
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Convenient and efficient synthesis of functionalized unsymmetrical Z-alkenyl disulfanes†
M. Musiejuk,
J. Doroszuk and
D. Witt *
*
Department of Organic Chemistry, Faculty of Chemistry, Gdansk University of Technology, Narutowicza 11/12, 80-233 Gdansk, Poland. E-mail: chemwitt@pg.gda.pl; Fax: +48 58 3472694
First published on 7th March 2018
Abstract
We developed a simple and efficient method for the synthesis of functionalized unsymmetrical Z-alkenyl disulfanes under mild conditions in moderate to good yields. The designed method is based on the reaction of Z-alkenyl thiotosylates with thiols in the presence of base. The developed method allows the preparation of unsymmetrical Z-alkenyl disulfanes bearing additional hydroxy, carboxy, or amino functionalities.
Introduction
Compounds with R-S-S-R structures, where the R groups are alkyl, vinyl or aryl, are known as symmetrical disulfides if the R groups are the same. A large number of unsymmetrical disulfides, in which the R groups are different, are also well known. In the literature, these compounds are often called organic disulfides; however, the IUPAC recommended nomenclature is disulfanes.1 The name disulfide should only be applied to ionic compounds, such as sodium disulfide (Na2S2). Moreover, the term disulfane is more widely applicable than disulfide because it facilitates naming even when the R groups are acyl and/or phosphoryl groups.The formation of unsymmetrical disulfanes is an important transformation in organic synthesis and medicinal chemistry.2 Recent developments in disulfide bond formation reactions have been reviewed.3 Although many different methods exist for the preparation of unsymmetrical disulfanes, the most common approach involves substitution of a sulfenyl derivative with a thiol or thiol derivative. To date, the most commonly utilized sulfenyl derivatives are sulfenyl chlorides,4 S-alkyl thiosulfates and S-aryl thiosulfates (Bunte salts),5 S-alkylsulfanylisothioureas,6 benzothiazol-2-yl disulfanes,7 benzotriazolyl sulfanes,8 dithioperoxyesters,9 (alkylsulfanyl)dialkylsulfonium salts,10 2-pyridyl disulfanes and derivatives,11 N-alkyltetrazolyl disulfanes,12 sulfenamides,13 sulfenyldimesylamines,14 sulfenyl thiocyanates,15 4-nitroarenesulfenanilides,16 thiolsulfinates and thiosulfonates,17 sulfanylsulfinamidines,18 thionitrites,19 sulfenyl thiocarbonates,20 thioimides,21 and thiophosphonium salts.22 Other practical procedures involve the reaction of a thiol with a sulfinylbenzimidazole,23 a rhodium-catalyzed disulfide exchange,24 an electrochemical method,25 the ring opening of an aziridine using tetrathiomolybdate in the presence of a symmetrical disulfane,26 or the use of diethyl azodicarboxylate (DEAD)27 or a solid support28 in a sequential coupling of two different thiol groups. Recently, the oxidation of a mixture of two different thiols by 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) to produce an unsymmetrical disulfane has also been reported.29
Earlier studies demonstrated the preparation of functionalized unsymmetrical molecules, such as dialkyl disulfanes,30 alkyl-aryl disulfanes,31 ‘bioresistant’ disulfanes,32 the unsymmetrical disulfanes of L-cysteine and L-cystine,33 and diaryl disulfanes34 based on the readily available 5,5-dimethyl-2-thioxo-1,3,2-dioxaphosphorinane-2-disulfanyl derivatives. These disulfanyl derivatives of phosphorodithioic acid were convenient for the preparation of α-sulfenylated carbonyl compounds,35 functionalized phosphorothioates,36 and unsymmetrical alkynyl sulfides37 as well as symmetrical38 and unsymmetrical39 trisulfanes.
Ajoene was first isolated by Block40 in 1984 as an E/Z-mixture of a rearrangement product of allicin produced from freshly crushed garlic. It was established to be an allyl sulfoxide containing an unusual vinyl disulfane functionality, which is rarely seen in the structures of natural products. Z-Ajoene is more active than its E-isomer as an anti-thrombotic agent,41 and some studies on anticancer treatments have focused primarily on the Z-isomer.42
Although many different synthetic methods exist for the preparation of unsymmetrical disulfanes, the preparation of unsymmetrical alkenyl disulfanes can be achieved by only two methods. The first method is based on the reaction of sulfenyl bromide with trityl-alkenyl sulfide.43 The second method involves the low temperature hydroxide-promoted cleavage of an alkenyl thioester followed by sulfenylation with an appropriate S-alkylated p-toluenethiosulfonate to afford vinyl disulfide in high yield after chromatography.44 Unfortunately, the methods provide exclusively E or a mixture of Z/E alkenyl disulfanes, respectively. In this context, we set out to investigate the feasibility of a more convenient and experimentally practical diastereoselective method to exclusively access Z-alkenyl disulfanes.
Results and discussion
Our synthetic strategy included the preparation of E-alkenylboronic acid45 2 from terminal alkyne 1 followed by its conversion to appropriate E-alkenyliodonium salt46 3 by known methods. Further reaction with sodium p-toluenethiosulfonate provided Z-1-octenyl p-toluenethiosulfonate 4 with inversion of configuration (Scheme 1).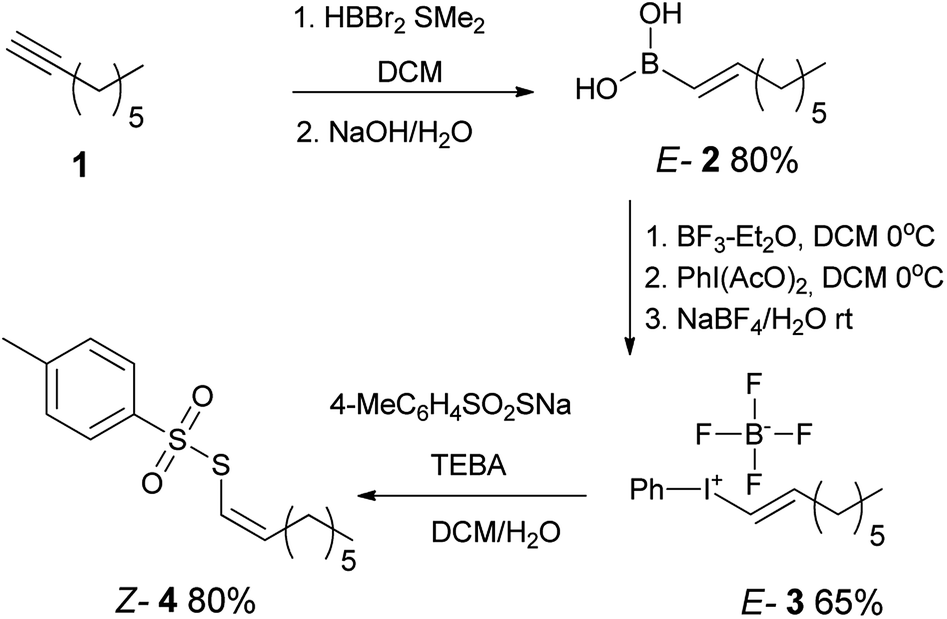 | ||
| Scheme 1 Preparation of Z-1-octenyl p-toluenethiosulfonate 4. | ||
The reaction of Z-1-octenyl p-toluenethiosulfonate 4 with a variety of thiols in the presence of NEt3 provided Z-alkenyl disulfanes 6 in good or very good yield (Table 1). All compounds have been fully characterized by 1H and 13C NMR spectroscopy (see the ESI†). E/Z-Stereochemistry was assigned based on the vinyl coupling constants in the 1H NMR spectra; 15 Hz was indicative of the E-isomer and 10 Hz for the Z-isomer.
|
|||||
|---|---|---|---|---|---|
| Entry | R | X | Solvent | Yieldb (%) | |
| a Performed with 4 (0.67 mmol), 5 (0.61 mmol), NEt3 (0.61 mmol) in solvent (5 mL), 15 min.b isolated yield.c Performed with 4 (1.22 mmol), 5 (0.61 mmol), NEt3 (0.61 mmol) in solvent (5 mL), 15 min. | |||||
| 1 | 5a | –C12H25 | H | CH2Cl2 | 6a (90) |
| 2 | 5b | –(CH2)11OH | H | CH2Cl2 | 6b (82) |
| 3 | 5c | (CH2)10CO2Me | H | CH2Cl2 | 6c (88) |
| 4 | 5d | –(CH2)11N3 | H | CH2Cl2 | 6d (70) |
| 5 | 5e | –(CH2)11NH2 | H | CH2Cl2 | 6e (77) |
| 6 | 5f | 4-MeC6H4- | H | CH2Cl2 | 6f (40) |
| 7 | 5f | 4-MeC6H4- | H | CH2Cl2 | 6f (62)c |
| 8 | 5g | 2-Furyl–CH2– | H | CH2Cl2 | 6g (71) |
| 9 | 5h | 4-Py– | H | CH2Cl2 | 6h (52) |
| 10 | 5i | CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2- CHCH2- |
H | CH2Cl2 | 6i (51) |
| 11 | 5i | CH2CHCH2- |
Ac | MeOH | 6i (80) |
| 12 | 5j | HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2- CCH2- |
Ac | MeOH | 6j (78) |
| 13 | 5k | H-CysOEt | H | CH2Cl2 | 6k (60) |
| 14 | 5l | BocCysOEt | H | CH2Cl2 | 6l (59) |
| 15 | 5m | 4-MeOC6H4- | H | CH2Cl2 | 6m (43) |
| 16 | 5m | 4-MeOC6H4- | H | CH2Cl2 | 6m (62)c |
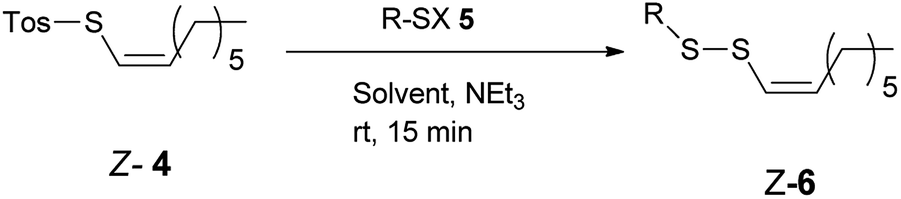
The reaction proceeded via the nucleophilic substitution of the thiolate anion (generated from 5) at the sulfur atom of thiotosylate 4, and the p-toluenesulfinate anion served as the leaving group, which is why the Z geometry of the alkene remained unchanged. The thiolate anion can also be generated in situ from the corresponding thioacetate and sodium methoxide in methanol (entries 11–12). Such an approach is very convenient when a high-purity or stable thiol is not readily available. The developed method seems to be very versatile. The presence of additional functional groups including carbon–carbon multiple bonds (entries 10–12) and hydroxy (entry 2), ester (entry 3), azide (entry 4), amino (entry 5) aryl or heteroaryl (entries 6–9 and 15) moieties did not interfere with the formation of Z-alkenyl disulfanes 6. Arylthiol 5 – disulfane Z-6 exchange reaction was responsible for the formation of corresponding diaryl disulfane and moderate yield of 6f and 6m (entries 6 and 15). The exchange reaction can be limited by the excess of Z-4, what resulted in higher yield of 6f and 6m respectively (entries 7 and 16). L-Cysteine derivatives were also converted to the corresponding Z-alkenyl disulfanes 6k and 6l (entries 13–14). The biological activities of these compounds are expected to be higher than their E-isomer analogs.43,44
Conclusions
We have developed the first simple and efficient diastereoselective method for the synthesis of functionalized unsymmetrical Z-alkenyl disulfanes under mild conditions in moderate to good yields. The developed method allows the preparation of unsymmetrical Z-alkenyl disulfanes bearing additional hydroxy, carboxy, or amino functionalities. Anti-fungal and anti-cancer activity studies of the Z-alkenyl L-cysteine disulfane derivatives are in progress.Experimental
A typical procedure for the preparation of Z-alkenyl disulfanes 6 and representative analytical data
A compound Z-4 (0.67 mmol, 200 mg) was dissolved in dry DCM (3 mL) in the round bottom flask. Then a solution of thiol 5 (0.61 mmol) and NEt3 (0.61 mmol) in dry DCM (2 mL) was added. Reaction was stirred for 15 min. After this time solvent was evaporate and Et2O (10 mL) was added. Slurry was washed with water (10 mL) and aqueous phase was extracted 2 times with Et2O (2 × 10 mL). Organic layers were dried with MgSO4 and evaporated. The residue was purified by column chromatography (SiO2).(Z)-1-(dodec-1-yldisulfanyl)-oct-1-ene Z-6a
Chromatography, PE, Rf = 0.6; a colorless oil, yield 188 mg (90%) IR (ATR): 510 (s), 625 (m), 1490 (w), 2875 (m), 2900 (m) cm−1. 1H NMR (400 MHz, CDCl3) δ 6.09 (dt, J = 9.3, 1.4 Hz, 1H), 5.65 (dt, J = 9.3, 7.4 Hz, 1H), 2.72 (t, J = 7.3 Hz, 2H), 2.19 (qd, J = 7.4, 1.3 Hz, 2H), 1.70 (dt, J = 14.9, 7.2 Hz, 2H), 1.47–1.24 (m, 26H), 0.93–0.88 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 132.66, 129.24, 39.07, 31.93, 31.67, 29.66, 29.64, 29.60, 29.51, 29.36, 29.23, 29.08, 28.91, 28.85, 28.80, 28.46, 22.70, 22.62, 14.13, 14.09. HRMS (ESI): m/z [M + H]+ calcd for C20H41S2: 345.2650; found: 345.2655.(Z)-11-(1-octen-1-yldisulfanyl)-undecan-1-ol Z-6b
Chromatography: PE:DCM 1:1; Rf = 0.4; a colorless oil, yield 173 mg (82%) IR (ATR): 755 (w), 1100 (w), 1500 (w), 1650 (w), 2875 (s), 2900 (s), 3300 (br) cm−1. 1H NMR (400 MHz, CDCl3) δ 6.09 (dt, J = 9.3, 1.3 Hz, 1H), 5.65 (dt, J = 9.3, 7.4 Hz, 1H), 3.66 (t, J = 6.6 Hz, 2H), 2.72 (t, J = 7.4 Hz, 2H), 2.19 (qd, J = 7.4, 1.3 Hz, 2H), 1.72–1.60 (m, 2H), 1.60–1.50 (m, 2H), 1.44–1.23 (m, 23H), 0.91 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 132.68, 129.22, 63.10, 39.06, 32.81, 31.67, 29.58, 29.51, 29.48, 29.42, 29.21, 29.07, 28.90, 28.84, 28.80, 28.44, 25.74, 22.62, 14.09. HRMS (ESI): m/z [M + H]+ calcd for C19H39OS2: 347.2442; found: 347.2438.(Z)-Methyl 11-(1-octen-1-yldisulfanyl)undecanoate Z-6c
Chromatography: PE:DCM 3:1; Rf = 0.4; a colorless oil, yield 201 mg (88%): IR (ATR): 500 (s), 625 (m), 1200 (w), 1490 (w), 1750 (m), 2875 (w), 2990 (m) cm−1. 1H NMR (400 MHz, CDCl3) δ 6.09 (dt, J = 9.3, 1.3 Hz, 1H), 5.65 (dt, J = 9.3, 7.4 Hz, 1H), 3.69 (s, 3H), 2.75–2.70 (m, 2H), 2.32 (t, J = 7.6 Hz, 2H), 2.19 (qd, J = 7.4, 1.3 Hz, 2H), 1.70–1.52 (m, 4H), 1.46–1.24 (m, 20H), 0.91 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 174.32, 132.67, 129.23, 51.45, 39.05, 34.11, 31.66, 29.43, 29.37, 29.23, 29.18, 29.14, 29.07, 28.89, 28.84, 28.79, 28.43, 24.95, 22.61, 14.09. HRMS (ESI): m/z [M + H]+ calcd for C20H39O2S2: 375.2391; found: 375.2396.(Z)-1-(11-azidoundec-1-yldisulfanyl)-oct-1-ene Z-6d
Chromatography: PE; Rf = 0.45; a colorless oil, yield 159 mg (70%) IR (ATR): 510 (s), 625 (m), 1260 (w), 1500 (w), 2110 (m), 2875 (m), 2900 (m)) cm−1. 1H NMR (400 MHz, CDCl3) δ 6.09 (dt, J = 9.3, 1.3 Hz, 1H), 5.65 (dt, J = 9.3, 7.4 Hz, 1H), 3.28 (t, J = 7.0 Hz, 2H), 2.73 (t, J = 7.4 Hz, 2H), 2.19 (qd, J = 7.4, 1.3 Hz, 2H), 1.75–1.55 (m, 4H), 1.47–1.24 (m, 22H), 0.91 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 132.68, 129.22, 51.50, 39.05, 31.67, 29.46, 29.20, 29.15, 29.07, 28.89, 28.85, 28.80, 28.43, 26.72, 22.62, 14.09. HRMS (ESI): m/z [M + H]+ calcd for C19H38N3S2: 372.2507; found: 372.2511.(Z)-1-(11-aminoundec-1-yldisulfanyl)-oct-1-ene Z-6e
Chromatography: DCM: MeOH 14:1; Rf = 0.3; a colorless oil, yield 162 mg (77%) IR (ATR): 510 (s), 625 (s), 800 (w), 1100 (w), 1490 (w), 1510 (w), 2875 (m), 2900 (s), 3500 (br) cm−1. 1H NMR (400 MHz, CDCl3) δ 6.09 (dt, J = 9.3, 1.3 Hz, 1H), 5.65 (dt, J = 9.3, 7.4 Hz, 1H), 2.87–2.81 (m, 2H), 2.75–2.70 (m, 2H), 2.18 (dt, J = 7.4, 4.2 Hz, 2H), 1.75–1.55 (m, 4H), 1.47–1.24 (m, 24H), 0.91 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 132.66, 129.25, 39.75, 39.07, 31.67, 29.50, 29.41, 29.25, 29.08, 28.92, 28.84, 28.48, 27.91, 26.53, 22.62, 14.11. HRMS (ESI): m/z [M + H]+ calcd for C19H40NS2: 346.2602; found: 346.2604.(Z)-1-(p-tolyldisulfanyl)-oct-1-ene Z-6f
Chromatography: PE; Rf = 0.8; a colorless oil, yield 65 mg (40%) IR (ATR): 500 (s), 625 (m), 780 (w), 1500 (w), 2875 (w), 2990 (m) cm−1. 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 8.1 Hz, 2H), 7.15 (d, J = 8.1 Hz, 2H), 6.16 (dt, J = 9.3, 1.3 Hz, 1H), 5.75–5.65 (m, 1H), 2.36 (s, 3H), 2.19 (m, 2H), 1.46–1.25 (m, 8H), 0.91 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 137.52, 134.02, 129.74, 129.12, 127.92, 31.66, 29.08, 29.02, 28.89, 28.82, 22.61, 21.08, 14.09. HRMS (ESI): m/z [M + H]+ calcd for C15H23S2: 267.1241; found: 267.1243.(Z)-1-(furan-2-ylmethyldisulfanyl)-oct-1-ene Z-6g
Chromatography: PE; Rf = 0.7; a colorless oil, yield 111 mg (71%) IR (ATR): 600 (m), 740 (m), 770 (w), 900 (m), 1010 (m), 1125 (m), 1500 (w), 2875 (w), 2950 (m) cm−1. 1H NMR (400 MHz, CDCl3) δ 7.40 (dd, J = 1.8, 0.8 Hz, 1H), 6.34 (dd, J = 3.2, 1.8 Hz, 1H), 6.28 (d, J = 0.6 Hz, 1H), 5.84 (dt, J = 9.3, 1.3 Hz, 1H), 5.61 (dt, J = 9.3, 7.4 Hz, 1H), 3.94 (s, 2H), 2.20–2.12 (m, 2H), 1.42–1.15 (m, 8H), 0.91 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 150.16, 142.46, 133.11, 128.24, 110.64, 108.97, 35.72, 31.66, 29.05, 28.84, 28.76, 22.61, 14.08. HRMS (ESI): m/z [M + H]+ calcd for C13H21OS2: 257.1034; found: 257.1031.(Z)-1-(pyridin-4-yldisulfanyl)-oct-1-ene Z-6h
Chromatography: DCM; Rf = 0.4; a colorless oil, yield 80 mg (52%) IR (ATR): 510 (s), 625 (m), 740 (m), 770 (m), 1450 (w), 1490 (w), 1600 (s), 2875 (w), 2990 (w) cm−1. 1H NMR (400 MHz, CDCl3) δ 8.50 (d, J = 6.1 Hz, 2H), 7.41 (dd, J = 4.6, 1.6 Hz, 2H), 6.00 (dt, J = 9.2, 1.3 Hz, 1H), 5.81 (dt, J = 9.2, 7.5 Hz, 1H), 2.32 (qd, J = 7.4, 1.3 Hz, 2H), 1.52–1.23 (m, 8H), 0.93 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 149.50, 148.78, 135.62, 125.93, 120.14, 31.65, 29.09, 29.00, 28.89, 22.63, 14.09. HRMS (ESI): m/z [M + H]+ calcd for C13H20NS2: 254.1037; found: 254.1033.(Z)-1-(allyldisulfanyl)-oct-1-ene Z-6i
Chromatography: PE; Rf = 0.6; a colorless oil, yield 106 mg (80%) IR (ATR): 510 (s), 650 (s), 990 (w), 1510 (w), 2875 (w), 2990 (m) cm−1. 1H NMR (400 MHz, CDCl3) δ 6.08 (dt, J = 9.3, 1.3 Hz, 1H), 5.87 (ddt, J = 17.1, 10.0, 7.3 Hz, 1H), 5.66 (dt, J = 9.3, 7.4 Hz, 1H), 5.25–5.14 (m, 2H), 3.37 (dd, J = 7.3, 0.9 Hz, 2H), 2.19 (qd, J = 7.4, 1.3 Hz, 2H), 1.52–1.15 (m, 8H), 0.91 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 133.07, 132.99, 128.74, 118.66, 41.95, 31.66, 29.05, 28.85, 28.83, 22.61, 14.08. HRMS (ESI): m/z [M + H]+ calcd for C11H21S2: 217.1085; found: 217.1088.(Z)-1-(propargyldisulfanyl)-oct-1-ene Z-6j
Chromatography: PE; Rf = 0.55; a colorless oil, yield 102 mg (78%) IR (ATR): 625 (s), 1250 (w), 1500 (w), 2875 (w), 2990 (m), 3240 (w) cm−1. 1H NMR (400 MHz, CDCl3) δ 6.21 (dt, J = 9.3, 1.4 Hz, 1H), 5.73 (dt, J = 9.3, 7.4 Hz, 1H), 3.49 (d, J = 2.6 Hz, 2H), 2.32 (t, J = 2.6 Hz, 1H), 2.21 (qd, J = 7.4, 1.3 Hz, 2H), 1.46–1.25 (m, 8H), 0.91 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 134.23, 127.62, 79.33, 72.41, 31.64, 29.04, 28.82, 28.76, 27.14, 22.61, 14.09. HRMS (ESI): m/z [M + H]+ calcd for C11H19S2: 215.0928; found: 215.0932.(Z)-Ethyl (R)-2-amino-3-(oct-1-ene-1-yldisulfanyl)propanoate Z-6k
Chromatography: DCM: MeOH 14:1; Rf = 0.35; a colorless oil, yield 107 mg (60%) IR (ATR): 510 (s), 625 (m), 1010 (w), 1200 (w), 1500 (w), 1750 (m), 2875 (w), 2990 (w), 3500 (br) cm−1. 1H NMR (400 MHz, CDCl3) δ 6.11 (dt, J = 9.3, 1.3 Hz, 1H), 5.70 (dq, J = 8.9, 7.5 Hz, 1H), 4.22 (q, J = 7.1 Hz, 2H), 3.81 (dd, J = 8.0, 4.5 Hz, 1H), 3.15 (dd, J = 13.7, 4.5 Hz, 1H), 2.89 (dd, J = 13.7, 8.0 Hz, 1H), 2.18 (qd, J = 7.4, 1.3 Hz, 2H), 1.57–1.07 (m, 8H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 173.66, 134.02, 127.86, 61.35, 53.43, 43.90, 31.63, 29.01, 28.85, 28.80, 22.58, 14.18, 14.07. HRMS (ESI): m/z [M + H]+ calcd for C13H26NO2S2: 292.1399; found: 292.1403.(Z)-Ethyl (R)-2-((tert-butoxycarbonyl)amino)-3-(oct-1-ene-1-yldisulfanyl)propanoate Z-6l
Chromatography: PE: DCM 1:2; Rf = 0.5; a colorless oil, yield 141 mg (59%) IR (ATR): 500 (s), 625 (m), 1010 (m), 1200 (s), 1375 (m), 1625 (m), 1750 (m), 2875 (w), 2990 (w), 3000 (w) cm−1. 1H NMR (400 MHz, CDCl3) δ 6.11 (dt, J = 9.3, 1.3 Hz, 1H), 5.72 (dt, J = 9.3, 7.4 Hz, 1H), 5.35 (d, J = 7.3 Hz, 1H), 4.61 (d, J = 6.1 Hz, 1H), 4.28–4.21 (m, 2H), 3.19 (ddd, J = 19.8, 14.0, 5.2 Hz, 2H), 2.17 (qd, J = 7.4, 1.2 Hz, 2H), 1.48 (s, 9H), 1.44–1.25 (m, 11H), 0.90 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.70, 155.06, 133.93, 127.97, 80.14, 61.79, 53.43, 53.07, 41.53, 31.64, 29.04, 28.86, 28.85, 28.31, 22.59, 14.13, 14.08. HRMS (ESI): m/z [M + H]+ calcd for C18H34NO4S2: 392.1929; found: 392.1934.(Z)-1-(4-methoxylphenyldisulfanyl)-oct-1-ene Z-6m
Chromatography: PE; Rf = 0.3; a colorless oil, yield 74 mg (43%) IR (ATR): 523 (w), 823 (m), 1031 (m), 1244 (s), 1490 (s), 1589 (m), 2853 (s), 2923 (s), 3333 (br) cm−1. 1H NMR (400 MHz, CDCl3) δ 7.52–7.47 (m, 2H), 6.91–6.83 (m, 2H), 6.22 (dt, J = 9.3, 1.3 Hz, 1H), 5.71 (dt, J = 9.2, 7.4 Hz, 1H), 3.83 (s, 3H), 2.19–2.07 (m, 2H), 1.44–1.17 (m, 8H), 0.90 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 159.86, 134.14, 132.49, 128.34, 127.90, 114.61, 55.38, 31.64, 29.00, 28.83, 28.80, 22.59, 14.09. HRMS (ESI): m/z [M + H]+ calcd for C15H23OS2: 283.4725; found: 283.4729.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Science Centre (NCN) for financial support (grant no. 2015/19/B/ST5/03359).Notes and references
- R. Steudel, Chem. Rev., 2002, 102, 3905 CrossRef CAS PubMed.
- (a) R. Cremlyn and J. An, Introduction to Organosulfur Chemistry, Wiley, New York, 1996 Search PubMed; (b) S. Oae, Organic Sulfur Chemistry: Structure and Mechanism, CRC Press, Boca Raton FL, 1991 Search PubMed; (c) V. M. Vrudhula, J. F. MacMaster, L. Zhengong, D. E. Kerr and P. D. Senter, Bioorg. Med. Chem. Lett., 2002, 12, 3591 CrossRef CAS PubMed; (d) Y. Mu, M. Nodwell, J. L. Pace, J. P. Shaw and J. K. Judice, Bioorg. Med. Chem. Lett., 2004, 14, 735 CrossRef CAS PubMed.
- (a) I. Shcherbakova and A. F. Pozharskii, in Comprehensive Organic Functional Group Transformations II, ed. A. R. Katritzky, R. Taylor and Ch. Ramsden, Pergamon, Oxford, 2004, vol. 2, pp. 177–187 Search PubMed; (b) R. Sato and T. Kimura, in Science of Synthesis, ed. N. Kambe, J. Drabowicz and G. A. Molander, Thieme, Stuttgart-New York, 2007, vol. 39, pp. 573–588 Search PubMed; (c) D. Witt, Synthesis, 2008, 2491 CrossRef CAS; (d) B. Mandal and B. Basu, RSC Adv., 2014, 4, 13854 RSC; (e) M. Musiejuk and D. Witt, Org. Prep. Proced. Int., 2015, 47, 95 CrossRef CAS.
- (a) D. N. Harpp, B. T. Friedlander, C. Larsen, K. Steliou and A. Stockton, J. Org. Chem., 1978, 43, 3481 CrossRef CAS; (b) C. Brown and G. R. Evans, Tetrahedron Lett., 1996, 37, 9101 CrossRef CAS.
- (a) J. M. Swan, Nature, 1957, 180, 643 CrossRef CAS; (b) P. Hiver, A. Dicko and D. Paquer, Tetrahedron Lett., 1994, 35, 9569 CrossRef CAS.
- K. Sirakawa, O. Aki, T. Tsujikawa and T. Tsuda, Chem. Pharm. Bull., 1970, 18, 235 CrossRef CAS.
- (a) A. L. Ternay, C. Cook and E. Brzezinska, Phosphorus, Sulfur Silicon Relat. Elem., 1994, 95, 351 CrossRef; (b) A. L. Ternay and E. Brzezinska, J. Org. Chem., 1994, 59, 8239 CrossRef.
- R. Hunter, M. Caira and N. Stellenboom, J. Org. Chem., 2006, 71, 8268 CrossRef CAS PubMed.
- C. Leriverend and P. Metzner, Synthesis, 1994, 761 CrossRef CAS.
- P. Dubs and R. Stuessi, Helv. Chim. Acta, 1976, 59, 1307 CrossRef CAS.
- (a) D. H. R. Barton, C. Chen and M. G. Wall, Tetrahedron, 1991, 47, 6127 CrossRef CAS; (b) D. H. R. Barton, A. C. O'Sullivan and M. M. Pechet, J. Org. Chem., 1991, 56, 6697 CrossRef CAS.
- M. Ohtani and N. Narisada, J. Org. Chem., 1991, 56, 5475 CrossRef CAS.
- M. Bao and M. Shimizu, Tetrahedron, 2003, 59, 9655 CrossRef CAS.
- A. Blaschette and M. Naveke, Chem.-Ztg., 1991, 115, 61 CAS.
- R. G. Hiskey and B. F. Ward Jr., J. Org. Chem., 1970, 35, 1118 CrossRef CAS PubMed.
- L. Benati, P. C. Montevecchi and P. Spagnolo, Tetrahedron Lett., 1986, 27, 1739 CrossRef CAS.
- (a) R. Cragg, J. P. N. Husband and A. F. Weston, J. Chem. Soc., Chem. Commun., 1970, 1701 RSC; (b) D. A. Armitage, M. J. Clark and C. C. Tsao, J. Chem. Soc., Perkin Trans. 1, 1972, 680 RSC; (c) G. Capozzi, A. Capperucci, A. Degl'Innocenti, R. DelDuce and S. Menichetti, Tetrahedron Lett., 1989, 30, 2995 CrossRef CAS; (d) A. Rajca and M. Wiessler, Tetrahedron Lett., 1990, 31, 6075 CrossRef CAS.
- I. V. Koval, Russ. J. Org. Chem., 2002, 38, 232 CrossRef CAS.
- S. Oae, Y. H. Kim, D. Fukushima and K. Shinhama, J. Chem. Soc., Perkin Trans. 1, 1978, 913 RSC.
- S. J. Brois, J. F. Pilot and H. W. Barnum, J. Am. Chem. Soc., 1970, 92, 7629 CrossRef CAS.
- (a) K. S. Boustang and A. B. Sullivan, Tetrahedron Lett., 1970, 11, 3547 CrossRef; (b) D. H. Harpp, D. K. Ash, T. G. Beck, J. G. Gleason, B. A. Orwig, W. F. VanHorn and J. P. Snyder, Tetrahedron Lett., 1970, 11, 3551 CrossRef; (c) J. Klose, C. B. Reese and Q. Song, Tetrahedron, 1997, 53, 14411 CrossRef CAS.
- M. Masui, Y. Mizuki, K. Sakai, C. Ueda and H. Ohmori, J. Chem. Soc., Chem. Commun., 1984, 843 RSC.
- D. R. Graber, R. A. Morge and J. C. Sih, J. Org. Chem., 1987, 52, 4620 CrossRef CAS.
- (a) M. Arisawa and M. Yamaguchi, J. Am. Chem. Soc., 2003, 125, 6624 CrossRef CAS PubMed; (b) K. Tanaka and K. Ajiki, Tetrahedron Lett., 2004, 45, 5677 CrossRef CAS.
- Q. T. Do, D. Elothmani, G. Le Guillanton and J. Simonet, Tetrahedron Lett., 1997, 38, 3383 CrossRef CAS.
- (a) D. Sureshkumar, V. Ganesh, R. S. Vidyarini and S. Chandrasekaran, J. Org. Chem., 2009, 74, 7958 CrossRef CAS PubMed; (b) D. Sureshkumar, S. M. Koutha and S. Chandrasekaran, J. Am. Chem. Soc., 2005, 127, 12760 CrossRef CAS PubMed.
- T. Mukaiyama and K. Takahashi, Tetrahedron Lett., 1968, 9, 5907 CrossRef.
- A. K. Galawde and A. F. Spatola, Org. Lett., 2003, 5, 3431 CrossRef PubMed.
- (a) J. K. Vandavasi, W.-P. Hu, Ch.-Y. Chen and J.-J. Wang, Tetrahedron, 2011, 67, 8895 CrossRef CAS; (b) R. Smith, X. Zeng, H. Müller-Bunz and X. Zhu, Tetrahedron Lett., 2013, 54, 5348 CrossRef CAS; (c) M. Musiejuk, T. Klucznik, J. Rachon and D. Witt, RSC Adv., 2015, 5, 31347 RSC.
- S. Lach, S. Demkowicz and D. Witt, Tetrahedron Lett., 2013, 54, 7021 CrossRef CAS.
- S. Antoniow and D. Witt, Synthesis, 2007, 363 CAS.
- J. Kowalczyk, P. Barski, D. Witt and B. A. Grzybowski, Langmuir, 2007, 23, 2318 CrossRef CAS PubMed.
- M. Szymelfejnik, S. Demkowicz, J. Rachon and D. Witt, Synthesis, 2007, 3528 CAS.
- S. Demkowicz, J. Rachon and D. Witt, Synthesis, 2008, 2033 CAS.
- E. Okragla, S. Demkowicz, J. Rachon and D. Witt, Synthesis, 2009, 1720 CAS.
- S. Lach and D. Witt, Synthesis, 2011, 3975 CrossRef CAS.
- J. Doroszuk, M. Musiejuk, S. Demkowicz, J. Rachon and D. Witt, RSC Adv., 2016, 6, 105449 RSC.
- (a) A. Kertmen, S. Lach, J. Rachon and D. Witt, Synthesis, 2009, 1459 CAS; (b) S. Lach and D. Witt, Heteroat. Chem., 2014, 25, 10 CrossRef CAS.
- (a) S. Lach, M. Sliwka-Kaszynska and D. Witt, Synlett, 2010, 2857 CAS; (b) S. Lach and D. Witt, Synlett, 2013, 24, 1927 CrossRef CAS.
- E. Block and S. Ahmad, J. Am. Chem. Soc., 1984, 106, 8295 CrossRef CAS.
- E. Block, S. Ahmad, J. L. Catalfamo, M. K. Jain and R. Apitz-Castro, J. Am. Chem. Soc., 1986, 108, 7045 CrossRef CAS.
- (a) M. Li, J.-R. Ciu, Y. Ye, J.-M. Min, L.-H. Zhang, K. Wang, M. Gares, J. Cros, M. Wright and J. Leung-Track, Carcinogenesis, 2002, 23, 573 CrossRef CAS PubMed; (b) M. Li, J. M. Min, J. R. Cui, L.-H. Zhang, K. Wang, A. Valette, C. Davrinche, M. Wight and J. Leung-Track, Nutr. Cancer, 2002, 42, 241 CrossRef CAS PubMed.
- G. Zhang and K. L. Parkin, J. Agric. Food Chem., 2013, 61, 1896 CrossRef CAS PubMed.
- (a) R. Hunter, C. H. Kaschula, I. M. Parker, M. R. Caira, P. Richards, S. Travis, F. Taute and T. Qwebani, Bioorg. Med. Chem. Lett., 2008, 18, 5277 CrossRef CAS PubMed; (b) C. H. Kaschula, R. Hunter, N. Stellenboom, M. R. Caira, S. Winks, T. Ogunleye, P. Richards, J. Cotton, K. Zilbeyaz, Y. Wang, V. Siyo, E. Ngarande and M. I. Parker, Eur. J. Med. Chem., 2012, 50, 236 CrossRef CAS PubMed.
- H. C. Brown and J. B. Campbell Jr, J. Org. Chem., 1980, 45, 389 CrossRef CAS.
- M. Ochiai, M. Toyonari, T. Nagaoka, D.-W. Chen and M. Kida, Tetrahedron Lett., 1997, 38, 6709 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00659h |
| This journal is © The Royal Society of Chemistry 2018 |