
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn silico functional and tumor suppressor role of hypothetical protein PCNXL2 with regulation of the Notch signaling pathway
Muhammad Naveed *ab,
Komal Imranb,
Ayesha Mushtaqb,
Abdul Samad Mumtazc,
Hussnain A. Janjuad and
Nauman Khalid*e
*ab,
Komal Imranb,
Ayesha Mushtaqb,
Abdul Samad Mumtazc,
Hussnain A. Janjuad and
Nauman Khalid*e
aDepartment of Biotechnology, Faculty of Life Sciences, University of Central Punjab, Lahore 5400, Pakistan. E-mail: naveed.quaidian@gmail.com; Tel: +92 301 5524624
bDepartment of Biotechnology and Biochemistry, University of Gujrat, Gujrat 50700, Pakistan
cDepartment of Plant Sciences, Quaid-i-Azam University, Islamabad 44500, Pakistan
dDepartment of Industrial Biotechnology, Att-Ur-Rahman School of Applied Biosciences, National University of Sciences and Technology, H-12, Islamabad, Pakistan
eSchool of Food and Agricultural Sciences, University of Management and Technology, Lahore 54000, Pakistan. E-mail: nauman.khalid@umt.edu.pk; Fax: +92 42 3518478; Tel: +92 333 5278329
First published on 12th June 2018
Abstract
Since the last decade, various genome sequencing projects have led to the accumulation of an enormous set of genomic data; however, numerous protein-coding genes still need to be functionally characterized. These gene products are called “hypothetical proteins”. The hypothetical protein pecanex-like protein 2 Homo sapiens (PCNXL2) is found to be mutated in colorectal carcinoma with microsatellite instability; therefore, annotation of the function of PCNXL2 in tumorigenesis is very important. In the present study, bioinformatics analysis of PCNXL2 was performed at the molecular level to assess its role in the progression of cancer for designing new anti-cancer drugs. The retrieved sequence of PCNXL2 was functionally and structurally characterized through the web tools Pfam, Batch CD (conserved domain) search, ExPASy, COACH and I-TASSER directed for pathway analysis and design to explore the intercellular interactions of PCNXL2 involved in cancer development. The present study has shown that PCNXL2 encodes multi-pass transmembrane proteins whose tumor suppressor function may involve regulating Notch signaling by transporting protons across the membrane to provide suitable membrane potential for γ secretase function, which may liberate the Notch intracellular domain NICD from the receptor to inside the cell. Furthermore, domain A of PCNXL2 may exhibit nuclear transport activity of NICD from the cytoplasm to the nucleus through interaction with a nuclear localization signal that may act as an activator for Notch signaling in the nucleus. Conclusively, the tumor suppressor role of PCNXL2 by regulation of the Notch signaling pathway and its functional and structural characteristics are important findings. However, further studies are required to validate the putative role of PCNXL2 as a cancer biomarker in cancer development.
Introduction
Although the genome sequences of many organisms are available in databases, there are some proteins whose functions are unknown; it is necessary to functionally characterize these proteins, which are known as hypothetical proteins.1 The sequences of these proteins are known, but no experimental studies have been conducted to explore their functions. This is due to the fact that the proteins are predicted using computational methods, which rely on signals in DNA sequences to predict them as genes or based on their similarity to genes in other organisms. Therefore, it is very important to classify hypothetical proteins in order to obtain new understanding of regulatory and molecular processes in cells.Functional annotations of hypothetical proteins and docking sites provide an extensive outlook of essential biochemical pathways or the cellular processes responsible for the development and prognosis of any disease in the body.2 The associations between biologically relevant molecules such as proteins, nucleic acids, carbohydrates, and lipids play central roles in signal transduction; however, the relative orientation of the two interacting partners may affect the type of signal produced (e.g., agonism vs. antagonism).3 Therefore, docking is useful for predicting both the strength and type of a signal produced by hypothetical protein–ligand interactions to the appropriate target binding sites.4 Therefore, characterization of the binding behavior of hypothetical proteins will play an important role in the rational design of drugs with the elucidation of fundamental biochemical processes.
Pecanex homolog 2 (PCNXL2) is a protein-coding gene, and homologs of this gene are present from Drosophila to humans.5 In Drosophila, it is observed that loss of pcx (pecanex) may cause a neurogenic phenotype that may involve the N signaling pathway. pcx resides in the endoplasmic reticulum in Drosophila, and its disruption causes enlargement of the ER; this confirms its characteristic role in ER functioning.6 Previous studies reported that frameshift mutations have been found in FLJ11383 (PCNXL2) in patients suffering from colorectal carcinoma.7 Although the incidence of mutations is high, the role of these mutations in tumorigenesis is not yet clear. A study demonstrated that PCNXL2 encodes a multipass transmembrane protein with unknown functions in Drosophila that may be required during activation of the N signaling pathway. Actually, pecanex (pcx) functionally encodes a multi-pass transmembrane protein with unknown functions that is widely found from Drosophila to humans; due to its maternal neurogenic effects, it may be involved in activation and regulation of N signaling pathways.8 A study reported in Drosophila described pcx as a component of the Notch signaling pathway that is required upstream during activation of the pathway.6 Because pcx residues are present in the endoplasmic reticulum and its disruption can cause ER enlargement, pcx may lead to lateral inhibition of neuroblasts. No known predicted secondary structures of PCNXL2 have been identified to date in humans. Therefore, the detailed structure and function of this protein must be determined to understand the signaling transduction pathway. In humans, the Notch signaling pathway has diverse consequences due to its cross-talk with other pathways or cell-specific regulatory molecules; its disruption can lead to pulmonary arterial hypertension, leukemia and several cancers by inhibiting its tumor suppressor role in a cellular context.9 Notch signaling involves direct cell to cell communication through interaction of the expressed ligand with the Notch receptor of a neighboring cell. Ligand–receptor interaction causes Notch activation by cleavage of the receptor through activated γ secretase to release the activated Notch intracellular domain, NICD, in the cytoplasm; this is followed by translocation to the nucleus, where it may associate with the DNA binding protein CSL and regulate the transcription of other tumor suppressor genes.10 During cellular maturation, the extracellular domain of N is cleaved by furin protease, ADAM10/ADAM17 and γ secretase to liberate the intracellular domain for cell–cell Notch mediating signaling. N requires several posttranslational modifications, such as O-glycosylation, for optimal disulfide bond formation in the extracellular domain of N for activation of N signaling.11
pcx is a component of the N signaling pathway; however, the exact molecular function of the pcx encoded multi-pass transmembrane protein is not known. The present study was conducted to characterize PCNXL2 functionally and structurally to understand its gene regulatory network; molecular docking was performed to provide insight into its exact molecular process in the N signaling pathway, which may be related to its tumor suppressor role and its putative role as a cancer biomarker for patient prognosis and therapeutic intervention.
Materials and methods
Sequence retrieval
The complete protein sequence of PCNXL2 was retrieved from the NCBI Protein Database (https://www.ncbi.nlm.nih.gov/) and GenBank (https://www.ncbi.nlm.nih.gov/genbank/) under the specified accession number (NP_055616.3) and gene ID (gi|134254443). A flow sheet of the methodology is provided in Fig. 1. | ||
| Fig. 1 Illustration of string analysis. The string analysis shows that PCNXL2 interacts with eight neighboring proteins as a result of biochemical events and/or electrostatic forces. Literature analysis found only three proteins in the string network: actin-like 6 protein (ACTL6A), TATA box binding associated factor RNA polymerase 1 B (TAF1B), and fibulin-5 (FBLN5) play important roles in the regulation of cell fate, causing cancer by interacting within the PCNXL2 signaling pathway. | ||
Conserved domain prediction
Pfam, Batch CD and ScanProsite were used to predict the conserved domains in PCNXL2 in order to characterize its protein functions at the molecular level. Pfam (https://pfam.xfam.org/) is a database containing a large collection of protein families; each protein family is represented by multiple sequence alignments (MSA) and hidden Markov models (HMMs).12 The family of the hypothetical protein was identified by searching consensus sequences in the structure coding conserved domains of the PCNXL2 protein.13A Batch CD search (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi) was also performed to highlight conserved domains in PCNXL2 for further confirmation of our results. This program uses the NCBI web interface to explore the conserved domain database within protein-coding sequences using RPSBLAST, a variant of PSI-BLAST, and Position-Specific Scoring Matrices (PSSMs).14 ScanProsite (https://prosite.expasy.org/scanprosite/) allows searching of domains, functional sites and protein families by comparing amino acid patterns in the PCNXL2 protein.15 These predictions assisted in the characterization of the biochemical function of our hypothetical protein.
Prediction of physiochemical parameters
All proteins are dependent upon their structures and functions and, in turn, upon their physical and chemical parameters in a biological system. We used the ExPASy-ProtParam tool (https://www.expasy.org/tools/protparam.html) and EMBOSSPepstats (https://www.ebi.ac.uk/Tools/seqstats/emboss_pepstats/) to investigate the physical and chemical properties of PCNXL2 for its functional validation in the lab.16 We submitted protein queries in the form of a SWISS ID or protein sequence.17,18 The server directly calculated the values of pI/MW (isoelectric point, molecular weight), percentage of each amino acid, extinction coefficient (EC), instability index (II), aliphatic index (AI) and GRAVY (Grand Average of Hydrophobicity) of the key protein query.19Nature and cellular location of protein
We used the SOSUI server (https://harrier.nagahama-i-bio.ac.jp/sosui/) to calculate the amphiphilicity index and hydropathy index to estimate the nature of the PCNXL2 protein.20 The nature of a protein indicates whether it resides in the cytoplasm or spans the transmembrane. The SOSUI server contains an arsenal of tools, such as SOSUI (Batch), SOSUIsignal, SOSUIgramN and SOSUImp1. These tools were also utilized to predict part of the secondary structure of PCNXL2 from its given amino acid sequence (AAS) to estimate the location of the hypothetical protein in a cell.21Protein–protein interaction network
A protein interacts with other proteins in the cell environment; we studied these interactions in silico using STRING v9.1 (Search Tool for Retrieval of Interacting Genes). STRING (https://string-db.org/) is a large repository of protein–protein interaction networks, including functional interactions, regulatory interactions and stable complexes of proteins.22 We searched the protein–protein interactions (PPIs) of PCNXL2 by submitting a protein query sequence in the search box of STRING. STRING executes protein interactions by molecular machines actively assembled by specific PPIs in the STRING database.23 Determining the protein–protein interaction network of PCNXL2 would empower our current study of biochemical signaling pathways in cancer development.Disulfide bonding in the protein
Disulfide bonds between cysteine residues of a protein play an important role in the folding of the protein into a functional and stable conformation. We used CYSPRED, DIANA and CYSCON to predict disulfide bonding within PCNXL2 to obtain some insight into the experimental structure determination and stability of the protein.CYSPRED (https://gpcr.biocomp.unibo.it/cgi/predictors/cyspred/pred_cyspredcgi.cgi) determines whether the cysteine residues in your query protein form disulfide bridges/bonds. CYSPRED is a neural network-based predictor that is trained to distinguish the bonding states of cysteine in proteins, starting from the residue chain in its non-binding state, with high efficiency.24
DIANA (https://clavius.bc.edu/%7Eclotelab/DiANNA/) was also used because it helps predict the disulfide connectivity of an input protein sequence. Correct prediction of the disulfide bonds in a hypothetical protein is of crucial interest for understanding the protein function and is key for tertiary prediction methods.25 The tertiary structure of a hypothetical protein will be helpful in identifying docking sites, moving one step closer towards designing drugs that target diseases caused by mutations in the PCNXL2 hypothetical gene.
One additional tool, CYSCON (https://www.csbio.sjtu.edu.cn/bioinf/Cyscon/), was used for disulfide bond prediction to enhance and confirm the knowledge we obtained from the abovementioned software. CYSCON first identified the most confident disulfide bonds within the hypothetical protein; then, the prediction focused on the remaining cysteine residues based on Support Vector Regression (SVR) training.26
Protein structure prediction
Protein structure prediction was performed to estimate the location of all the atoms in the PCNXL2 molecule using its amino acid sequence through the computational methods RaptorX (https://raptorx.uchicago.edu/) and Iterative Threading Assembly Refinement (I-TASSER) (https://zhanglab.ccmb.med.umich.edu/I-TASSER/). First, RaptorX was used due to its excellent secondary and tertiary structure predictions for longer hypothetical protein molecules. The PCNXL2 protein sequence was submitted to RaptorX to obtain the predicted 3D model, contact map and solvent accessibility.27 The best model was selected on the basis of P-value, GDT (global distance test), uGDT (un-normalized GDT) and RMSD (root mean square deviation) by estimating the absolute global quality and the absolute local quality of each residue in the model.28 Second, I-TASSER was used for detailed functional annotation of domains predicted in our best model. I-TASSER cannot run longer protein sequences, so we first predicted domains in the complete 3D model of PCNXL2 and then used I-TASSER to elucidate the biological function of the protein. I-TASSER uses a hierarchical approach to protein structure and function prediction.26 I-TASSER was used to elucidate the biological functions of PCNXL2, including its gene ontology, by comparing its structure and function profile with those of known proteins; ligand binding site prediction, solvent accessibility and enzyme commission number were determined on the basis of a sequence-to-structure-to-function paradigm.29Ligand binding site prediction
COACH (https://zhanglab.ccmb.med.umich.edu/COACH/) was used for protein–ligand binding site prediction in PCNXL2. COACH is a meta-server approach that uses two comparative methods, TM-SITE and S-SITE, for ligand binding site predictions. For the final ligand binding site predictions, the TM-SITE and S-SITE predictions were combined with COFACTOR (https://zhanglab.ccmb.med.umich.edu/COFACTOR/), FindSite (https://cssb.biology.gatech.edu/findsite) and ConCavity (https://compbio.cs.princeton.edu/concavity/).30 Domain sequences of PCNXL2 were submitted in COACH, and 3D models were constructed by I-TASSER. COACH outputs hot spot sites on PCNXL2 for ligand binding after ranking their physicochemical properties, such as van der Waals potentials, hydrophobicity, desolvation and electrostatic interactions.miRNA target prediction
We further determined the potential role of PCNXL2 protein in the treatment of cancer by filtering the PCNXL2 protein sequence in miRTarBase. miRTarBase searched a list of microRNAs targeting PCNXL2.31 miRTarBase (https://mirtarbase.mbc.nctu.edu.tw/) is the experimentally validated microRNA target interaction database.32 The miRNA-target expression profile of the enlisted microRNAs was also studied in miRTarBase to evaluate which microRNA expression is high in a particular cancer based upon the Pearson coefficient (r) and probability value (P). A Pearson coefficient ranging from −1 to +1 indicates the type of correlation between the microRNA expression level and the occurrence of cancer. 0 indicates no relationship, while +1 indicates a strong positive correlation and −1 indicates a negative correlation. The probability value is the criterion for the approval or rejection of a null hypothesis. If the probability value is less than or equal to 0.005, our null hypothesis of ‘‘high expression level of a particular microRNA causes cancer by inhibiting PCNXL2 expression in a cell’’ is rejected.Pathway analysis and design
Finally, pathway analysis was performed by viewing gene expression in the Cancer Genome Atlas and its involvement in biological pathways of different cancers.33 The Cancer Genome Atlas (TCGA) was used in a comprehensive and coordinated effort to accelerate our understanding of the molecular basis of cancer by visualizing the expression of the PCNXL2 gene in a particular cancer. Reactome (https://www.reactome.org/), a curated and peer-reviewed pathway database, was also used for detailed study of the protein interactions playing important roles in the transduction of a signaling pathway.34 Literature and KEGG pathways (https://www.genome.jp/kegg/pathway.html) were also reviewed in order to generate a hypothesis that these relevant molecular and regulatory processes are controlled by selected microRNAs targeting PCNXL2 in the progression of cancer.35 DAVID (https://david.ncifcrf.gov/home.jsp) was also used to provide comprehensive information by using a set of computational tools to evaluate the biological meaning of the list of genes interacting with our hypothetical protein PCNXL2.36 We did not find any biological pathways related to PCNXL2 or any other related gene pathways in the KEGG and Reactome databases. To address this issue, we manually designed the pathway by reviewing the literature to deepen our understanding of the function of PCNXL2 in cancerous tissues.Results
PCNXL2 contains the pecanex_c conserved protein domain family
Proteins are generally composed of one or more functional regions, commonly named domains; characterization of these domains is necessary to confirm the structure and functions of a protein at the cellular level. The Pfam database suggests that the hypothetical protein pecanex-like protein 2 Homo sapiens (PCNXL2) contains the pecanex_c conserved protein domain family (pfam05041) with the terminal region of the pecanex protein homologs (pecanex protein C-terminus), as described in Table 1. The pecanex protein is a maternal-effect neurogenic gene found in Drosophila. Searching with the Batch CD search tool, a specific hit with a bit score of 384.928 and an E value of 9.69802 × 10−122 indicates that clancl04904 (pecanex C superfamily) contains one domain and one cluster.| ProtParam tool | EMBOSS Pepstats | ||
|---|---|---|---|
| Sequence ID | gi|134254443|r | Charge | 9.5 |
| Family (Pfam) | Pecanex_C superfamily | Improbability of expression in inclusion bodies | 0.747 |
| Domain (ScanProsite) | DNAJ_1 | Average residue weight | 111.033 |
| Alignment | 1619–1844 | A280 extinction coefficients 1 mg ml−1 | 1.039 |
| HMM length | 229 | A280 molar extinction coefficients | 246![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 635 635 |
| Bit score | 369.2 | Tiny (A + C + G + S + T) | 662 |
| E value | 6.3 × 10−111 | Small (A + B + C + D + G + N + P + S + T + V) | 1084 |
| Number of AA | 2137 | Aliphatic (A + I + L + V) | 608 |
| MW | 237276.9 |
Aromatic (F + H + W + Y) | 238 |
| pI | 6.29 | Non-polar (A + C + F + G + I + L + M + P + V + W + Y) | 1108 |
| EC | 246635 |
Polar (D + E + H + K + N + Q + R + S + T + Z) | 1029 |
| II | 48.98 | Charged (B + D + E + H + K + R + Z) | 487 |
| AI | 87.45 | Basic (H + K + R) | 265 |
| GRAVY | −0.204 | Acidic (B + D + E + Z) | 222 |
ScanProsite predicted that DNAJ_1 PS00636 (Nt-dnaJ domain signature) belongs to the J protein family in PCNXL2, ranging from 1270 to 1289 amino acids. DNAJ_1 regulates the activity of hsp70, which is a molecular chaperone responsible for the translocation of polypeptides across organelle membranes, folding of nascent proteins, targeting proteins for degradation and inferring responses to stress.37
PCNXL2 is a hydrophilic protein
The ProtParam tool computed various physiochemical properties of PCNXL2, such as its molecular weight, theoretical pI, amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index and grand average of hydropathicity (GRAVY), as shown in Table 1. The ProtParam tool calculated a pI of 6.9 and a MW of 237276.9 for protein PCNXL2; these parameters assist in protein separation through 2-D gel electrophoresis in the lab. The extinction coefficient (EC) shows the amount of light absorbed by a protein relative to its amino acid composition at a specific wavelength (280 nm); this enables estimation of the amount of pure protein in a sample. The instability index (II) refers to the stability of a protein in a test tube; for PCNXL2, the value is 48.98, which classifies the protein as unstable. The aliphatic index indicates the relative volume of the aliphatic side chains. The aliphatic index is regarded as a positive factor for increased thermostability of proteins. A higher AI value indicates increased thermostatic stability of a protein. Sequence ID gi|134254443|r PCNXL2 reflects a moderate AI value of 87.45. The smaller the grand average of hydropathy (GRAVY), the more hydrophilic the protein. For PCNXL2, the GRAVY value is −0.204; this suggests that PCNXL2 is a hydrophilic protein (Table 1). In the 3D structure of a protein, hydrophilic domains tend to be found at the exterior surface of the protein while hydrophobic domains form the internal core of the protein.
EMBOSS Pepstats shows statistics of the physiochemical properties of PCNXL2, such as the number and molar percent of each type of amino acid; by applying DayhoffStat, the number and molar percent of each physiochemical class of amino acid, the extinction coefficient at 1 mg ml−1 (A280) and the molar extinction coefficient (A280) can be obtained. The EMBOSS Pepstats results showed that PCNXL2 exhibits 9.5 charges and consists of up to 608 aliphatic, 238 aromatic, 1108 nonpolar and 1029 polar amino acids (Table 1). The aliphatic amino acids compose the core of the protein, while polar and charged amino acids are present on the surface of the protein and interact with solvent molecules. The positive and negative amino acids form salt bridges and maintain the three-dimensional structure of the protein. The molar percentage of each amino acid reflects the ability of the protein to form secondary structures. The amino acids M, A, L, E, K, G, Y, S, H, R, and Q prefer to adopt helical conformations, while T, W, Y, F, I, and V prefer to adopt β conformations. DayhoffStat predicted that most of the amino acids in PCNXL2 (A, R, N, Q, E, K, H, P, S, T, W, and Y) have negative scores; this characterizes PCNXL2 as hydrophilic.
PCNXL2 is a multispanning membrane protein
The SOSUI server predicted that PCNXL2 is an endoplasmic reticulum (ER) membrane protein consisting of 16 transmembrane helices (Table 2). Membrane proteins represent 50% of the targets of all modern medicinal drugs; thus, PCNXL2 is demonstrated to be a vital candidate for drug design against cancers.| No. | N-terminal | Transmembrane region | C-terminal | Type | Length |
|---|---|---|---|---|---|
| 1 | 63 | TNSCHLYLWLFLLLLPLALHLAF | 85 | PRIMARY | 23 |
| 2 | 92 | VFFYCSAVTIFFTIIKLVSYRLH | 114 | PRIMARY | 23 |
| 3 | 881 | IVLVSLLGFLTLSQGFCKDMWVL | 903 | PRIMARY | 23 |
| 4 | 936 | TYSRPIYFCVLCGLILLLDTGA | 957 | PRIMARY | 22 |
| 5 | 982 | RDYLIVFLYCFPAISLLGLFPQI | 1004 | PRIMARY | 23 |
| 6 | 1008 | CTYLLEQIDMLFFGGSAVSGITS | 1030 | SECONDARY | 23 |
| 7 | 1034 | SVARSVLAAALLHAVCFSAVKEP | 1056 | PRIMARY | 23 |
| 8 | 1059 | MQHIPALFSAFCGLLVALSYHLS | 1081 | SECONDARY | 23 |
| 9 | 1129 | WDLIVCAVVAVLSFAVSASTVFL | 1151 | PRIMARY | 23 |
| 10 | 1157 | LSIVLFALAGAVGFVTHYVLPQL | 1179 | PRIMARY | 23 |
| 11 | 1224 | KYILYPALILNALTIDAFLISN | 1245 | PRIMARY | 22 |
| 12 | 1249 | LGTHWDIFLMIIAGMKLLRTSFC | 1271 | SECONDARY | 23 |
| 13 | 1273 | PVYQFINLSFTVIFFHFDYKDIS | 1295 | SECONDARY | 23 |
| 14 | 1301 | DFFMVSILFSKLGDLLHKLQFVL | 1323 | SECONDARY | 23 |
| 15 | 1333 | WGSSFHVFAQLFAIPHSAMLFFQ | 1355 | SECONDARY | 23 |
| 16 | 1360 | SIFSTPLSPFLGSVIFITSYVRP | 1382 | SECONDARY | 23 |
Other built-in software in the SOSUI server, such as SOSUI (Batch), confirmed that PCNXL2 is a 100% membrane protein. SOSUIsignal predicted that PCNXL2 has no signal peptide, which shows that PCNXL2 is not involved in the secretory pathways of the cell. SOSUIgramN predicted that the inner membrane is the subcellular localization site of PCNXL2. SOSUImp1 showed that PCNXL2 is a multi-spanning membrane protein with potential to perform functions such as signal transduction, immunological reaction and cell adhesion (Table 3).
| SOSUI (Batch) | SOSUI signal | SOSUIgramN | SOSUImp1 | ||
|---|---|---|---|---|---|
| Nature | Percentage | Signal peptide | Nature | Subcellular localization site | Spanning membrane protein |
| Membrane protein | 100% | No | Soluble | IM (inner membrane) | Multi |
PCNXL2 interacts with tumor suppressor proteins
STRING analysis showed that PCNXL2 experiences interactions with neighboring proteins through various biochemical events and/or electrostatic forces to perform various molecular processes involved in homeostasis of the cell (Fig. 2). After a through literature review, a comprehensive understanding of the functioning of genes in cancer development was proposed. The following proteins were observed in the string network: actin-like 6 protein (ACTL6A), TATA box binding associated factor, RNA polymerase 1 B (TAF1B), and fibulin-5 (FBLN5); these play major roles in the regulation of cell fate, suggesting that cancer in cells involves their interactions with the PCNXL2 signaling pathway. | ||
| Fig. 2 Predicted model for the whole sequence of PCNXL2. (RaptorX results: current status of the model of PCNXL2 is complete). | ||
Actin-like 6-A (ACTK6A) is a member of the SWI/SNF like BRG1/brm associated factor (BAF) chromatin remodeling complex that encodes BAF53a, the 53 kDa subunit of the BAF complex in mammals.38 ACTL6A is involved in diverse cellular processes, such as nuclear migration, chromatin remodeling and transcriptional regulation. Recently, a study by Bao et al.39 demonstrated that SOX2 is a potential target of ACTL6A and ACTL6A ectopic expression; enhanced SOX2 expression up-regulates Notch 1 and triggers the Notch signaling pathway in the cell. Hence, ACTL6A can enhance Notch activity in cells, exhibiting a tumor suppressor function by targeting reduced proliferation and apoptosis of cancerous cells. Another study by Rowland and Peeper40 suggests that ACTL6A regulates the SWI/SNF chromatin remodeling complex to suppress differentiation in the epidermis by preventing SWI/SNF from binding to promoters of KLF4 and other differentiation genes. KLF4 is a transcription regulator that dictates diverse biological processes, including pluripotency, cell differentiation, tumorigenesis and proliferation.40
TATA box binding associated factor RNA polymerase 1 B (TAF1B) is a component of the RNA polymerase 1 core factor complex; it plays active roles in multiple steps during transcription initiation, such as pre-initiation complex (PIC) assembly and post polymerase recruitment events. TAF1B, the second largest component of TATA-binding protein (TBP), binds to ribosomal DNA by recruiting POL1 to the SL1/TIF-IB complex through its interaction with RRN3.41 A study demonstrated that MARCK2, PCNXL2 and TAF1B are putative target genes in colorectal carcinoma with microsatellite instability.7 PCNXL2 (74%) and TAF1B (82%) were found to be mutated in MSI-H colorectal carcinomas. This study reported that frameshift mutations of the three abovementioned target genes are related to functional inactivation by generating truncated proteins and may be related to tumorigenesis. Cell cycling and protein synthesis are key regulatory tasks for cancerous cells. TAF1B is involved in the synthesis of proteins and mutations and has been found in many cancers; therefore, we can conclude that mutations result in truncated proteins which are unable to perform their functional roles in the homeostasis of cells. Furthermore, these mutations may inactivate tumor suppressor genes and DNA repairing genes in cells by providing a suitable cancerous microenvironment which initiates tumorigenesis.
Fibulin-5 (FBLN5) is a member of the fibulin family that is widely expressed as an extracellular matrix protein (ECM) and is involved in cell-to-matrix or cell-to-cell communication, elastogenesis, cell adhesion and cell mobility.42 Fibulin-5 is a 66 kDa glycoprotein that is localized in elastic fiber and is involved in regulating elastic fiber assembly. It has a unique RGD domain which can bind to integrins on cell surface receptors. Integrins regulate intracellular signaling through extracellular signal-regulated kinase (ERK), and focal adhesion kinase (FAK) mediates cell adhesion to the extracellular matrix.43 The Notch signaling pathway performs its tumor suppressor role by inhibiting Wnt/β catenin signaling through increased p21 expression, which may act as a negative modulator of Wnt-4. Fubilin-5 also acts as a metastasis suppressor by inhibiting the Wnt/β catenin pathway through inhibition of ERK via its unique RGD domain, leading to activation of GSK3β. GSK3β downregulates β-catenin by proteasomal degradation, preventing its translocation to the nucleus; this leads to suppression of the oncoproteins metalloproteinase-7 (MMP-7) and c-Myc.44
PCNXL2 is a cysteine-rich protein
CYSPRED predicted that cysteines which are capable of forming disulfide bonds within PCNXL2 are responsible for the stability of the protein (Table 4). When more disulfides are present in a protein, the protein will be more stable, with a positive signal for the 3-D structure of the hypothetical protein. DIANA predicted that the disulfide bond topology in PCNXL2 is of crucial importance for understanding its function and its tertiary prediction methods. DIANA showed the bonds that are predicted to be present between respective cysteine residues within PCNXL2, which represents a further step towards the design of anti-cancer drugs by targeting hypothetical protein molecular docking sites (Table 4).| CYSPRED | DIANA | CYSCON | ||||
|---|---|---|---|---|---|---|
| Cysteine | Prediction | Reliability | Sequence ID | Bonded cysteine | SSBOND | Cysteine residues |
| CYS 144 | Bonding state | 8 | gi|134254443|r | 34–1239, 64–337, 144–1402, 290–880, 312–614, 315–874, 331–797, 445–467, 519–1634, 652–715, 736–1472, 865–1473, 912–1587, 915–2086, 959–1418, 976–1475, 1017–1038, 1064–1781, 1102–2101, 1189–1873, 1454–2114, 1474–1924, 1486–1821, 1597–1704, 1607–1728 | SSBOND#1 | 34–2114 |
| CYS 290 | Bonding state | 1 | SSBOND#2 | 64–2086 | ||
| CYS 312 | Bonding state | 2 | SSBOND#3 | 144–2101 | ||
| CYS 315 | Bonding state | 8 | SSBOND#4 | 290–1924 | ||
| CYS 331 | Bonding state | 8 | SSBOND#5 | 312–1821 | ||
| CYS 337 | Bonding state | 2 | SSBOND#6 | 315–1873 | ||
| CYS 467 | Bonding state | 8 | SSBOND#7 | 331–1781 | ||
| CYS 519 | Bonding state | 1 | SSBOND#8 | 337–1728 | ||
| CYS 715 | Bonding state | 1 | SSBOND#9 | 445–1704 | ||
| CYS 736 | Bonding state | 1 | SSBOND#10 | 467–1634 | ||
| CYS 1418 | Bonding state | 8 | SSBOND#11 | 614–652 | ||
| CYS 1472 | Bonding state | 8 | SSBOND#12 | 715–736 | ||
| CYS 1473 | Bonding state | 8 | SSBOND#13 | 1017–1038 | ||
| CYS 1474 | Bonding state | 4 | SSBOND#14 | 1402–1418 | ||
| CYS 1475 | Bonding state | 4 | SSBOND#15 | 1587–1597 | ||
| CYS 1607 | Bonding state | 4 | ||||
| CYS 2086 | Bonding state | 2 | ||||
CYSCON, a disulfide bond predicting software program, predicted SSBonds for the cysteine residues of PCNXL2 (Table 4). These results display a new path to improve the ab initio structure modeling of cysteine-rich proteins such as PCNXL2.
PCNXL2 structure and function
A web-based tool, RaptorX, generated the best domain of our PCNXL2 protein sequence and predicted the secondary and tertiary structures for the complete sequence of PCNXL2 (Fig. 3). RaptorX modulated two domain structures solely by taking the FASTA format of the PCNXL2 protein. The software results for PCNXCL2 comprise three sections which contain important information about its secondary structure modeling. The quality of the predicted model depends on three critical parameters: P-value, Score and uGDT (GDT). The results were divided into three section boxes; Section 1 contains the input protein sequence and domain prediction, Section 2 contains a summary of the predicted results in detail along with the most promising predicted complete structure of PCNXL2, and Section 3 is the main hotspot section of our study because it contains structural and functional annotations of crucial predicted domains. These sections were examined in sequence: the 1st section divides the whole sequence of the protein into its residues, and each of the amino acids exhibits a domain assignment; each residue is given a particular number, and the residues marked by ‘0’ are not part of the modeling process due to the absence of matches in the PDB templates. The total no. of residues that were generated by the software for PCNXL2 is 2131. | ||
| Fig. 3 Predicted structure of the selected domain. | ||
The second section contains information about the domain number. The best template is 3m1iC; it has a probability P-value of 7.8 × 10−7, which is less than the threshold and is considered to be a good model. The current model comprises 2131 residues with uGDT (136) and GDT (6), further confirming the viability of the model (Table 5).
| Summary of predicted model | |
|---|---|
| Domains | 2 |
| Best template | 3m1iC |
| Probability value ‘P-value’ | 7.8 × 10−7 |
| Overall uGDT | 136 |
| GDT | 6 |
| Modeled residue count | 1144 |
| Disordered position count | 913 |
| Secondary structure details | 31% H (helix), 8% E (beta sheet), 60% C (loop) |
The most important and targeted section of our study is the 3rd section, ‘detailed prediction results’, which contains relevant functional and structural information about the two predicted domains. The best model was selected on the basis of P-value using the templates 3m1iC, 3gjxA, 4gmxC, 4fgvA, and 4c0oA for the first domain and 4he8: I for the second domain; all the templates have different functions in the cell. To select the best-predicted domain, various parameters were used, such as rank, P-value, uGDT (GDT), uSeqId (SeqId), and Score. The first domain (domain A) has rank: 0, P-value: 7.8 × 10−7, uGDT (GDT): 98 (9), uSeqId (SeqId): 93 (9), and Score: 237; it contains 1054 amino acids, with a sequence from 821 to 1875. In contrast, the second domain has rank: 1, P-value: 4.55 × 10−3, uGDT (GDT): 38 (43), uSeqId (SeqId): 10 (11), and Score: 53; it contains 89 amino acids, from 1 to 89. All values for the first domain A are close to the threshold, with P-values less than 0.005; the uGDT must be greater than 50 and the Score must be in the range of 0 to the domain sequence length. This domain was considered to be the best domain compared to a second domain, whose values varied considerably from this threshold value (Fig. 4).
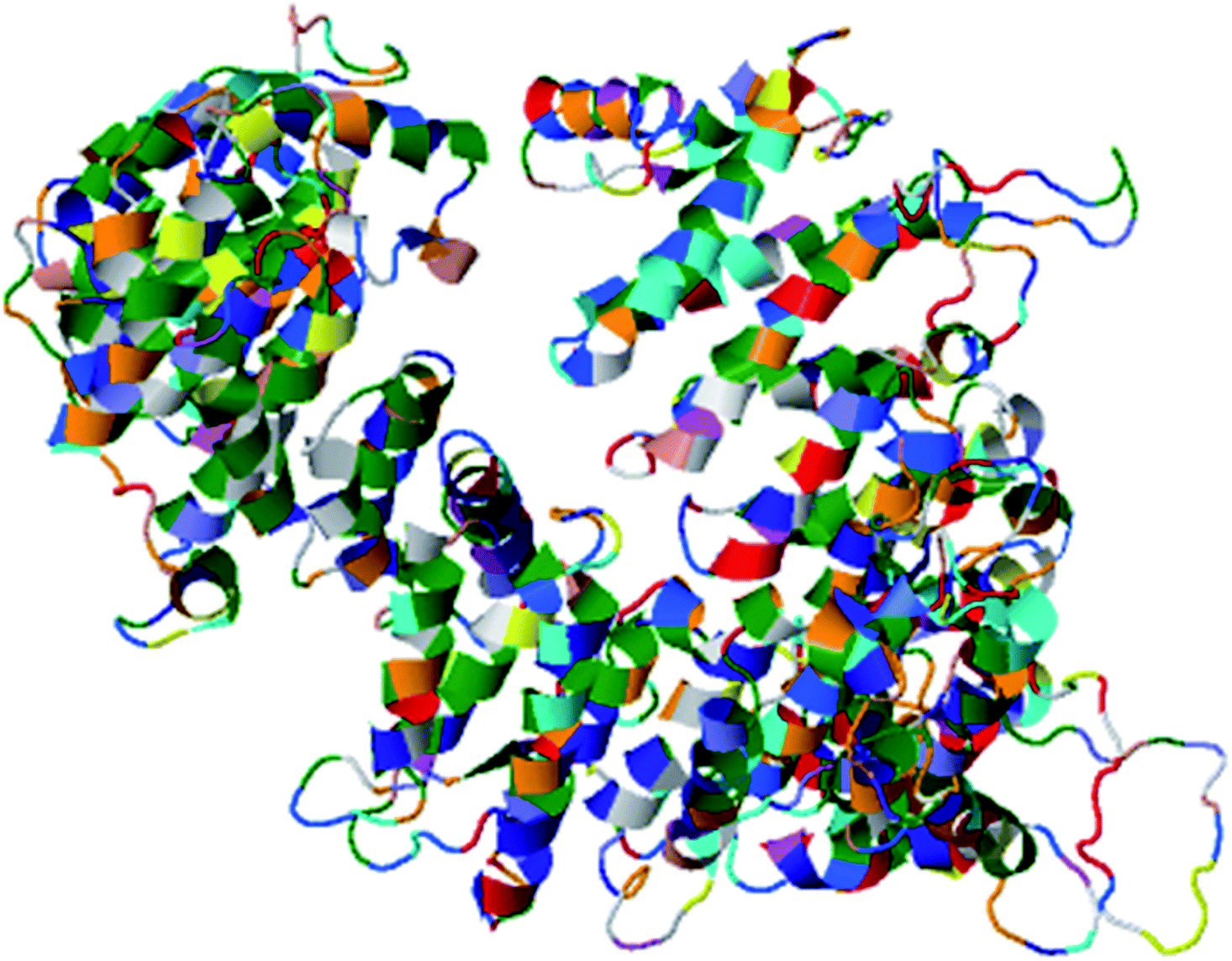 | ||
| Fig. 4 Illustration of the best predicted model for domain A. Because the C score of model 1 is higher (−1.18) than those of the other 4 predicted models, with an estimated TM score of 0.57 ± 0.15 and an RSMD of 11.9 ± 4.4 Å, it is considered to be the best predicted model for domain A. | ||
I-TASSER was applied to obtain comprehensive structural and functional understanding of the RaptorX-predicted domain. Domain A consists up of 1054 amino acids, from amino acids 821 to 1875 in the PCNXL2 protein. Pfam revealed the presence of a pcx domain from amino acids 1619 to 1844 in PCNXL2; thus, the pcx domain can be considered to be part of domain A. Therefore, the sequences of domain A and pcx were submitted separately in I-TASSER for structure and function predictions.
The secondary structure of domain A showed that it is enriched with long stretches of α helixes (M, A, L, E, K, G, Y, S, H, R, Q) and loops with a confidence score of 9, followed by a few short stretches of β sheets (T, W, Y, F, I, V), suggesting that the core region in domain A is an α helix secondary structure element. The remaining amino acids (P, G, S, D, and N) disrupt the secondary structure and give rise to loops or turns called secondary structure breakers. The remaining amino acids (P, G, S, D, and N) disrupt the secondary structure and give rise to loops or turns called secondary structure breakers. Prediction of the solvent accessibility discovered buried and exposed regions of domain A with values ranging from 0 (buried residue) to 9 (exposed residue). Most of the residues in the domain have 0 values, showing their involvement in the core region of the protein; residues with higher values are hydrophilic and can potentially be involved in hydration processes. The predicted normalized B factor estimates the thermal mobility of residues in a protein. The residues at the N and C termini have positive normalized B factors, indicating that these regions are relatively stable; meanwhile, the α and β regions have values that are either negative or close to 0, revealing that these regions are structurally stable. The arrangements of particular amino acids in a primary structure determine the secondary and tertiary globular structures of a protein by sequestration of hydrophobic amino acids in the core and enrichment of hydrophilic amino acids on the surface. The top five models were generated by I-TASSER. The model with the highest confidence score (C score; −1.18), TM score (0.57 ± 0.15) and RMSD (11.9 ± 4.4 Å) was considered to be the best-predicted model for domain A (Fig. 5). After selection, the best-predicted model was aligned against all known protein structures in the PDB database to find proteins that are structurally close to our target protein. Ten protein structures close to the target were predicted. One PDB structure (3w3tA) with a C score close to 0.9 was observed and was selected for further functional study of the target protein. The model (2vdcA) with the highest EC score (0.122) and TM score (0.338) was selected; it was found to be a glutamate synthase with an EC number of 1.4.1.13. Finally, gene ontology (GO) was performed to determine the function of domain A. This section comprises two parts; the first shows 10 homologous GO templates, while the second is a consensus prediction of GO terms, including molecular function, biological process, and cellular components. A true positive template was selected by analyzing the functional homology score (Fh score) between the target and template proteins and by estimating the confidence level. The first two templates, 1qgkA and 2z5kA, were selected with Fh scores of 0.08 and C scores of 0.09 and 0.08, respectively. The GO term predicted that the cellular component of domain A is the cytosol. A literature review of these proteins revealed that domain A exhibits nuclear transport activity; thus, it may transport macromolecules or substrates from the cytoplasm to the nucleus by interacting with nuclear localization signals.45 This domain may also play roles in biological processes by phosphodiester bond hydrolysis.46
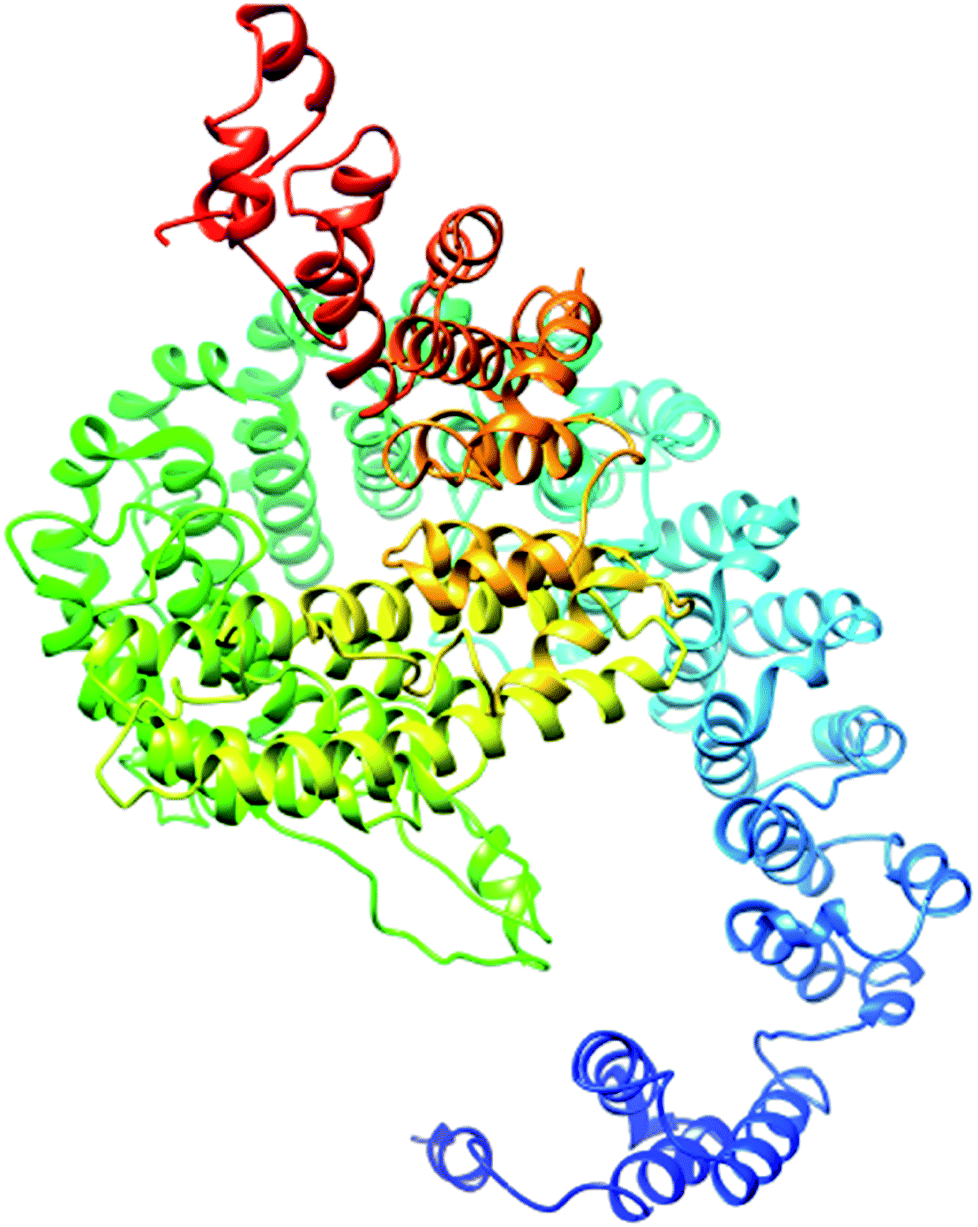 | ||
| Fig. 5 Illustration of the best predicted model for the pcx domain. Because the C score of model 1 is higher (−4.20) than those of the other 4 predicted models, with an estimated TM score of 0.27 ± 0.08 and an RSMD of 15.9 ± 3.2 Å, it is considered to be the best predicted model for the pcx domain. | ||
The secondary structure of the pcx domain showed that it is enriched with long stretches of α helixes (M, A, L, E, K, G, Y, S, H, R, Q) with a confidence score of 9, followed by a very small number of short stretches of β sheets (T, W, Y, F, I, V), suggesting that the core region in the pcx domain is an α helix secondary structure element. Most of the residues in the pcx domain have scores of 9, showing that they are hydrophilic and may undergo hydration or act as active sites. In the pcx domain, the residues at the N and C termini were predicted with positive normalized B factors, indicating that these regions are relatively stable; meanwhile, the values in the α and β regions are either negative or close to 0, revealing that these regions are structurally more stable. The best-predicted model by I-TASSER for the pcx domain has a higher C score (−4.20), TM score (0.27 ± 0.08) and RMSD (15.9 ± 3.2 Å) (Fig. 6). After selection, the best-predicted model was aligned against all known protein structures in the PDB database, and ten protein structures close to the target were predicted. Three closer protein structures, 5cwcA, 5cwoA and 2zxeA, with TM scores greater than 5 were selected for further functional study of our target protein. The enzyme commission number and active site predicted 5 models for the target protein. The model (imhsA) was selected with the highest EC score of 0.065 and TM score of 0.503, illustrating that it is a proton transport ATPase with an EC number of 3.6.3.6. Gene ontology (GO) demonstrated the molecular function of the pcx domain using 10 homologous GO templates. True positive templates, 3ixzA and 2zxeA, were selected by analyzing their Fh scores (0.07 and 0.06, respectively). The GO term predicted that the cellular component of the pcx domain is an intrinsic component of the cell membrane which spans or embeds both leaflets around the membrane. A literature review of these proteins revealed that the pcx domain exhibits ATPase activity coupled to transmembrane movement of ions (protons) by establishing membrane potential for essential biological processes, such as the liberation of an activated protein from receptors. A domain can play roles in biological processes by performing particular functions in the cell.
 | ||
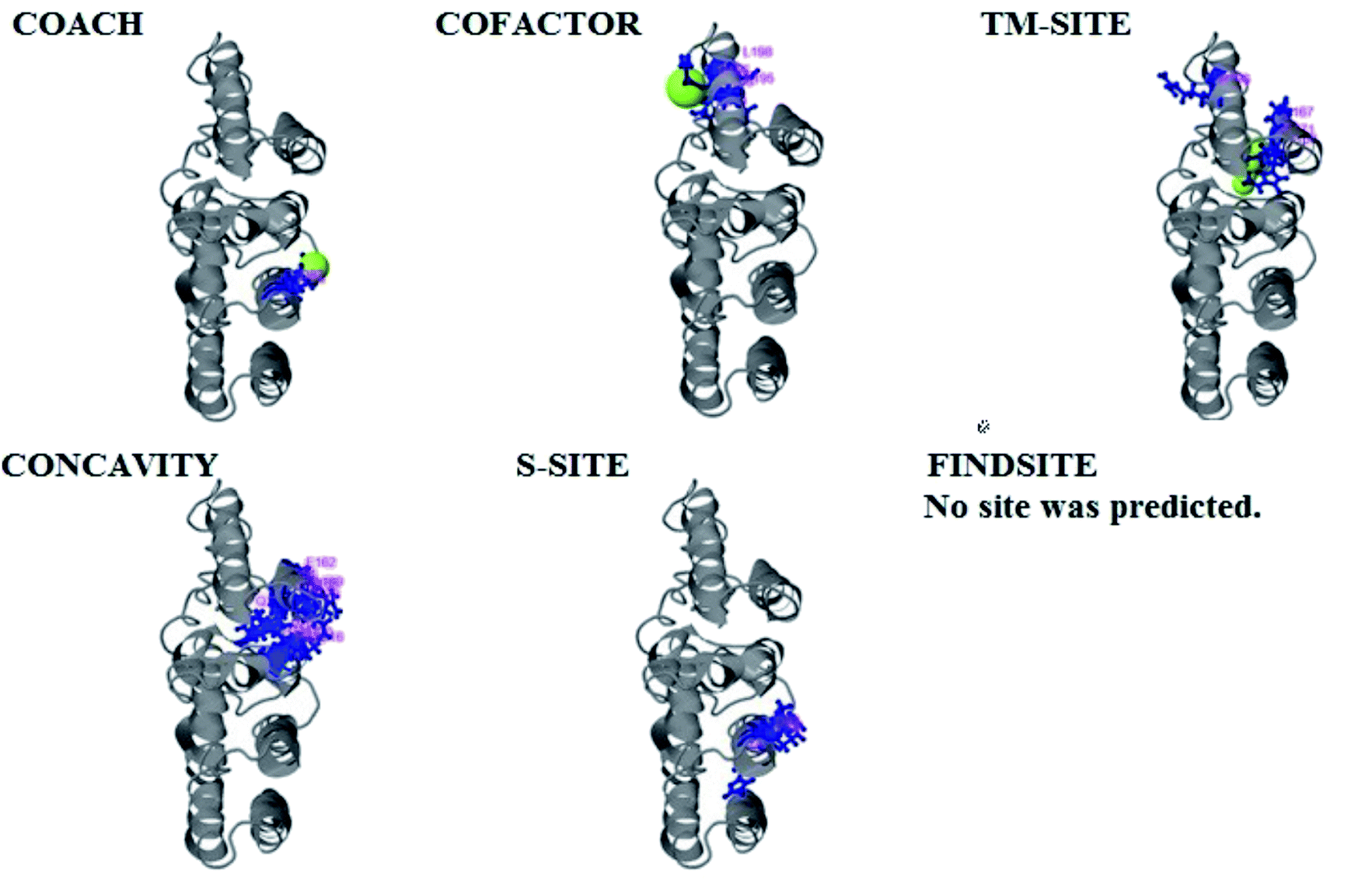
| Fig. 6 Illustration of the ligand binding site predictions of domain A. The ligand binding site predictions of domain A were executed by COACH, combining the results from five algorithms: COFACTOR, TM-SITE, S-SITE, FindSite and ConCavity. The first PDB hit, 3y3yA, was selected with a C score of 0.06, a cluster size of 4 and binding to the peptide ligand. The predicted ligand binding is indicated with yellow-green spheres, and the corresponding binding residues are shown with blue balls and sticks. | ||
PCNXL2 ligand binding interactions
The PDB file of the best predicted domain-A model was submitted to COACH for ligand binding site predictions by matching target models in the BioLip database. 10 models were predicted by COACH with different ligands. Selection of a model was conducted on the basis of C score; the higher the C score, the more reliable the model. The first PDB hit, 3y3yA, was selected with a C score of 0.06 and a cluster size of 4. This model binds to the peptide ligand. In addition to the peptide ligand, it can bind with other ligands available in the “mult” link. COACH also combines the results of five algorithms, including COFACTOR, TM-SITE, S-SITE, FindSite and ConCavity, as illustrated in Fig. 7. | ||
| Fig. 7 Illustration of the ligand binding site predictions for the pcx domain. The ligand binding site predictions of the pcx domain were executed by COACH, combining the results from five algorithms: COFACTOR, TM-SITE, S-SITE, FindSite and ConCavity. The best model was considered to be 3f1uA due to its higher C score (0.05) and cluster size of 3; also, it binds to the Zn ligand. The predicted ligand binding is indicated with yellow-green spheres, and the corresponding binding residues are shown with blue balls and sticks. | ||
COACH also predicted 10 models for ligand binding interactions of the pcx domain. The best model was considered to be 3fu1A, with a C score of 0.05 and a cluster size of 3; it binds to the zinc (Zn) ligand. The RAP ligand model can also bind with other ligands available in the “mult” link. The combined results of five algorithms, including COFACTOR, TM-SITE, S-SITE, FindSite and ConCavity, are illustrated in Fig. 8.
 | ||
| Fig. 8 Illustration of PCNXL2 regulating the Notch signaling pathway. PCNXL2 present in the ER membrane releases a protein consisting of up of 1054 amino acids containing domain A and the pcx domain. The pcx domain exhibits ATPase activity coupled to transmembrane movement of ions (protons) by establishing a membrane potential for essential biological processes, such as liberation of the activated protein NICD from receptors. Domain A exhibits nuclear transport activity; it transports NICD from the cytoplasm to the nucleus to transcriptional factor CSL to activate the expression of Notch targeted tumor suppressor genes and to downregulate tumor oncogenes, including p21, p53, p27, Smad, p63, p65, as well as WNT signaling and AP1\HPV\MRCKα. | ||
miRNAs down-regulating expression of PCNXL2
miRTarBase, the experimentally validated microRNA-target interactions database, predicted microRNAs targeting PCNXL2 with their expression profiles in various cancers, such as prostate cancer.47,48 MicroRNAs with r values close to 1 have a linear relationship between their expression and cancer incidence. MicroRNAs with P values greater than 0.005 accept the null hypothesis that states that a higher level of expression of microRNAs targeting PCNXL2 is the cause of cancer in humans. After searching with miRTarBase, 58 microRNAs were found in humans targeting PCNXL2, which were further shortlisted on the basis of the hypothesis.The miRNA-target expression profiles of the enlisted microRNAs were also studied in miRTarBase to evaluate which microRNA expression is high in a particular cancer based upon the Pearson coefficient (r) and probability value (P).49 In this expression profile, miRNA was checked against the PCNXL2 mRNA level in a particular cell by viewing and studying graphical charts. miRNAs which have positive R values in particular cancers are considered to have a positive linear relationship, justifying that targeted PCNXL2 expression is directly proportional to miRNA expression for a particular cell. Meanwhile, miRNAs which have negative R values are considered to have a negative linear relationship, justifying that targeted PCNXL2 expression is down-regulated with increased expression of the miRNA in a particular cancer. To accept the null hypothesis, the miRNA must have a P-value greater than 0.005, justifying that a high expression level of a particular microRNA causes cancer by inhibiting PCNXL2 expression in a particular cell type. The expression of selected miRNAs has been experimentally validated to have a negative linear relationship with PCNXL2 mRNA in different cancers and cells, as shown in Table 6.
| MicroRNA | Colorectal carcinoma | Breast cancer | Pancreatic cancer | Prostate cancer | CNS germ cell tumor | Multiple myeloma | Differentiated embryonic stem cell tumor |
|---|---|---|---|---|---|---|---|
| hsa-miR-154-5p | ✓ | ✓ | ✓ | ■ | ✓ | ✓ | ✓ |
| hsa-miR-382-5p | ✓ | ✓ | ■ | ✓ | ✓ | ■ | ✓ |
| hsa-miR-335-3p | ✓ | ■ | ■ | ✓ | ■ | ■ | ■ |
| hsa-miR-548d-3p | ✓ | ✓ | ■ | ■ | ✓ | ■ | ■ |
| hsa-miR-512-3p | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ■ |
| hsa-miR-520e | ✓ | ✓ | ✓ | ■ | ✓ | ✓ | ■ |
| hsa-miR-520d-3p | ✓ | ■ | ■ | ✓ | ✓ | ✓ | ■ |
| hsa-miR-520c-3p | ✓ | ✓ | ■ | ■ | ■ | ✓ | ■ |
| hsa-miR-520b | ✓ | ✓ | ✓ | ■ | ■ | ■ | ■ |
| hsa-miR-520a-3p | ■ | ✓ | ■ | ■ | ■ | ■ | ■ |
| hsa-miR-373-3p | ✓ | ✓ | ✓ | ■ | ■ | ✓ | ■ |
| hsa-miR-302c-3p | ■ | ✓ | ✓ | ■ | ■ | ✓ | ■ |
| hsa-miR-302b-3p | ■ | ✓ | ■ | ■ | ■ | ✓ | ■ |
| hsa-miR-302a-3p | ✓ | ✓ | ✓ | ■ | ■ | ✓ | ■ |
| hsa-miR-93-5p | ✓ | ✓ | ■ | ■ | ✓ | ✓ | ■ |
| hsa-miR-20b-5p | ✓ | ✓ | ✓ | ■ | ✓ | ■ | ■ |
| hsa-miR-20a-5p | ■ | ✓ | ✓ | ■ | ■ | ■ | ■ |
| hsa-miR-17-5p | ✓ | ■ | ■ | ■ | ■ | ■ | ■ |
| hsa-miR-106b-5p | ✓ | ✓ | ■ | ■ | ■ | ■ | ■ |
| hsa-miR-106a-5p | ■ | ■ | ■ | ■ | ✓ | ■ | ■ |
| hsa-miR-1283 | ■ | ✓ | ✓ | ■ | ✓ | ✓ | ■ |
PCNXL2 regulates the Notch signaling pathway
The hypothetical protein pecanex-like protein 2 Homo sapiens (PCNXL2) is an endoplasmic membrane protein consisting of up to 16 transmembrane helixes and containing a pecanex_c conserved protein domain. PCNXL2 is mostly present in the ER and contributes to some biological processes that occur in the ER. In a previous study, Yamakawa et al.8 described that pecanex (pcx) encodes a multipass transmembrane protein with unknown functions that may be involved in the N signaling pathway. In this study, we hypothesize that pcx has functions in the Notch signaling pathway and cancer.Proper folding of Notch (N) protein is essential for its activation; therefore, the formation of disulfide bonds in the extracellular domain of N is required for its proper folding by Ero1L. When an unfolded and misfolded protein appears in the ER, the organelle suffers stress known as “ER stress”. This stress can induce apoptosis. The cell can reduce its stress level through a response known as unfolded protein response (UPR), described by Yamakawa et al.8 The unfolded protein response (UPR) is a response that enables the body to cope with ER stress. In stressed cells, Ire-1 is an ER tethered endonuclease that acts as a sensor of ER stress and splices Xbp1 mRNA, which promotes protein folding and results in the transcriptional induction of genes such as Ero1L.50 ER-associated degradation (ERAD), aggresome formation and autophagic processes are also coping responses of the body to ER stress.
Therefore, the disruption of the N-signaling pathway in the absence of pcx function may be partially due to misfolding of N because pcx acts upstream of the activated form of N and probably functions in signal receiving in cells by providing stability to the N protein to aid its proper functioning. The structure of pcx suggests that it plays a role in the formation of disulfide bridges in the extracellular domain of N by providing cohesion and stability for its proper folding by enabling its expression in the cell. Pecanex-like protein 2 Homo sapiens (PCNXL2) releases a multi-pass transmembrane protein with 1054 amino acids comprised of domain A and the pcx domain from the inner membrane of the ER into the cytosol. Because PCNXL2 also contains a DNA_J domain, it facilitates the translocation of polypeptides across organelle membranes.51 Domain A is found to be present in the cytosol, while the pcx domain is an intrinsic component of the cell membrane. Notch signaling is activated by binding Notch ligands to Notch receptors on signal-receiving cells, which leads to activation of γ secretase for cleavage of the receptor to release and translocate the Notch intracellular domain NICD to the nucleus. For proper function of γ secretase, the active site must contain water to carry out hydrolysis in the hydrophobic environment of the membrane; however, it is not understood how water and proton exchange are effected. Here, we demonstrate that the pcx domain may exhibit ATPase activity coupled to transmembrane movement of ions (protons) by establishing membrane potential for essential biological processes such as the liberation of an activated protein (NICD) from receptors. The pcx domain may play its role by establishing localization of the activated domain in a cell. Furthermore, domain A exhibits nuclear transport activity of macromolecules or substrates (NICD) from the cytoplasm to the nucleus by interacting with a nuclear localization signal (Fig. 9). Domain A may also play a role in biological processes by phosphodiester bond hydrolysis. In the nucleus, NICD binds to transcriptional factor CSL to activate the expression of Notch signaling.52
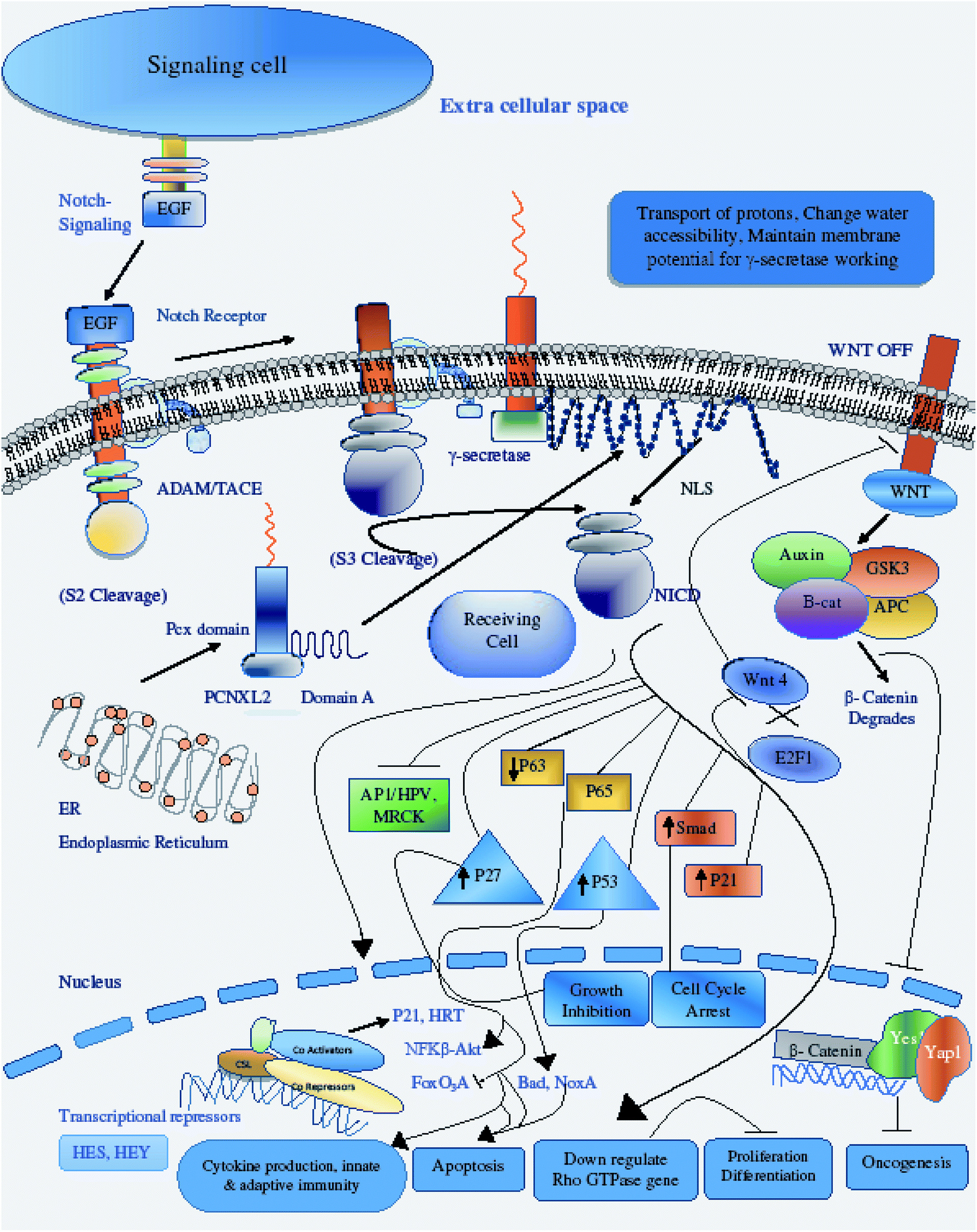 | ||
| Fig. 9 Illustration of the methodology flow sheet. Functional and structural characterization of PCNXL2 was achieved by performing 10 steps. The sequence was retrieved from NCBI. Physiochemical properties and conserved domains were predicted using the Pfam and Protpram tools. SOUSI server was used to identify the location and nature of the protein. The interactions of PCNXL2 with other proteins were investigated by string analysis. CYSPRED, CYSCON and DIANA were used to estimate disulphide bonding patterns within the protein. The structure was predicted using Raptor X and ITASSER. miRTarBase was used to identify miRNAs targeting PCNXL2 in different cancers. COACH was used to identify protein ligand binding sites. Finally, the pathway was designed by reviewing literature studies and KEGG pathways. | ||
There are diverse consequences of the Notch signaling pathway; among these, cyclin/CDK inhibitor p21WAF1/CLIP1 is an important target of Notch signaling. Increased Notch 1 activity leads to increased p21 expression, a key modulator of negative regulation of Wnt-4 expression of the Wnt/β catenin signaling pathway.53 The crosstalk between the p21WAF1/CLIP1, Notch and the Wnt/β catenin pathway is important for regulation of tumor development by preventing the proliferation of tumor cells through down-regulating the Wnt signaling pathway and expression of its oncogenes.54 Another target of Notch activation is induction of the NF-κB pathway, promoting cytokine production for the development of innate and adaptive immunity in cells for growth control and apoptosis.55 A negative feedback loop is present between Notch and p63, and p63 overexpression has been found in many cancers; p63 expression is suppressed by the activation of Notch through selective modulation of interferon-responsive genes. Like p63, the small GTPases Rho, Rac and CD42 have inverse relationships with Notch activation in the control of stem cell potential and tumor development.56 Notch CBF-dependent mechanisms achieve tumor suppression by downregulating ROCK1/2 and MRCKα kinases, which are key regulators of overexpressed Rho GTPase in tumors. Another consequence of Notch activation is the down-regulation of HPV oncogene expression; Notch 1 is under the direct effect of p53, so increased levels of p53 upregulate the expression of Notch in respective cells either through the inhibition of endogenous AP-1 activity or through other indirect mechanisms.57 P53 and its relatives are also key regulators of noxa expression; the P53 status of a cell regulates noxa expression. Noxa activation is followed by its interaction with Bcl-2 family members, freeing Bax/Bak and promoting apoptosis to mitochondria through caspase cascade activation.58 An additional target of Notch activation is the suppression of FoxO3a, which is a key transcriptional factor responsible for deciding the fate of a cell either by promoting tumor metastasis or increased cancer cell invasion. The canonical Notch/HERP pathway is a promising negative regulator of FoxO3a by binding of the HSE/HRE/Tle transcription repressor complex to the promoter of FoxO3a.59 Cross-talk between the Notch and transforming growth factor-β (TGF-β) signaling pathways plays critical roles in the control of cell fate by regulating Hes-1 expression, an important target of the Notch pathway. NICD and Smad3 interact directly in a ligand-dependent manner; Smad3 can be recruited to CSL-binding sites on DNA in the presence of CSL and NICD, indicating that Notch and TGF-β signaling are integrated through direct protein–protein interactions.60
We conclude that the pecanex protein is a tumor suppressor protein regulating Notch signaling to perform its tumor-suppressing function in developing cells, preventing misfolding of N proteins that can cause proliferation, cell survival, tumor invasion and expression of oncogenes.
Discussion
Pecanex-like protein 2 Homo sapiens (PCNXL2) is a member of the evolutionarily conserved pecanex_c conserved protein domain family. The pecanex_c conserved protein domain family comprises multi-pass transmembrane domains of large transmembrane proteins.8 In previous literature studies, pcx was suggested to be part of the N signaling pathway, with its mutant in Drosophila based upon its neurogenic phenotype.61 Recently, a study published by Yamakawa et al.8 demonstrated that pcx is a component of the N signaling pathway in Drosophila by encoding a multi-pass transmembrane protein of unknown function; the possible functions of this protein have not yet been explored. In this study, we have provided evidence that PCNXL2 encodes a multi-pass transmembrane protein that regulates the Notch signaling pathway in humans as a cancer biomarker.Previously, frame shift mutations were found in FLJ11383 (PCNXL2) in patients suffering from colorectal carcinoma. Although the incidence of mutations was high, the role of these mutations in tumorigenesis was not clear.7 The current study suggests that pcx is independently involved in activation of the Notch signaling pathway by transporting protons across the membrane, thus establishing a membrane potential gradient to release embedded water to the active site of γ secretase channeling; this provides a suitable environment for hydrolysis to proceed, with liberation of NICD from the receptors. Therefore, it was not previously understood how water and proton exchange were effected to carry out hydrolysis in the hydrophobic environment of the membrane, which is the focus of the current research.62 Furthermore, domain A induced interaction with nuclear localization to transport NICD in the nucleus for the activation of tumor repressor genes in signal receiving cells.
However, the results suggest that the downregulation of PCNXL2 in signal receiving cells may inhibit the proper function of γ secretase. It may also partially cause truncated N protein synthesis with deregulation of the Notch signaling pathway, resulting in cancer development; this is consistent with our hypothesis that pcx regulates the N signaling pathway by providing a suitable membrane environment for the genesis of activated N protein in signal-receiving cells.
Other possible reasons include the induction of several downstream events by the activated N protein synthesis, including regulation of the Notch signaling pathway, overexpression of tumor suppressor genes and inhibition of Wnt signaling. Therefore, Notch signaling cross-talk with other pathways may enhance the complex sub branched mechanism of PCNXL2, with compensated up-regulation of Notch targeted tumor suppressor genes p21, p53, and p27; this can lead to down-regulation of oncogenes, proliferation, differentiation, and cell cycle and growth arrest.63
Previous literature studies also confirmed that improper or misfolded PCNXL2 protein-induced ER stress can lead to apoptosis.11 ER stress due to misfolded proteins can cause cell death and dysfunction, which can pave the road for other diseases such as diabetes, ischemia, neurodegeneration and reperfusion injury by disturbing cell homeostasis.64 Here, we suggest that PCNXL2 is involved in regulation of endoplasmic reticulum architecture by facilitating regulatory machinery. As is evident from this study, PCNXL2 constitutes a J protein family domain (DNA_J) that is a member of the hsp40 molecular chaperone family; this may correspond to stress by degradation of misfolded proteins. Therefore, it can be concluded that aberrant expression of PCNXL2 may lead to accumulation of misfolded proteins, and dysregulation of UPR can affect not only the stress responses of the ER but also the regulation of the architecture of ER.65
This work has shown that interference with the gene regulatory network can challenge the expression of tumor suppressor genes; especially, PCNXL2 can up-regulate the Notch signaling pathway, which determines cell fate by avoiding the deregulation of N signaling. The current results challenge the role of the pecanex protein as a cancer biomarker for patient prognosis and therapeutic intervention by resolving the tumor suppressor function of Notch in many cancers, such as prostate and colorectal cancer, and other biological processes. Therefore, in future experiments, it will be important to determine the link between human hereditary diseases and ER structural dysregulation and the cancer biomarker role of PCNXL2 in the absence of pcx function.
Conclusion
This study concludes that PCNXL2 is a tumor suppressor protein that plays a valuable role in regulating the Notch signaling pathway in humans. We also speculate that the PCNXL2 DNA_J domain is involved in the regulation of ER architecture by coping with ER stress. PCNXL2 encodes a multi-pass transmembrane protein of 1054 amino acids in humans comprising the pcx domain and domain A. Structural and functional characterization of the domains demonstrated that the pcx protein may regulate Notch signaling by providing suitable membrane potential to γ secretase through liberation of the Notch intracellular domain, NICD, inside the cell. The pcx protein also facilitates transportation of NICD from the cytoplasm to the nucleus to express tumor suppressor proteins in fate-determining cells. Several cancers, including prostate and colorectal, develop in humans due to disruption of Notch signaling; however, PCNXL2 is found to be directly linked with activation of Notch signaling, designating its role as a cancer biomarker in cancerous cells. To our knowledge, this is the first report of the molecular function of PCNXL2 based upon its 3-dimensional structure; it opens up new perspectives for analysis of the cancer biomarker role of PCNXL2. Future research should be conducted to elucidate the association of ER biology with cellular environment changes.Conflicts of interest
There are no conflicts to declare.References
- K. P. S. Adinarayana, T. S. Sravani and C. Hareesh, Bioinformation, 2011, 6, 128 CrossRef PubMed.
- M. Shahbaaz, M. ImtaiyazHassan and F. Ahmad, PLoS One, 2013, 8, e84263 Search PubMed.
- S. M. Naveed, S. K. Kamzi, F. Anwar, F. Arshad, T. Zafar Dar and M. Zafar, Journal of Applied Bioinformatics & Computational Biology, 2016, 5(2) DOI:10.4172/2329-9533.1000127.
- M. Naveed, S. Tehreem, S. Mubeen, F. Nadeem, F. Zafar and M. Irshad, Open Life Sci., 2016, 11, 402–416 Search PubMed.
- A. Geisinger, M. Alsheimer, A. Baier, R. Benavente and R. Wettstein, Biochim. Biophys. Acta, Gene Struct. Expression, 2005, 1728, 34–43 CrossRef PubMed.
- T. Yamakawa, K. Yamada, T. Sasamura, N. Nakazawa, M. Kanai, E. Suzuki, M. E. Fortini and K. Matsuno, Development, 2012, 139, 558–567 CrossRef PubMed.
- N.-G. Kim, H. Rhee, L. S. Li, H. Kim, J.-S. Lee, J.-H. Kim, N. K. Kim and H. Kim, Oncogene, 2002, 21, 5081–5087 CrossRef PubMed.
- S. G. LaBonne, I. Sunitha and A. P. Mahowald, Dev. Biol., 1989, 136, 1–16 CrossRef PubMed.
- M. Nicolas, A. Wolfer, K. Raj, J. A. Kummer, P. Mill, M. van Noort, C.-c. Hui, H. Clevers, G. P. Dotto and F. Radtke, Nat. Genet., 2003, 33, 416–421 CrossRef PubMed.
- J. S. Mumm and R. Kopan, Dev. Biol., 2000, 228, 151–165 CrossRef PubMed.
- A.-C. Tien, A. Rajan, K. L. Schulze, H. D. Ryoo, M. Acar, H. Steller and H. J. Bellen, J. Cell Biol., 2008, 182, 1113–1125 CrossRef PubMed.
- R. D. Finn, P. Coggill, R. Y. Eberhardt, S. R. Eddy, J. Mistry, A. L. Mitchell, S. C. Potter, M. Punta, M. Qureshi and A. Sangrador-Vegas, Nucleic Acids Res., 2016, 44, D279–D285 CrossRef PubMed.
- M. Punta, P. C. Coggill, R. Y. Eberhardt, J. Mistry, J. Tate, C. Boursnell, N. Pang, K. Forslund, G. Ceric and J. Clements, Nucleic Acids Res., 2011, 40, D290–D301 CrossRef PubMed.
- A. Marchler-Bauer, M. K. Derbyshire, N. R. Gonzales, S. Lu, F. Chitsaz, L. Y. Geer, R. C. Geer, J. He, M. Gwadz and D. I. Hurwitz, Nucleic Acids Res., 2014, 43, D222–D226 CrossRef PubMed.
- E. De Castro, C. J. Sigrist, A. Gattiker, V. Bulliard, P. S. Langendijk-Genevaux, E. Gasteiger, A. Bairoch and N. Hulo, Nucleic Acids Res., 2006, 34, W362–W365 CrossRef PubMed.
- M. Naveed, I. Ahmed, N. Khalid and A. S. Mumtaz, Braz. J. Microbiol., 2014, 45, 603–611 CrossRef PubMed.
- E. Gasteiger, C. Hoogland, A. Gattiker, S. e. Duvaud, M. R. Wilkins, R. D. Appel and A. Bairoch, Protein identification and analysis tools on the ExPASy server, Springer, 2005 Search PubMed.
- W. Li, A. Cowley, M. Uludag, T. Gur, H. McWilliam, S. Squizzato, Y. M. Park, N. Buso and R. Lopez, Nucleic Acids Res., 2015, 43, W580–W584 CrossRef PubMed.
- R. Mohan and S. Venugopal, Bioinformation, 2012, 8, 722 CrossRef PubMed.
- M. Ikeda, M. Arai and T. Shimizu, Genomics Inform., 2000, 11, 426–427 Search PubMed.
- T. Hirokawa, S. Boon-Chieng and S. Mitaku, Bioinformatics, 1998, 14, 378–379 CrossRef PubMed.
- A. C. Lewis, R. Saeed and C. M. Deane, Mol. BioSyst., 2010, 6, 55–64 RSC.
- L. J. Jensen, M. Kuhn, M. Stark, S. Chaffron, C. Creevey, J. Muller, T. Doerks, P. Julien, A. Roth and M. Simonovic, Nucleic Acids Res., 2009, 37, D412–D416 CrossRef PubMed.
- P. Fariselli, P. Riccobelli and R. Casadio, Proteins: Struct., Funct., Bioinf., 1999, 36, 340–346 CrossRef.
- F. Ferrè and P. Clote, Nucleic Acids Res., 2006, 34, W182–W185 CrossRef PubMed.
- J. Yang, B.-J. He, R. Jang, Y. Zhang and H.-B. Shen, Bioinformatics, 2015, 31, 3773–3781 Search PubMed.
- J. Ma, S. Wang, F. Zhao and J. Xu, Bioinformatics, 2013, 29, i257–i265 CrossRef PubMed.
- J. Peng and J. Xu, Proteins: Struct., Funct., Bioinf., 2011, 79, 161–171 CrossRef PubMed.
- J. Yang and Y. Zhang, Nucleic Acids Res., 2015, 43, W174–W181 CrossRef PubMed.
- J. Yang, A. Roy and Y. Zhang, Bioinformatics, 2013, 29, 2588–2595 CrossRef PubMed.
- C.-H. Chou, N.-W. Chang, S. Shrestha, S.-D. Hsu, Y.-L. Lin, W.-H. Lee, C.-D. Yang, H.-C. Hong, T.-Y. Wei and S.-J. Tu, Nucleic Acids Res., 2016, 44, D239–D247 CrossRef PubMed.
- S. M. Peterson, J. A. Thompson, M. L. Ufkin, P. Sathyanarayana, L. Liaw and C. B. Congdon, Front. Genet., 2014, 5, 23 Search PubMed.
- K. A. Hoadley, C. Yau, D. M. Wolf, A. D. Cherniack, D. Tamborero, S. Ng, M. D. Leiserson, B. Niu, M. D. McLellan and V. Uzunangelov, Cell, 2014, 158, 929–944 CrossRef PubMed.
- A. Fabregat, K. Sidiropoulos, P. Garapati, M. Gillespie, K. Hausmann, R. Haw, B. Jassal, S. Jupe, F. Korninger and S. McKay, Nucleic Acids Res., 2016, 44, D481–D487 CrossRef PubMed.
- K. Nishida, K. Ono, S. Kanaya and K. Takahashi, F1000Research, 2014, 3, 144 Search PubMed.
- D. W. Huang, B. T. Sherman and R. A. Lempicki, Nat. Protoc., 2009, 4, 44–57 CrossRef PubMed.
- W.-L. Tsou, M. Ouyang, R. R. Hosking, J. R. Sutton, J. R. Blount, A. A. Burr and S. V. Todi, Neurobiol. Dis., 2015, 82, 12–21 CrossRef PubMed.
- K. Zhao, W. Wang, O. J. Rando, Y. Xue, K. Swiderek, A. Kuo and G. R. Crabtree, Cell, 1998, 95, 625–636 CrossRef PubMed.
- X. Bao, J. Tang, V. Lopez-Pajares, S. Tao, K. Qu, G. R. Crabtree and P. A. Khavari, Cell Stem Cell, 2013, 12, 193–203 CrossRef PubMed.
- B. D. Rowland and D. S. Peeper, Nat. Rev. Cancer, 2006, 6, 11–23 CrossRef PubMed.
- W. Zhai and L. Comai, Mol. Cell. Biol., 2000, 20, 5930–5938 CrossRef PubMed.
- W. S. Argraves, L. M. Greene, M. A. Cooley and W. M. Gallagher, EMBO Rep., 2003, 4, 1127–1131 CrossRef PubMed.
- H. Yanagisawa, M. K. Schluterman and R. A. Brekken, Cell Commun. Signaling, 2009, 3, 337–347 CrossRef PubMed.
- X. Chen, X. Song, W. Yue, D. Chen, J. Yu, Z. Yao and L. Zhang, Oncotarget, 2015, 6, 15022 Search PubMed.
- T. Imasaki, T. Shimizu, H. Hashimoto, Y. Hidaka, S. Kose, N. Imamoto, M. Yamada and M. Sato, Mol. Cell, 2007, 28, 57–67 CrossRef PubMed.
- A. Cook, E. Fernandez, D. Lindner, J. Ebert, G. Schlenstedt and E. Conti, Mol. Cell, 2005, 18, 355–367 CrossRef PubMed.
- K. I. Muhammad Naveed, A. Mushtaq, S. Tehreem and S. Ullah Khan, Int. J. Biosci., 2017, 10, 22–42 CrossRef.
- N. M. Ashraf, K. Imran, D. W. Kastner, K. Ikram, A. Mushtaq, A. Hussain and N. Zeeshan, Mol. Cell. Probes, 2017, 36, 21–28 CrossRef PubMed.
- K. I. Muhammad Naveed, A. Mushtaq, S. Tehreem and S. Ullah Khan, Int. J. Biosci., 2017, 10, 22–42 CrossRef.
- C. Xu, B. Bailly-Maitre and J. C. Reed, J. Clin. Invest., 2005, 115, 2656–2664 CrossRef PubMed.
- P. Walsh, D. Bursać, Y. C. Law, D. Cyr and T. Lithgow, EMBO Rep., 2004, 5, 567–571 CrossRef PubMed.
- S. Chikara and K. M. Reindl, Transl. Lung Cancer Res., 2013, 2, 449 Search PubMed.
- M. Hofmann, K. Schuster-Gossler, M. Watabe-Rudolph, A. Aulehla, B. G. Herrmann and A. Gossler, Genes Dev., 2004, 18, 2712–2717 CrossRef PubMed.
- V. Devgan, C. Mammucari, S. E. Millar, C. Brisken and G. P. Dotto, Genes Dev., 2005, 19, 1485–1495 CrossRef PubMed.
- B. Nickoloff, J.-Z. Qin, V. Chaturvedi, M. Denning, B. Bonish and L. Miele, Cell Death Differ., 2002, 9(8), 842–855 CrossRef PubMed.
- B.-C. Nguyen, K. Lefort, A. Mandinova, D. Antonini, V. Devgan, G. Della Gatta, M. I. Koster, Z. Zhang, J. Wang and A. T. di Vignano, Genes Dev., 2006, 20, 1028–1042 CrossRef PubMed.
- K. Lefort, A. Mandinova, P. Ostano, V. Kolev, V. Calpini, I. Kolfschoten, V. Devgan, J. Lieb, W. Raffoul and D. Hohl, Genes Dev., 2007, 21, 562–577 CrossRef PubMed.
- C. Ploner, R. Kofler and A. Villunger, Oncogene, 2008, 27, S84–S92 CrossRef PubMed.
- A. Mandinova, K. Lefort, A. T. di Vignano, W. Stonely, P. Ostano, G. Chiorino, H. Iwaki, J. Nakanishi and G. P. Dotto, EMBO J., 2008, 27, 1243–1254 CrossRef PubMed.
- A. Blokzijl, C. Dahlqvist, E. Reissmann, A. Falk, A. Moliner, U. Lendahl and C. F. Ibáñez, J. Cell Biol., 2003, 163, 723–728 CrossRef PubMed.
- S. G. Labonne and A. Furst, J. Neurogenet., 1989, 5, 99–104 CrossRef PubMed.
- M. S. Wolfe, Biochemistry, 2006, 45, 7931–7939 CrossRef PubMed.
- K. G. Leong and A. Karsan, Blood, 2006, 107, 2223–2233 CrossRef PubMed.
- A. H. Schönthal, Scientifica, 2012 DOI:10.6064/2012/857516.
- M. Puhka, H. Vihinen, M. Joensuu and E. Jokitalo, J. Cell Biol., 2007, 179, 895–909 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |