
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ACI/EG eutectic mixture mediated synthesis, characterization and in vitro osteoblast differentiation assessment of spiropyrrolo[1,2-b]isoquinoline analogues†
Govindasami Periyasami *ab,
Natarajan Arumugama,
Mostafizur Rahamana,
Raju Suresh Kumara,
Muthurangan Manikandanc,
Musaad A. Alfayezc,
Dhanaraj Premnathd and
Ali Aldalbahi*a
*ab,
Natarajan Arumugama,
Mostafizur Rahamana,
Raju Suresh Kumara,
Muthurangan Manikandanc,
Musaad A. Alfayezc,
Dhanaraj Premnathd and
Ali Aldalbahi*a
aDepartment of Chemistry, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia. E-mail: aaldalbahi@ksu.edu.sa; pkandhan@ksu.edu.sa
bDepartment of Organic Chemistry, University of Madras, Guindy Campus, Chennai 600 025, India
cStem Cell Unit, Department of Anatomy, College of Medicine, King Saud University, Riyadh, Saudi Arabia
dDepartment of Oral Biology, Dentistry Building-D325, Winnipeg, Manitoba, Canada
First published on 2nd May 2018
Abstract
An eco-friendly acetylcholine iodide–ethylene glycol (ACI/EG) deep eutectic mixture mediated green protocol has been developed for the synthesis of hitherto unexplored multi-functionalized linear tricyclic spiropyrrolo[1,2-b]isoquinoline analogues. The effects of the synthesized compounds on the osteoblast differentiation of hBMSC-TERT cell lines were investigated and promising results were observed with significant IC50 values. In addition, molecular modeling simulations were also performed with the 3D structure of BMP-2 to reveal binding interactions and orientations of highly potent spiropyrrolo[1,2-b]isoquinoline analogues.
1. Introduction
Healthy bones are important for a safe and happy life. Bone growth is controlled by cells called osteoclasts (OCs) and osteoblasts (OBs) during skeletal development throughout the human life. Removal of mineralized bone by OCs followed by the formation of the bone matrix through the OBs is the systematic approach taken during bone remodeling. There are various factors that control bone remodeling including insulin-like growth factors (IGFs), tumor growth factor-beta (TGF-β), bone morphogenetic proteins (BMPs) and cytokines.1 Among the various inductive growth factors of bone morphogenetic proteins (BMPs) including kidney development, limb formation, angiogenesis, tissue fibrosis and tumor development, osteogenic differentiation is one of the well-known and notable regulating processes that involve BMP-2.2 It increases the number of mature OBs by enhancing the differentiation capability of various stem cells. Also, biological signal pathways for inducing osteogenic differentiation are regulated by the binding of BMP-2 with microdomains on cell surfaces.3 Thus, BMP-2 is a potent inducer of bone formation and it also enhances the efficiency of osteogenic differentiation. Lee et al.4 demonstrated that isoquinoline alkaloids induce OB differentiation of stromal cells without the loss of cell viability. But, to date, there is no report in the literature on the effects of pyrroloisoquinoline and its analogues on osteogenic differentiation.The pyrroloisoquinoline structural framework is present in a number of alkaloids including the ecteinascidin family, and cactus and Hippeastrum genus alkaloids (Fig. 1), and it has remarkable activities in the central nervous system5 besides possessing antitubercular,6a antihypertensive,6b anti HIV-1,6c anti-leukemic,6d and anti-cancer activities.6e Its analogues can be used as radiotracers in positron emission tomography (PET) for imaging serotonin uptake sites.7 The tendency of lycorine alkaloids to inhibit cell growth and cell division is utilized to study their activity against a number of cancer cell lines, showing a decrease in tumor cell growth and an increase in survival rates with no observable adverse effects in animal treatment. Isoquinoline analogues containing spiro centers are frequently encountered in natural products and they possess diverse pharmacological properties.8 Also the spiro structural motif, including polycyclic spiro heterocycles, which intercalates easily by its nature into DNA, can be used as a class of effective anti-tumor pharmacophore units.9 The structural rigidity and complexity of spiro compounds are highly favorable for increasing affinity towards proteins by reducing the loss of conformational entropy, which is a topic of interest in drug discovery research.10
 | ||
| Fig. 1 Pyrrolo[1,2-b]isoquinoline core and spiro-center natural products. | ||
Our research aim is to synthesize spiropyrrolo[1,2-b]isoquinolines and study their effects on OB differentiation, particularly the significant increase in alkaline phosphatase activity (ALP) of ecteinascidin-743 (alkaloid in the ecteinascidin family), a marker of osteoblastic differentiation.11
2. Results and discussion
2.1. Chemistry
Interesting synthetic methodologies have been reported in the literature for the synthesis of various pyrrolo[1,2-a]isoquinoline derivatives including one-pot or multi-step reactions including solid phase supported,12a photoredox-catalyzed,12b metal-catalyzed,12c acid/base catalyzed,12d and Pictet–Spengler cyclization12e reactions. However, the syntheses of linear tricyclic spiropyrrolo[1,2-b]isoquinoline ring systems and their biological applications are relatively unexplored. The results of our recent report13 on the multicomponent reaction of an inexpensive quaternary alkylammonium class of eutectic mixture mediated heterocyclic molecules encouraged us to synthesize spiropyrrolo[1,2-b]isoquinoline analogues using a similar methodology. An inexpensive room temperature eutectic mixture,14 acetylcholine iodide–ethylene glycol (ACI/EG) mediated synthesis of spiro heterocycles, particularly pyrrolo[1,2-b]isoquinoline analogues, through azomethine ylide 1,3-dipolar cycloaddition reactions is yet to be reported. The retrosynthetic strategy explains the construction of regioselective spiropyrrolo[1,2-b]isoquinoline molecules from easily available starting materials in a single synthetic step as shown in Fig. 2. | ||
| Fig. 2 Synthetic strategy for linear tricyclic spiropyrrolo[1,2-b]isoquinoline cycloadducts. | ||
During the synthesis, the chalcone group is retained in the spiro adduct which may be due to the stability of the mono adduct and to avoid the formation of the sterically hindered bis cycloadduct. One of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in the dipolarophiles, (1E,4E)-1,5-p-substituted diphenylpenta-1,4-dien-3-one derivatives,15 may preferably undergo an addition reaction with an azomethine ylide which leads to the unreacted prop-2-en-1-one unit in spiro heterocyclic hybrids. Spiropyrroloisoquinoline with a chalcone group is an excellent candidate in medicinal chemistry particularly in Alzheimer’s disease treatment,16 and it is also capable of inducing apoptosis17 and uncoupling mitochondrial respiration.18 In general, chalcone compounds did not show genotoxic effects and they may be devoid of this significant side effect.19
C bonds in the dipolarophiles, (1E,4E)-1,5-p-substituted diphenylpenta-1,4-dien-3-one derivatives,15 may preferably undergo an addition reaction with an azomethine ylide which leads to the unreacted prop-2-en-1-one unit in spiro heterocyclic hybrids. Spiropyrroloisoquinoline with a chalcone group is an excellent candidate in medicinal chemistry particularly in Alzheimer’s disease treatment,16 and it is also capable of inducing apoptosis17 and uncoupling mitochondrial respiration.18 In general, chalcone compounds did not show genotoxic effects and they may be devoid of this significant side effect.19
The spiro heterocycle tethered pyrrolo[1,2-b]quinoline analogues were obtained in good to excellent yields (85–92%) through 1,3-dipolar cycloaddition reactions of azomethine ylides generated in situ from an equimolar amount of a cyclic amino acid (1), a diketone (2 or 6) and various substituted dipolarophiles (4a–c) in ACI/EG at 40 °C. The required ACI/EG eutectic mixture, as a reaction medium, was prepared by mixing acetylcholine iodide and ethylene glycol in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 molar ratio and then the mixture was heated at 70 °C.20 Although our group has previously established and reported reaction conditions, we examined the synthesis of our target linear tricyclic spiropyrrolo[1,2-b]isoquinoline analogues under different reaction conditions (Table 1). As a result, it is noteworthy that the cycloaddition reaction in ACI/EG at 40 °C furnished an excellent yield of cycloadducts in 12 h (entry 8). Under the eutectic solvent medium, the interaction of ACI/EG via hydrogen bonding with the carbonyl group of the substrates, namely isatin/(1E,4E)-1,5-diphenylpenta-1,4-dien-3-one, would enhance the polarity of the carbon and oxygen. As a result, the electrophilicity of the isatin carbonyl would increase which in turn may influence the nucleophilic attack by the NH group of isoquinoline-3-carboxylic acid, then the subsequent dehydration and decarboxylation furnishes the 1,3-dipolar azomethine ylide intermediate. Similarly, the polarized enol form of (1E,4E)-1,5-diphenylpenta-1,4-dien-3-one is presumably activated by the interaction of the eutectic mixture with the dipolarophile via hydrogen bonding and then this facilitates the addition of the azomethine ylide. During this catalytic process, the dipolarophile underwent facile interaction with the intermediate azomethine ylide yielded cycloadducts significantly. Under different temperatures and times of the reaction, the yield of the product did not change significantly compared to the reaction at 40 °C for 12 h. Hence, all of the subsequent reactions (Scheme 1) were carried out using the optimized reaction conditions. After completion of the reaction (monitored by TLC), the pure product was obtained by column chromatography.
:9 molar ratio and then the mixture was heated at 70 °C.20 Although our group has previously established and reported reaction conditions, we examined the synthesis of our target linear tricyclic spiropyrrolo[1,2-b]isoquinoline analogues under different reaction conditions (Table 1). As a result, it is noteworthy that the cycloaddition reaction in ACI/EG at 40 °C furnished an excellent yield of cycloadducts in 12 h (entry 8). Under the eutectic solvent medium, the interaction of ACI/EG via hydrogen bonding with the carbonyl group of the substrates, namely isatin/(1E,4E)-1,5-diphenylpenta-1,4-dien-3-one, would enhance the polarity of the carbon and oxygen. As a result, the electrophilicity of the isatin carbonyl would increase which in turn may influence the nucleophilic attack by the NH group of isoquinoline-3-carboxylic acid, then the subsequent dehydration and decarboxylation furnishes the 1,3-dipolar azomethine ylide intermediate. Similarly, the polarized enol form of (1E,4E)-1,5-diphenylpenta-1,4-dien-3-one is presumably activated by the interaction of the eutectic mixture with the dipolarophile via hydrogen bonding and then this facilitates the addition of the azomethine ylide. During this catalytic process, the dipolarophile underwent facile interaction with the intermediate azomethine ylide yielded cycloadducts significantly. Under different temperatures and times of the reaction, the yield of the product did not change significantly compared to the reaction at 40 °C for 12 h. Hence, all of the subsequent reactions (Scheme 1) were carried out using the optimized reaction conditions. After completion of the reaction (monitored by TLC), the pure product was obtained by column chromatography.
| Entry | Solvent system | Time (h) | Yield (%) |
|---|---|---|---|
| a General conditions: all reactions were performed under vigorous stirring at room temperature (except entry 8); optimized reaction conditions are in bold (entry 8); eutectic mixtures were subjected to high vacuum before use (entry 7 and 8); trace*: identifying product Rf by TLC. | |||
| 1 | Ethanol | 48 | Trace* |
| 2 | Methanol | 48 | Trace* |
| 3 | Methanol/H2O | 48 | Trace* |
| 4 | Acetonitrile | 48 | Trace* |
| 5 | Acetonitrile/Et3N | 48 | 15 |
| 6 | Dioxane/MeCN (1:1)/Et2NH |
48 | 10 |
| 7 | ACI/EG, rt | 48 | 60 |
| 8 | ACI/EG, 40 °C | 12 | 90 |
 | ||
| Scheme 1 Synthesis of oxindole substituted monospiropyrrolo[1,2-b]isoquinolines 5a–c. | ||
ACI/EG was recovered from the reaction mixture by fractional distillation and dried overnight under high vacuum at 40 °C. It was further utilized for five consecutive reactions and the results revealed that the eutectic mixture is stable and recyclable without much significant loss of activity (Table 2).
| Medium | Yield percentage of isoxazolidine 5a in experiments | ||||
|---|---|---|---|---|---|
| First | Second | Third | Fourth | Fifth | |
| Recovered ACI/EG | 90 | 88 | 88 | 87 | 86 |
The FT-IR spectrum of compound 5a showed a band at 1673 cm−1 due to the α,β-unsaturated carbonyl and a band at 1686 cm−1 due to the amide carbonyl functional group. The NH group stretching was observed at 3382 cm−1. The 1H NMR spectrum of compound 5a exhibited a doublet at δ 4.19 (J = 9.0 Hz) for the Ha proton, a distorted doublet at δ 3.53 due to the benzylic proton and a multiplet at δ 3.95–4.01 due to the Hc proton. Two broad singlets observed at δ 2.89 and 3.94 corresponded to Hd and He protons, respectively. The trans vinyl protons of the olefinic system appeared as two doublets at δ 6.12 (J = 16.2 Hz) and δ 7.12 (J = 16.2 Hz). The NH proton of the oxindole ring resonated at δ 8.25 as a singlet. In the 13C NMR spectrum, the spiro quaternary carbon displayed a peak at 72.79 ppm. The oxindole carbonyl carbon was observed at 180.78 ppm. The α,β-unsaturated carbonyl carbon exhibited a peak at 196.06 ppm. A DEPT-135 study confirmed that the two methylene carbons appeared at 35.30 and 47.90 ppm. The mass spectrum of 5a exhibited a molecular ion peak at m/z 496.61 and the compound gave acceptable elemental analysis results. Finally, the structure and stereochemistry of cycloadduct 5a were elucidated unambiguously by single crystal X-ray diffraction analysis21 (Fig. 3). NMR spectra of all synthesized compounds are provided in the ESI.†
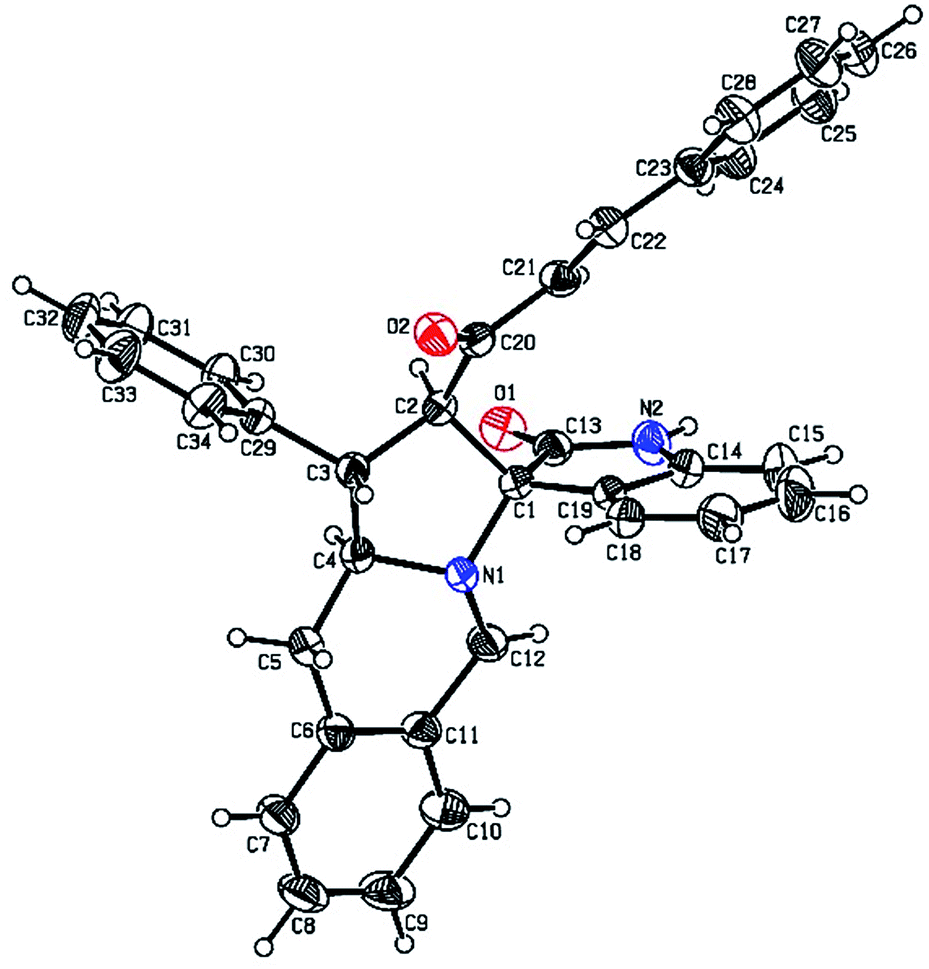 | ||
| Fig. 3 Oak Ridge Thermal Ellipsoid Plot (ORTEP) of cycloadduct 5a. | ||
To explore the synthetic utility of the optimized protocol, we have investigated the cycloaddition reaction with a different diketone viz, acenaphthoquinone as this structural motif possesses strong antioxidant properties, free radical scavenging activity and the ability to reduce lipid peroxidation.22 The generated azomethine ylide (7) readily undergoes cycloaddition with dipolarophiles (4a–c) affording the cycloadducts (8a–c) in good yields (Scheme 2).
 | ||
| Scheme 2 Synthesis of acenaphthoquinone substituted monospiropyrrolo[1,2-b]isoquinolines 8a–c. | ||
The structure and regiochemistry of the cycloadducts (8a–c) were also unambiguously established by their spectroscopic and elemental analysis data.† The stability of formation of the azomethine ylide supports the relative stereochemistry of the synthesized novel spiropyrrolo[1,2-b]isoquinolines. In Schemes 1 and 2, formation of syn-ylides23 3a and 7a may not be observed due to the unfavorable steric repulsion between the carbonyl group of the diketone and the isoquinoline ring. Most probably, anti-ylides22 3 and 7, generated from tetrahydroisoquinoline-3-carboxylic acid, isatin (2) and acenaphthoquinone (6), respectively, are involved in the transition state which undergoes addition to dipolarophiles (4a–c) to give the observed cycloadducts (Scheme 3). In both Schemes 1 and 2, the cycloaddition proceeded via an endo-transition state24 and the possibility of formation of the other isomer via an exo-transition state was ruled out.
 | ||
| Scheme 3 Formation of stable anti-ylides in the transition state. | ||
Under the optimized reaction conditions, stirring the ACI/EG medium at 40 °C, we obtained linear tricyclic spiropyrrolo[1,2-b]isoquinoline analogues in good yields. The yields and results are summarized in Table 3.
| Entry | Secondary amino acid | Diketone | Dipolarophile | Spiropyrrolo[1,2-b] isoquinolinea,b | Time (h) | Yieldc (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: vigorous stirring at 40 °C in ACI/EG eutectic mixture medium.b Completion of the reaction monitored by TLC.c Yield of the isolated product after flash column chromatography. | ||||||
| 1 | 1 | 2 | 4a | 5a | 12 | 90 |
| 2 | 1 | 2 | 4b | 5b | 12 | 92 |
| 3 | 1 | 2 | 4c | 5c | 12 | 86 |
| 4 | 1 | 6 | 4a | 8a | 12 | 85 |
| 5 | 1 | 6 | 4b | 8b | 12 | 86 |
| 6 | 1 | 6 | 4c | 8c | 12 | 88 |
2.2. Biology
 | ||
| Fig. 4 Plots (A) and (B) show IC50 values of representative compounds 5a (20 μM) and 8a (8 μM), respectively. | ||
 | ||
| Fig. 5 Qualitative analysis of alizarin red staining of mineralized nodule formation of 5a and 8a on day 7 in osteogenic differentiation media. | ||
In different concentrations of 5a at 15 μM and 8a at 5 μM, both the cycloadducts show significant effects on osteogenic differentiation of hBMSC-TERT (Fig. 5). Osteoimage staining of representative compounds was also carried out, and the observed nodule images (Fig. 6a) and the percentage of mineralization (Fig. 6b) are shown in Fig. 6.
 | ||
| Fig. 6 (a) Quantitative analysis of osteoimage mineralized nodule formation of 5a and 8a on day 7 in osteogenic differentiation media. (b) Quantitative mineralization of representative compounds 5a and 8a with DMSO as the standard {data are presented as the means ± SD from three independent experiments, n = 6; *p < 0.05; **p < 0.01, ***p < 0.001}. | ||
 | ||
| Fig. 7 Osteoimage staining mineralization of spiropyrrolo[1,2-b]isoquinoline analogues 5a–c and 8a–c with DMSO as the standard. The data are presented as the means ± SD from three independent experiments, n = 6; *p < 0.05; **p < 0.01, ***p < 0.001. | ||
2.3. Docking studies
Molecular docking study results of compounds 5a and 8b (Fig. 8) clearly reveal that the molecular interactions with the biologically proved active site of the BMP-2 protein with docking scores of −2.261 (5a) and −0.781 (8b) and the energy of −51.512 (5a) and −47.780 (8b). The molecular interaction studies demonstrate that ligands were bound on the same active site cavity in the molecule, as biologically proved and deposited in the scientific database. This is evident from the superposition of residues in active site molecules (12–114) and amino acid residues of the BMP-2 protein, in Fig. 9 and 10. At a distance of 4 Å, THR 58, ASN 56, SER 85, LEU 84, GLN 83, THR 82, PRO 81, VAL 80, ARG 114, GLN 109, ASP 105, and GLN 104 amino acids were found in the active sites and involved in biochemical interactions with compounds 5a and 8b. Docking of 5a with the BMP-2 protein is shown with two hydrogen bonding interactions with the respective active site molecules of ARG 114 (H–O–C) along with π–π stacking. Similarly 8b has also shown moderate hydrogen bonding interactions with the active site and has shown one hydrogen bonding interaction with ARG 114 with π–π stacking and another hydrogen bonding interaction with the LEU 84 amino acid. Compound 8b has shown a good hydrogen bonding backbone interaction with positively charged amino acids. Moreover, compound 5a has shown surface molecular interactions with strong hydrogen bonding with good energy scores. Computational docking study results reveal that the molecular biological activities shown are based on the molecular interaction and energy and structural orientation between the compounds and the active site amino acid polymer. | ||
| Fig. 8 Optimized structure of cycloadducts 5a and 8b using the LigPrep module. | ||
 | ||
| Fig. 9 Orientation and binding interactions of (a) 5a with the BMP-2 active site and (b) 8b with the BMP-2 active site. | ||
 | ||
| Fig. 10 In silico packing illustration of representative compounds (a) 5a and (b) 8b with their appropriate binding packets of BMP-2. | ||
2.4. Evaluation of BMP-2 gene expression of spiropyrrolo[1,2-b]isoquinoline analogues
To authenticate the interactions between synthesized cycloadducts and the BMP-2 protein, based on the docking studies, the cycloadducts 5a–c and 8a–c have been subjected to BMP-2 expression evaluation after OS differentiation in OS differentiation media. In the spiroindolin-2-one series 5a–c, compound 5a maintained BMP-2 expression equal to the control, whereas 5b and 5c showed subdued expression. In the acenaphthylen-1-one series 8a–c, cycloadduct 8a resulted in lower expression of BMP-2, while 8b and 8c maintained BMP-2 expression level equal to the control. The results are presented in Fig. 11. | ||
| Fig. 11 BMP-2 gene expression pattern of compounds 5a–c and 8a–c using qRT-PCR. | ||
3. Conclusion
In conclusion, two series of spiropyrrolo[1,2-b]isoquinoline analogues were synthesized employing an ACI/EG eutectic mixture as an eco-friendly reaction medium through 1,3-dipolar cycloaddition methodology. To the best of our knowledge, this is the first report of the synthesis of spiropyrrolo[1,2-b]isoquinoline analogues in the ACI/EG benign reaction medium and their effects on OB differentiation of the hBMSC-TERT cell line. All of the synthesized compounds possessed the capability of enhancing the osteoblast differentiation of hBMSC-TERT cell lines with significant IC50 values of less than 20 μM. The structural and functional insights of spiropyrrolo[1,2-b]isoquinoline analogues with the BMP-2 protein were studied by molecular docking simulations using the Schrödinger Glide program. In addition, the hypothesis has been proved by aRT-PCR studies. These results emphasized the role of spiropyrrolo[1,2-b]isoquinolines in OB differentiation and could have myriad applications in bone biology.4. Materials and methods
4.1. Cell culture
We employed hBMSC-TERT cell lines created from primary normal human bone marrow BMSCs by over-expressing the human telomerase reverse transcriptase gene (hTERT). We are grateful to our collaborator Prof. Moustapha Kassem, University of Odense, Southern Denmark for providing the hBMSC-TERT cell line.25 These cells have been extensively characterized and exhibit cellular and molecular phenotypes similar to those of primary hBMSCs and were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 4.5 g L−1 D-glucose, 4 mM L-glutamine, 110 mg L−1 sodium pyruvate, 10% fetal bovine serum (FBS), 1× penicillin–streptomycin (Pen–Strep), and non-essential amino acids.4.2. In vitro osteoblast differentiation
Cells were grown in 48-well plates in standard DMEM at a density of 1 × 105 cells per mL. When cells reached 70–80% confluence, they were cultured in DMEM supplemented with osteogenic induction supplements containing 10% FBS, 1% Pen–Strep, 50 μg mL−1 L-ascorbic acid (Wako Chemicals, Germany), 10 mM β-glycerophosphate (Sigma-Aldrich), 10 nM calcitriol (1α, 25-dihydroxyvitamin D3; Sigma), and 10 nM dexamethasone (Sigma-Aldrich). The medium was replaced three times per week. Compounds 5a and 8a were mixed with osteogenic differentiation media at 15 μM and 5 μM, respectively, to evaluate their osteogenic differentiation potential.4.3. Cytochemical staining
4.4. Molecular docking
Molecular docking studies were performed using the Schrödinger Glide program (2017-4, Schrödinger LLC, New York 2014). The selected compounds 5a and 8b structures are drawn using ChemSketch software. In order to prepare ligand molecules with high quality, all-atom 3D structures for large numbers of drug-like molecules, starting with the 3D structures in SD Maestro format, LigPrep was used (Fig. 8). LigPrep produced a single, low-energy, 3D structure with corrected chiralities for each successfully processed input structure. The optimised ligands were docked into the bone morphogenetic protein-2 (BMP-2) structure using the Glide (SP & XP) module. The required typical structure file of BMP-2 protein structure was downloaded from the protein data bank (PDB). The structural file from experimental techniques may not be suitable for immediate use in molecular modelling calculations. PDB structures may miss information on connectivity that has been assigned along with bond orders and formal charges using the Protein Preparation Wizard. However, the receptor grid generation requires a “prepared” structure with all atoms and appropriate bond orders with formal charges. The shape and properties of receptor BMP-2 is generated on a grid by several different sets of fields around BMP-2 binding sites that provide progressively more accurate scoring of the ligand presence. To confirm the best molecular interaction and validate the docking score, the protocol was evaluated by re-docking.4.5. Real time qRT-PCR
Total RNA was extracted from differentiating cells with a series of cycloadducts 5a–c and 8a–c using the innuPREP RNA Mini Kit (Cat no: 845-KS-2040250, Analytik Jena, Berlin, Germany) as recommended by the manufacturer. Total RNA was quantified using a Nanodrop spectrophotometer (Nanodrop 2000, Thermo Scientific). cDNA was synthesized from 1 μg of the RNA samples using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA) using a ProFlex PCR System, Applied Biosystems according to the manufacturer’s instructions. Relative levels of mRNA were determined from cDNA using real time PCR (Applied Biosystem-Real Time PCR Detection System) with a Power SYBR Green PCR kit (Applied Biosystems, UK), according to the manufacturer’s instructions. Following normalisation to the reference gene GAPDH, quantification of gene expression was carried out using a comparative Ct method where ΔCt is the difference between the Ct values of the target and reference gene. (Primer sequence: GAPDH – F 5′ CTGGTAAAGTGGATATTGTTGCCAT 3′, R 5′ TGGAATCATATTGGAACATGTAAACC 3′, BMP2 – F 5′ GGAACGGACATTCGGTCCTT 3′, R 5′ CACCATGGTCGACCTTTAGGA 3′).5. Experimental
5.1. General procedure for the preparation of (1E,4E)-1,5-bis(p-substituted-phenyl)penta-1,4-dien-3-one, (4a–c)
A solution of p-substituted benzaldehyde (2.0 mol) and acetone (1.0 mol) was stirred at 15–20 °C in a 250 mL round bottom flask. 10% sodium hydroxide solution prepared and maintained at 15–20 °C was added slowly. The rate of addition was adjusted to make sure that the reaction temperature did not exceed 25–30 °C. The reaction mixture was vigorously stirred and then the precipitate was formed in 5 min. The stirring was continued for a further 1 hour. After completing the reaction as evidenced by TLC, dilute hydrochloric acid was added to neutralize the reaction mixture. The resulting precipitate was filtered and washed with cold water. The crude product was dried and recrystallized in an ethyl acetate/hexane mixture yielding 90–95% of pure (1E,4E)-1,5-bis(p-substituted-phenyl)penta-1,4-dien-3-one (4a–c).5.2. General procedure for the synthesis of monospiropyrrolo[1,2-b]isoquinolines (5a–c)
A mixture of 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid (1) (1.2 equiv.), isatin (2) (1.2 equiv.) and (1E,4E)-1,5-bis(p-substituted-phenyl)penta-1,4-dien-3-one (4a–c) (1.0 equiv.) was stirred in an ACI/EG eutectic mixture for 1 hour. After the completion of the reaction as evidenced by TLC, in an adequate test, the eutectic mixture was removed by vacuum distillation and the residue was chromatographed over silica gel with the hexane–ethyl acetate mixture (8:2) to give novel monospiropyrrolo[1,2-b]isoquinolines (5a–c) in good to excellent yields.
Yellow solid, mp 136 °C; yield: 90%; IR (KBr) 3382, 1686, 1673 cm−1; 1H (300 MHz, CDCl3) δ 2.89 (br s, 2H, Hd), 3.53 (dis d, 1H, Hb), 3.94 (br s, 2H, He), 3.95–4.01 (m, 1H, Hc), 4.19 (d, 1H, Ha, J = 9.0 Hz), 6.12 (d, 1H, J = 16.2 Hz), 6.72–7.58 (m, 18H, ArH), 7.12 (d, 1H, J = 16.2 Hz), 8.25 (s, 1H, NH). 13C (75 MHz, CDCl3) 35.30, 47.90, 52.38, 62.42, 63.71, 72.79, 109.30, 123.39, 125.67, 126.15, 126.47, 126.87, 127.11, 127.18, 127.82, 128.04, 128.65, 128.73, 128.75, 129.03, 129.31, 130.35, 133.58, 133.76, 134.30, 139.89, 140.58, 142.46, 180.78, 196.06. EI-MS m/z 496.61 (M+). Anal. calcd for C34H28N2O2: C, 82.23; H, 5.68; N, 5.64%. Found: C, 82.31; H, 5.59; N, 5.71%.

Yellow solid, mp 144 °C; yield: 92%; IR (KBr) 3384, 1686, 1674 cm−1; 1H (300 MHz, CDCl3) δ 2.81 (br s, 2H, Hd), 3.16 (s, 3H, Me), 3.48 (dis d, 1H, Hb), 3.91 (br s, 2H, He), 3.96–4.10 (m, 1H, Hc), 4.23 (d, 1H, Ha, J = 9.0 Hz), 6.09 (d, 1H, J = 16.0 Hz), 6.64–7.53 (m, 16H, ArH), 7.14 (d, 1H, J = 16.0 Hz), 8.29 (s, 1H, NH). 13C (75 MHz, CDCl3) 29.81, 34.28, 39.64, 56.38, 61.31, 62.69, 72.22, 110.52, 122.38, 126.49, 126.52, 126.59, 126.76, 127.09, 127.27, 127.86, 127.99, 128.29, 128.64, 128.70, 129.41, 129.49, 130.51, 133.58, 133.69, 134.48, 138.92, 140.11, 142.43, 180.99, 197.60. EI-MS m/z 556.65 (M+). Anal. calcd for C36H32N2O2: C, 82.41; H, 6.15; N, 5.35%. Found: C, 82.66; H, 6.22; N, 5.28%.

Pale yellow solid, mp 122 °C; yield: 86%; IR (KBr) 3382, 1683, 1676 cm−1; 1H (300 MHz, CDCl3) δ 2.88 (br s, 2H, Hd), 3.51 (dis d, 1H, Hb), 3.87 (br s, 2H, He), 3.91–4.19 (m, 1H, Hc), 4.35 (d, 1H, Ha, J = 9.0 Hz), 6.21 (d, 1H, J = 16.2 Hz), 6.98–8.16 (m, 16H, ArH), 7.20 (d, 1H, J = 16.2 Hz), 8.16 (s, 1H, NH). 13C (75 MHz, CDCl3) 34.29, 39.02, 49.62, 58.31, 61.64, 70.22, 111.12, 121.00, 125.09, 126.31, 126.38, 126.86, 127.12, 127.89, 128.22, 128.29, 128.58, 128.66, 128.89, 129.39, 129.59, 129.91, 133.19, 133.64, 134.84, 138.89, 140.56, 142.73, 181.52, 197.85. EI-MS m/z 654.36 (M+). Anal. calcd for C34H26Br2N2O2: C, 62.40; H, 4.00; N, 4.28%. Found: C, 62.59; H, 3.89; N, 5.01%.

5.3. General procedure for the synthesis of monospiropyrroloisoquinolines (8a–c)
A mixture of 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid (1) (1.2 equiv.), acenaphthoquinone (6) (1.2 equiv.) and (1E,4E)-1,5-bis(p-substituted-phenyl)penta-1,4-dien-3-one (4a–c) (1.0 equiv.) was stirred in an ACI/EG eutectic mixture for 1 hour. After the completion of the reaction as evidenced by TLC, in an adequate test, the eutectic mixture was removed by vacuum distillation and the residue was chromatographed over silica gel with the hexane–ethyl acetate mixture (8:2) to give novel monospiropyrrolo[1,2-b]isoquinolines (8a–c) in good yields.
Yellow solid, mp 186 °C; yield: 85%; IR (KBr) 1716, 1678 cm−1; 1H (300 MHz, CDCl3) δ 2.93 (br s, 2H, Hd), 3.42 (dis d, 1H, Hb), 3.94 (d, 1H, Ha), 4.08–4.09 (m, 2H, Hd), 4.13–4.19 (m, 1H, Hc), 5.69 (d, 1H, J = 16.2 Hz), 6.65–8.05 (m, 21H, ArH with alkene H). 13C (75 MHz, CDCl3) 35.44, 48.00, 52.67, 63.17, 64.25, 72.81, 121.09, 123.82, 125.02, 125.62, 126.13, 126.35, 126.42, 127.09, 127.67, 127.95, 128.44, 128.70, 128.75, 129.07, 129.12, 130.09, 130.23, 131.50, 132.44, 133.67, 133.78, 134.00, 137.64, 140.23, 141.84, 142.53, 196.42, 210.06. EI-MS m/z 531.64 (M+). Anal. calcd for C38H29NO2: C, 85.85; H, 5.50; N, 2.63%. Found: C, 85.91; H, 5.43; N, 2.70%.

Colourless solid, mp 160 °C; yield: 86%; IR (KBr) 1716, 1678 cm−1; 1H (300 MHz, CDCl3) δ 2.96 (br s, 2H, Hd), 3.13 (dis d, 1H, Hb), 3.68 (s, 3H, OMe), 3.98 (d, 1H, Ha), 4.00–4.03 (m, 2H, Hd), 4.11–4.14 (m, 1H, Hc), 5.55 (d, 1H, J = 16.2 Hz), 6.61–8.13 (m, 19H, ArH with alkene H). 13C NMR (75 MHz, CDCl3) 32.52, 46.21, 52.33, 56.62, 62.95, 64.19, 73.01, 120.85, 123.60, 125.42, 125.39, 125.90, 126.33, 126.44, 127.09, 127.67, 127.91, 128.49, 128.65, 128.71, 129.00, 129.09, 130.05, 130.53, 131.17, 132.46, 133.61, 133.68, 134.12, 137.66, 140.28, 141.68, 142.98, 196.19, 210.00. EI-MS m/z 591.69 (M+). Anal. calcd for C40H33NO4: C, 81.20; H, 5.62; N, 2.37%. Found: C, 81.26; H, 5.58; N, 2.42%.

Colourless solid, mp 172 °C; yield: 88%; IR (KBr) 1720, 1679 cm−1; 1H (300 MHz, CDCl3) δ 2.98 (br s, 2H, Hd), 3.32 (dis d, 1H, Hb), 3.89 (d, 1H, Ha), 4.01–4.03 (m, 2H, Hd), 4.09–4.13 (m, 1H, Hc), 5.62 (d, 1H, J = 16.2 Hz), 6.76–8.16 (m, 19H, ArH with alkene H). 13C NMR (75 MHz, CDCl3) 31.25, 45.95, 52.13, 62.91, 64.18, 73.09, 120.83, 123.44, 125.38, 125.54, 125.89, 126.30, 126.41, 126.98, 127.59, 127.88, 128.52, 128.64, 128.73, 129.07, 129.10, 130.00, 130.42, 131.24, 132.50, 133.61, 133.68, 134.17, 137.68, 140.30, 141.72, 142.96, 196.23, 210.15. EI-MS m/z 600.53 (M+). Anal. calcd for C38H27Cl2NO2: C, 76.00; H, 4.53; N, 2.33%. Found: C, 76.07; H, 4.59; N, 2.29%.

Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through research group no. RG-1438-038. Also, GPS thanks the Council of Scientific and Industrial Research (CSIR), New Delhi.References
- A. Yamaguchi, T. Komori and T. Suda, Endocr. Rev., 2000, 21, 393–411 CrossRef CAS PubMed.
- (a) S. Scarfì, World J. Stem Cell., 2016, 8, 1–12 CrossRef PubMed; (b) S. P. Alexander, H. E. Benson, E. Faccenda, A. J. Pawson, J. L. Sharman, M. Spedding, J. A. Peters and A. J. Harmar, Br. J. Pharmacol., 2013, 170, 1676–1705 CrossRef CAS PubMed; (c) P. Ducy and G. Karsenty, Kidney Int., 2000, 57, 2207–2214 CrossRef CAS PubMed.
- B. Hu, A. J. El Haj and J. Dobson, Int. J. Mol. Sci., 2013, 9, 19276–19293 CrossRef PubMed.
- J.-W. Lee, N. Mase, T. Yonezawa, H.-J. Seo, W. B. Jeon, B. Y. Cha, K. Nagai and J.-T. Woo, Biol. Pharm. Bull., 2010, 33, 1733–1739 CAS.
- C. A. Demerson, A. H. Philipp, L. G. Humber, M. J. Kraml, M. P. Charest, H. Tom and I. Vavra, J. Med. Chem., 1974, 17, 1140–1145 CrossRef CAS PubMed.
- (a) R. R. Kumar, S. Perumal, S. C. Manju, P. Bhatt, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem. Lett., 2009, 19, 3461–3465 CrossRef PubMed; (b) J. M. Ferland, C. A. Demerson and L. G. Humber, Can. J. Chem., 1985, 63, 361–365 CrossRef CAS; (c) X. Z. Zhao, K. Maddali, C. Marchand, Y. Pommier and T. R. Burke, Bioorg. Med. Chem., 2009, 17, 5318–5324 CrossRef CAS PubMed; (d) E. C. Taylor, P. Zhou, L. D. Jenning, Z. Mao, B. Hu and J.-G. Jun, Tetrahedron Lett., 1997, 38, 521–524 CrossRef CAS; (e) K. R. Senwar, P. Sharma, T. S. Reddy, M. K. Jeengar, V. L. Nayak, V. G. M. Naidu, A. Kamal and N. Shankaraiah, Eur. J. Med. Chem., 2015, 102, 413–424 CrossRef CAS PubMed.
- M. Suehiro, J. H. Greenberg, C. Y. Shine, C. González, B. Dembowski and M. Reivich, Nucl. Med. Biol., 1996, 4, 407–412 CrossRef.
- B.-Q. Tang, W.-J. Wang, X.-J. Huang, G.-Q. Li, L. Wang, R.-W. Jiang, T.-T. Yang, L. Shi, X.-Q. Zhang and W.-C. Ye, J. Nat. Prod., 2014, 77, 1839–1846 CrossRef CAS PubMed.
- (a) H. Ihmels, J. Mattav, F. May and L. Thomas, Org. Biomol. Chem., 2013, 11, 5184–5188 RSC; (b) C. S. Becker, G. I. Roshchupkina, T. V. Rybalova, Y. V. Gatilov, G. V. Romanenkoc and V. A. Reznikova, Mendeleev Commun., 2008, 18, 297–299 CrossRef CAS.
- A. V. Campoy, M. J. Todd and E. Freire, Biochemistry, 2000, 39, 2201–2207 CrossRef.
- K. Scotlandi, S. Perdichizzi, M. C. Manara, M. Serra, S. Benini, V. Cerisano, R. Strammiello, M. Mercuri, G. Reverter-Branchat, G. Faircloth, M. D’Incalci and P. Picci, Clin. Cancer Res., 2002, 8, 3893–3903 CAS.
- (a) T. E. Nielsen and M. Meldal, J. Comb. Chem., 2005, 7, 599–610 CrossRef CAS PubMed; (b) C. Vila, J. Lau and M. Rueping Beilstein, J. Org. Chem., 2014, 10, 1233–1238 Search PubMed; (c) C. Feng, J.-H. Su, Y. Yan, F. Guo and Z. Wang, Org. Biomol. Chem., 2013, 11, 6691–6694 RSC; (d) M. Leonardi, M. Villacampa and J. C. Menéndez, J. Org. Chem., 2017, 82, 2570–2578 CrossRef CAS PubMed; (e) E. D. Cox and J. M. Cook, Chem. Rev., 1995, 95, 1797–1842 CrossRef CAS.
- G. Periyasami, N. Arumugam and A. Aldalbahi, Tetrahedron, 2017, 73, 322–330 CrossRef CAS.
- Q. Zhang, K. D. O. Vigier, S. Royer and F. Jerome, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- A. I. Vogal, A Textbook of Practical Organic Chemistry, 5th edn, 1989, p. 1033 Search PubMed.
- (a) B. Zhou and C. Xing, Med. Chem., 2015, 5, 388–404 Search PubMed; (b) V. Koneni, K. V. Sashidhara, R. K. Modukuri, P. Jadiya, R. P. Dodda, M. Kumar, B. Sridhar, V. Kumar, R. Haque, M. I. Siddiqi and A. Nazir, ChemMedChem, 2014, 9, 2671–2684 CrossRef PubMed.
- T. Sakai, R. N. Eskander, Y. Euo, J. J. Kim, J. Mefford, J. Hopkins, N. N. Bhatia, X. Zi, B. H. Hoang, B. Flavokawain and B. Flavokawain, J. Orthop. Res., 2011, 30, 1045–1050 CrossRef PubMed.
- D. Chakraborty, A. Maity, C. K. Jain, A. Hazra, Y. P. Bharitkar, T. Jha, H. K. Majumder, S. Roychoudhury and N. B. Mondal, Med. Chem. Commun., 2015, 6, 702–707 RSC.
- H. J. Shang, Y. Qian, D. D. Zhu, X. G. Yang and H. L. Zhu, Eur. J. Med. Chem., 2011, 46, 4702–4708 CrossRef PubMed.
- S.-Y. Ku and S.-Y. Lu, Int. J. Electrochem. Sci., 2011, 6, 5219–5227 CAS.
- D. Gayathri, R. M. Sethukkarasi, D. Velmurugan, K. Ravikumar and G. Periyasami, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o3490 CAS.
- Y. Z. Zhu, S. H. Huang, K. H. Tan, J. Sun, M. Whiteman and Y. C. Zhu, Nat. Prod. Rep., 2004, 21, 478–489 RSC.
- H. Ardill, X. L. R. Fontaine, R. Grigg, D. Henderson, J. Montgomery, V. Sridharan and S. Surendrakumar, Tetrahedron, 1990, 46, 6449–6466 CrossRef CAS.
- (a) L. R. Domingo, J. Org. Chem., 1999, 64, 3922–3929 CrossRef CAS; (b) K. V. Gothelf and K. A. Jorgensen, Chem. Rev., 1998, 98, 863–909 CrossRef CAS PubMed.
- (a) J. L. Simonsen, C. Rosada, N. Serakinci, J. Justesen, K. Stenderup, S. I. Rattan, T. G. Jensen and M. Kassem, Nat. Biotechnol., 2002, 20, 592–596 CrossRef CAS PubMed; (b) M. Elsafadi, M. Manikandan, M. Atteya, J. A. Hashmi, Z. Iqbal, A. Aldahmash, M. Alfayez, M. Kassem and A. Mahmood, Stem Cells Int., 2016, 2016, 9378081 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00646f |
| This journal is © The Royal Society of Chemistry 2018 |