 169.9, CHP), 111.2, 111.3, 113.9, 120.5, 120.6, 126.2, 128.3, 128.6, 129.0, 138.7, 146.9, 149.7 (C-Ar); 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 18.9 (P). Found, %: C, 63.69; H, 6.72. For C23H29O6P (432.2). Calculated, %: C, 63.88; H, 6.76.
Diethyl((5-methyl-2-phenyl-1,3-dioxan-5-yl)methoxy)(4-methoxyphenyl)methyl phosphonate (10a)
0.29 g, yield 62% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 0.87 (s, 3H, CH3), 1.20–1.33 (m, 6H, 2CH3), 3.59–3.73 (m, 4H, 2CH2), 3.81 (s, 3H, OCH3), 3.88–4.16 (m, 6H, 3CH2), 4.65 (d, JHP = 15.0 Hz, 1H, CHP), 5.39 (s, IH, CH), 6.90 (d, J = 9.0 Hz, 2H, H-Ar), 7.32–7.43 (m, 7H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.4 (d, JCP = 5.3, CH3), 16.5 (d, JCP = 5.3, CH3), 19.2 (CH3), 34.6 (C), 55.7 (OCH3), 62.6 (d, JCP = 7.8, CH2OP), 63.1 (d, JCP = 7.8, CH2OP), 72.8 (OCH2), 73.0 (d, JCP = 14.3, CH2OCHP), 73.7 (OCH2), 79.0 (d, JCP = 170.6, CHP), 101.7 (OCH), 113.9, 114.0, 126.1, 126.2, 128.2, 128.9, 129.5, 129.6, 138.3, 159.8 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.2 (P). Found, %: C, 61.95; H, 7.14. For C24H33O7P (464.2). Calculated, %: C, 62.06; H, 7.16.Diethyl((5-methyl-2-phenyl-1,3-dioxan-5-yl)methoxy)(3,4-dimethoxyphenyl)-methyl phosphonate (10b)
0.35 g, yield 70% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 0.87 (s, 3H, CH3), 1.19 (t, J = 6.0 Hz, 3H, CH3), 1.26 (t, J = 6.0 Hz, 3H, CH3), 3.63–3.75 (m, 4H, 2CH2), 3.59–3.79 (m, 2H, CH2), 3.88 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 4.01–4.15 (m, 4H, 2CH2), 4.63 (d, JHP = 15.0 Hz, 1H, CHP), 5.38 (s, IH, CH), 6.84–7.00 (m, 2H, H-Ar), 7.05 (s, 1H, H-Ar), 7.28–7.37 (m, 5H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.4 (d, JCP = 6.0, CH3), 16.5 (d, JCP = 6.0, CH3), 19.1 (CH3), 35.0 (C), 55.8 (OCH3), 55.9 (OCH3), 62.8 (d, JCP = 7.8, CH2OP), 63.1 (d, JCP = 7.8 CH2OP), 72.9 (d, JCP = 14.3, CH2OCHP), 73.2 (OCH2), 73.3 (OCH2), 79.0 (d, JCP = 170.6, CHP), 101.8 (OCH), 110.8, 111.1, 111.2, 120.8, 120.9, 126.1, 128.2, 128.3, 128.9, 138.3, 149.0, 149.3 (C-Ar). 31P-NMR (194.4 MHz, CDCl3), δ, ppm: 19.2 (P). Found, %: C, 60.62; H, 6.85. For C25H35O8P (494.2). Calculated, %: C, 60.72; H, 7.13.Diethyl 2-(N-methyl-N-phenylamino)ethyloxy(4-methoxyphenyl)methyl phosphonate (12)
0.12 g, yield 29% colorless oil. 1H NMR spectrum, (300 MHz, CDCl3), δ, ppm (J, Hz): 1.21 (t, J = 6.0 Hz, 3H, CH3), 1.26 (t, J = 6.0 Hz, 3H, CH3), 2.98 (s, 3H, CH3), 3.57–3.68 (m, 4H, 2CH2), 3.82 (s, 3H, OCH3), 3.93–4.11 (m, 4H, 2CH2), 4.58 (d, JHP = 15.0 Hz, 1H, CHP), 6.68 (d, J = 9.0 Hz, 2H, H-Ar), 6.87 (d, J = 9.0 Hz, 2H, H-Ar), 7.18–7.35 (m, 5H, H-Ar). 13C-NMR (75.0 MHz, CDCl3), δ, ppm: 16.3 (d, JCP = 6.0, CH3), 16.4 (d, JCP = 6.0, CH3), 36.2 (CH3), 55.7 (OCH3), 55.8 (NCH2), 62.8 (d, JCP = 7.8, CH2OP), 63.6 (d, JCP = 7.8, CH2OP), 71.4 (d, JCP = 14.3, CH2OCHP), 78.7 (d, JCP = 163.1, CHP), 110.7, 110.8, 110.9, 120.7, 120.8, 126.1, 127.1, 128.2, 128.9, 138.7, 149.0 (C-Ar). 31P-NMR (194.4 MHz, CDCl3) δ, ppm: 19.1 (P). Found, %: C, 61.87; H, 7.38; N, 3.44. For C21H30NO5P (407.2). Calculated, %: C, 61.90; H, 7.42; N, 3.44.Conflicts of interest
There are no conflicts to declare.References
- R. Engel, Chem. Rev., 1977, 77, 349–367 CrossRef CAS.
- R. L. Hilderbrand, The Role of Phosphonates in Living Systems, CRC press, Boca Raton, FL, 1983 Search PubMed.
- D. J. Schauer and P. Helquist, Synthesis, 2006, 3654–3660 CAS.
- D. C. Reuter, J. E. McIntosh, A. C. Guinn and A. M. Madera, Synthesis, 2003, 2321–2324 CrossRef CAS.
- U. S. Dakarapu, A. Bokka, P. Asgari, G. Trog, Y. Hua, H. H. Nguyen, N. Rahman and J. Jeon, Org. Lett., 2015, 17, 5792–5795 CrossRef CAS PubMed.
- A. K. Bhattacharya and G. Thyagarajan, Chem. Rev., 1981, 81, 415–430 CrossRef CAS.
- B. A. Arbuzov, Pure Appl. Chem., 1964, 9, 307–336 CAS.
- Y. Belabassi, S. Alzghari and J. L. Montchamp, J. Organomet. Chem., 2008, 693, 3171–3178 CrossRef CAS PubMed.
- R. A. Dhokale and S. B. Mhaske, Org. Lett., 2013, 15, 2218–2221 CrossRef CAS PubMed.
- H. Firouzabadi, N. Iranpoor, S. Sobhani and Z. Amoozgar, Synthesis, 2004, 1771–1774 CrossRef CAS.
- B. Kaboudin, Tetrahedron Lett., 2003, 44, 1051–1053 CrossRef CAS.
- R. L. Hilderbrand, The Role of Phosphonates in Living Systems, CRC press, Boca Raton, FL, 1983 Search PubMed.
- R. U. Pokalwar, R. V. Hangarge, P. V. Maske and M. S. Shingare, ARKIVOC, 2006, xi, 196–204 Search PubMed.
- D. Q. Shi, Z. L. Sheng, X. P. Liu and H. Wu, Heteroat. Chem., 2003, 14, 266–268 CrossRef CAS.
- A. J. Ganzhorn, J. Hoflack, P. D. Pelton, F. Strasser, M. C. Chanal and S. R. Piettre, Bioorg. Med. Chem., 1998, 6, 1865–1874 CrossRef CAS PubMed.
- J. Guin, Q. Wang, M. Gemmeren and B. List, Angew. Chem., Int. Ed., 2015, 54, 355–358 CrossRef CAS PubMed.
- H. I. Hansen and J. Kehler, Synthesis, 1999, 1925–1927 CrossRef CAS.
- R. R. Schmidt and W. Kinzy, Adv. Carbohydr. Chem. Biochem., 1994, 50, 21–123 CrossRef CAS PubMed.
- R. R. Schmidt and K.-H. Jung, in Preparative Carbohydrate Chemistry, ed. S. Hannessian, Marcel Dekker, Inc., New York, 1997, pp. 283–312 Search PubMed.
- I. A. I. Ali and W. Fathalla, ARKIVOC, 2009, xiii, 193–199 Search PubMed.
- W. Fathalla, Sci. J. Chem., 2014, 2, 38–43 CrossRef CAS.
- I. A. I. Ali and W. Fathalla, Curr. Org. Chem., 2013, 17, 1903–1909 CrossRef CAS.
- D. Rhodes and R. F. Newton, US Pat. 4085222 A, US 05/523,238, 1978.
- L. Gavara, T. Boisse, J.-P. Hénichart, A. Daïch, B. Rigo and P. Gautret, Tetrahedron, 2010, 66, 7544–7561 CrossRef CAS.
- J. Manfred, A. Dieter, L. Reinhard, F. Rainer, R. Hans-Jochem, S. Rolf and H. Horst, US Pat. 4327025 A1, Bayer Aktiengesellschaft, 1982.
| This journal is © The Royal Society of Chemistry 2018 |