
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic enantioselective Henry reaction of α-keto esters, 2-acylpyridines and 2-acylpyridine N-oxides†
Feilong He‡
 a,
Guanghui Chen‡a,
Junxia Yanga,
Guojuan Lianga,
Ping Denga,
Yan Xiongb and
Hui Zhou*a
a,
Guanghui Chen‡a,
Junxia Yanga,
Guojuan Lianga,
Ping Denga,
Yan Xiongb and
Hui Zhou*a
aSchool of Pharmaceutical Science, Chongqing Medical University, Chongqing 400016, PR China. E-mail: hzhou@cqmu.edu.cn
bSchool of Chemistry and Chemical Engineering, Chongqing University, Chongqing 401331, PR China
First published on 5th March 2018
Abstract
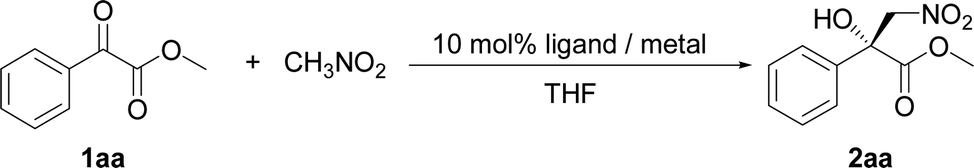
A pre-prepared Ni–PyBisulidine complex has been developed for the catalytic asymmetric Henry reaction of α-keto esters, 2-acylpyridines and 2-acylpyridine N-oxides. The corresponding β-nitro-α-hydroxy esters were obtained in good to excellent yields (up to 99%) with a high enantiomeric excess (ee) (up to 94%) with a catalyst loading of 1–2 mol%. The desired products of 2-acylpyridines and 2-acylpyridine N-oxides, which were simple methyl ketones, were obtained in medium to excellent yields (up to 94%) with medium to good ee (up to 86%) by using 2 mol% of catalyst.
Introduction
The Henry reaction is one of the important methods for C–C bond formation.1 The resulting products, β-nitroalcohols, are key intermediates and building blocks for the synthesis of bioactive natural products and pharmaceutical agents.1 Thus, increasing efforts have been directed towards developing a catalytic asymmetric Henry reaction.2 Compared with the well developed asymmetric Henry reaction of aldehydes, the asymmetric Henry reaction of ketones with the formation of a quaternary stereogenic center is more challenging because it often suffers from low reactivity and poor stereoselectivity.3 Although Tosaki et al. reported the kinetic resolution of racemic derivatives,4 the catalytic asymmetric Henry reaction of simple ketones is rarely reported. At present, the study mainly focused on reactive substrates such as trifluoromethyl ketones,5 α-keto esters,6 α-keto amides,7 α-keto-phosphonates,8 and glyoxal hydrates.9 Holmquist et al. expanded the scope of this reaction to 2-acylpyridine N-oxide, simple ketones, for the first time.10 Although great progress has been achieved, several factors, including the relatively high catalyst loading (5–20 mol%) or catalyst preparation, limit the use of existing catalytic methods. At the same time, developing new catalysts for the enantioselective Henry reaction of ketones is still necessary. Recently a sulfonylated pyridine bisimidazolidine: nickel–pyridine bisulidine (Ni–PyBisulidine) complex was introduced for the asymmetric hydrophosphonylation of aldehydes.11–13 In this paper, the use of pre-prepared Ni–PyBisulidine complexes for the asymmetric Henry reaction of α-keto esters, 2-acylpyridines and 2-acylpyridine N-oxides with low catalyst loading (down to 1 mol%) is reported.Results and discussion
The initial studies of the catalytic asymmetric Henry reaction focused on the addition of nitromethane (CH3NO2) to methyl phenyloxoacetate in the presence of the complex of chiral PyBisulidine L1 as ligand (Fig. 1). The complexes of nickel(II) acetate–L1 [Ni(OAc)2–L1], cobalt(II) acetate–L1 [Co(OAc)2–L1], zinc(II) acetate–L1 [Zn(OAc)2–L1] promoted the reaction in a 70–86% enantiomeric excess (ee) with low yields at room temperature (rt; Table 1, entries 1, 3 and 5). When nickel(II) acetylacetonate [Ni(acac)2] and copper(II) acetate [Cu(OAc)2] were used as the central metal, a low chiral induction was observed (Table 1, entries 2 and 4). When the complexes of iron(II) acetate–L1 [Fe(OAc)2–L1] and palladium(II) acetate–L1 [Pd(OAc)2–L1] were used as the catalysts, the corresponding products were not detected (Table 1, entries 6 and 7). Fortunately, the Ni(OAc)2–L1 complex could catalyze this reaction smoothly with 83% ee with a 85% yield when the reaction temperature rose to 35 °C (Table 1, entry 8). However, further increasing the reaction temperature did not improve the reactivity (Table 1, entry 9). After screening the benzenesulfonyl moiety of the ligands (Table 1, entries 8, 10–12), Ni(OAc)2–L1 was selected for further optimization which considered the reactivity and economy.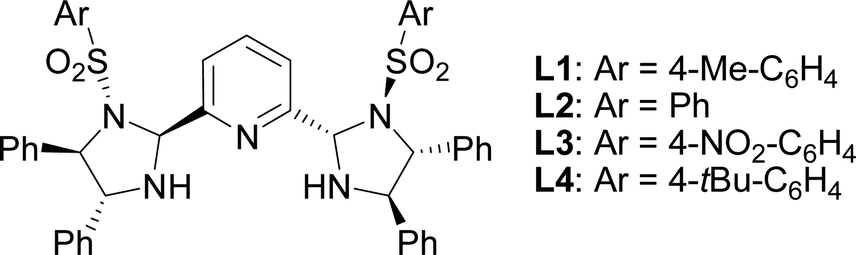 | ||
| Fig. 1 Chiral PyBisulidine used as ligands for the asymmetric Henry reaction. | ||
|
|||||
|---|---|---|---|---|---|
| Entry | Metal | Ligand | T (°C) | Yieldb (%) | eec (%) |
| a Reactions were carried out using methyl phenyloxoacetate (0.2 mmol) with CH3NO2 (0.2 mL) in THF (0.8 mL) in the presence of metal–PyBisulidines prepared in situ for 20 h.b Isolated yield.c Determined using HPLC analysis on a chiral stationary phase.d The reaction time was 65 h.e ND: not detected. | |||||
| 1 | Ni(OAc)2 | L1 | rt | 16 | 86 |
| 2 | Ni(acac)2 | L1 | rt | 76 | 8 |
| 3 | Co(OAc)2 | L1 | rt | 37 | 75 |
| 4 | Cu(OAc)2 | L1 | rt | 18 | 8 |
| 5 | Zn(OAc)2 | L1 | rt | 19 | 70 |
| 6d | Fe(OAc)2 | L1 | rt | NDe | — |
| 7d | Pd(OAc)2 | L1 | rt | NDe | — |
| 8 | Ni(OAc)2 | L1 | 35 | 85 | 83 |
| 9 | Ni(OAc)2 | L1 | 50 | 84 | 80 |
| 10 | Ni(OAc)2 | L2 | 35 | 84 | 69 |
| 11 | Ni(OAc)2 | L3 | 35 | 82 | 88 |
| 12 | Ni(OAc)2 | L4 | 35 | 62 | 72 |
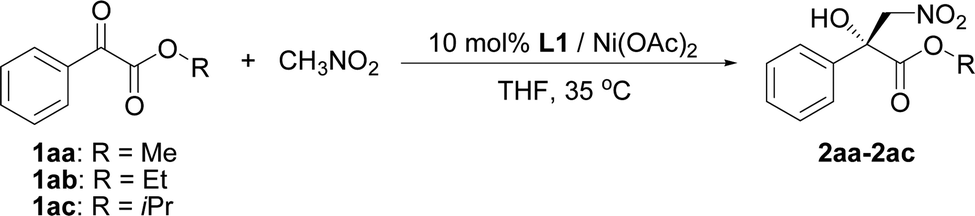
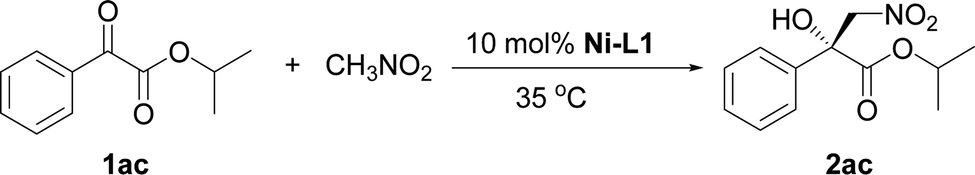
The influence of the ester group in the substrate was tested next (Table 2, entries 1–3). The best result in terms of the conversion and enantioselectivity was obtained with the isopropyl ester (Table 2, entry 3). The pre-prepared complex14 gave better results than the complex prepared in situ (Table 2, compare entries 3 and 4).
|
||||
|---|---|---|---|---|
| Entry | R | Catalyst preparation methodb | Yieldc (%) | eed (%) |
| a Reactions were carried out using an α-keto ester (0.2 mmol) in a mixture of THF (0.8 mL) and CH3NO2 (0.2 mL) for 20 h.b For details, see ref. 11.c Isolated yield.d Determined using HPLC analysis on a chiral stationary phase. | ||||
| 1 | Me | In situ | 85 (2aa) | 83 |
| 2 | Et | In situ | 62 (2ab) | 86 |
| 3 | i-Pr | In situ | 83 (2ac) | 88 |
| 4 | i-Pr | Pre-prepared | 85 (2ac) | 91 |
Encouraged by the initial results in the asymmetric Henry reaction, various solvents were screened in the presence of 10 mol% pre-prepared Ni–L1 (Table 3). Whereas small or trace amounts of products were detected in chloroform (CHCl3) or 1,2-dimethoxyethane (DME; Table 3, entries 2 and 5). Other solvents tested, such as methanol (CH3OH, MeOH), toluene and diethylene glycol dimethyl ether, gave moderate yields (Table 3, entries 3, 4 and 6). Tetrahydrofuran (THF) exhibited the best performance (Table 3, entry 1).
|
|||
|---|---|---|---|
| Entry | Solvent | Yieldb (%) | eec (%) |
| a Reactions were carried out on a 0.2 mmol scale of isopropyl phenyloxoacetate in the mixture of THF (0.8 mL) and CH3NO2 (0.2 mL) for 20 h. The catalyst was pre-prepared.b Isolated yield.c Determined by HPLC analysis on a chiral stationary phase.d Diglyme = diethylene glycol dimethyl ether. | |||
| 1 | THF | 85 | 91 |
| 2 | CHCl3 | 37 | 89 |
| 3 | CH3OH | 56 | 43 |
| 4 | Toluene | 68 | 90 |
| 5 | DME | Trace | — |
| 6 | Diglymed | 62 | 91 |
When the catalyst loading was reduced, the reactivity decreased sharply with slightly increasing ee (Table 4, entries 1–4). To increase the reactivity, some additives were screened in this reaction (Table 4, entries 5–8). The yields of Henry products were improved when using a 4 Å molecular sieve (MS; Table 4, entries 7 and 11) and addition of tert-amines (Table 4, entries 5, 6, 9 and 10). However, some acidic additives, such as benzoic acid (PhCOOH), were harmful for the reaction (Table 4, entry 8).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst loading | Base (mol%) | Additive | Yieldb (%) | eec (%) |
| a Reactions were carried out using scale of isopropyl phenyloxoacetate (0.2 mmol) in a mixture of THF (0.8 mL) and CH3NO2 (0.2 mL) for 20 h. The catalyst was pre-prepared.b Isolated yield.c Determined using HPLC analysis on a chiral stationary phase.d N,N-Diisopropylethylamine.e Molecular sieve. | |||||
| 1 | 10 mol% | None | None | 85 | 91 |
| 2 | 5 mol% | None | None | 80 | 92 |
| 3 | 2 mol% | None | None | 49 | 93 |
| 4 | 1 mol% | None | None | 28 | 94 |
| 5 | 10 mol% | iPr2NEtd (10) | None | 92 | 86 |
| 6 | 10 mol% | N-Methyl-morpholine (10) | None | 90 | 89 |
| 7 | 10 mol% | None | 4 Å MSe (38 mg) | 95 | 86 |
| 8 | 10 mol% | None | PhCOOH (10 mol%) | 46 | 90 |
| 9 | 2 mol% | iPr2NEt (10) | None | 69 | 88 |
| 10 | 2 mol% | N-Methyl-morpholine (10) | None | 66 | 92 |
| 11 | 2 mol% | None | 4 Å MS (38 mg) | 56 | 93 |
| 12 | 2 mol% | N-Methyl-morpholine (10) | 4 Å MS (20 mg) | 80 | 93 |
| 13 | 2 mol% | N-Methyl-morpholine (10) | 4 Å MS (30 mg) | 92 | 93 |
| 14 | 2 mol% | N-Methyl-morpholine (10) | 4 Å MS (50 mg) | 90 | 93 |
When the catalyst loading was reduced to 2 mol%, N-methylmorpholine showed a better performance than iPr2NEt in terms of enantioselectivity (Table 4, entries 9 and 10). The amount of 4 Å MS was screened together with 10 mol% N-methylmorpholine (Table 4, entries 12–14). Under the optimized conditions (at 35 °C, in the presence of 2 mol% Ni–L1, 10 mol% N-methylmorpholine, and 150 mg mmol−1 4 Å MS in THF), 2ac was obtained with a 92% yield with 93% ee (Table 4, entry 13).
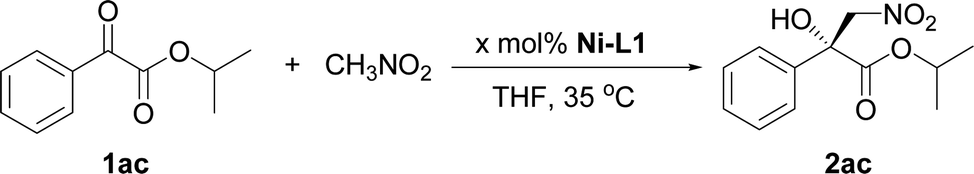
The current catalytic system was applied to various α-keto esters (Table 5). In all cases, the reactions were clean and proceeded and gave good to excellent yields with high enantioselectivities. Aromatic keto esters bearing the electron-donating groups gave smaller yields but the high enantioselectivities were maintained (72–83% yield, Table 5, 2bc–2dc, and 2gc). Aromatic keto esters bearing the electron-withdrawing group gave excellent yields (Table 5, 2ec, and 2ic–2mc) and the catalyst loading could be reduced to 1 mol% with high to excellent yields and high enantioselectivities (Table 5, 2ic–2mc). The keto esters derived from the bulkier ketone, such as β-acetonaphthone, also gave an excellent yield and high enantioselectivity (Table 5, 2fc). The heteroaromatic and alkyl keto esters gave smaller ee values (Table 5, 2hc and 2nb). The configuration of 2ac was identified as R using single crystal diffraction analysis,15 and the configuration of the other products were inferred to be analogous with that of 2ac. It should be noted that the pre-prepared complex Ni–L1 can be stored in air at 4 °C for at least three months without any loss of activity.16
|
|---|
| a Unless otherwise noted, all reactions were carried out with α-keto ester (0.2 mmol) with N-methylmorpholine (10 mol%) and 4 Å MS (30 mg) in a mixture of THF (0.8 mL) and CH3NO2 (0.2 mL) for 20 h. The catalyst was pre-prepared. The reaction time for 2dc, 2gc, and 2hc was 36 h. |
 |
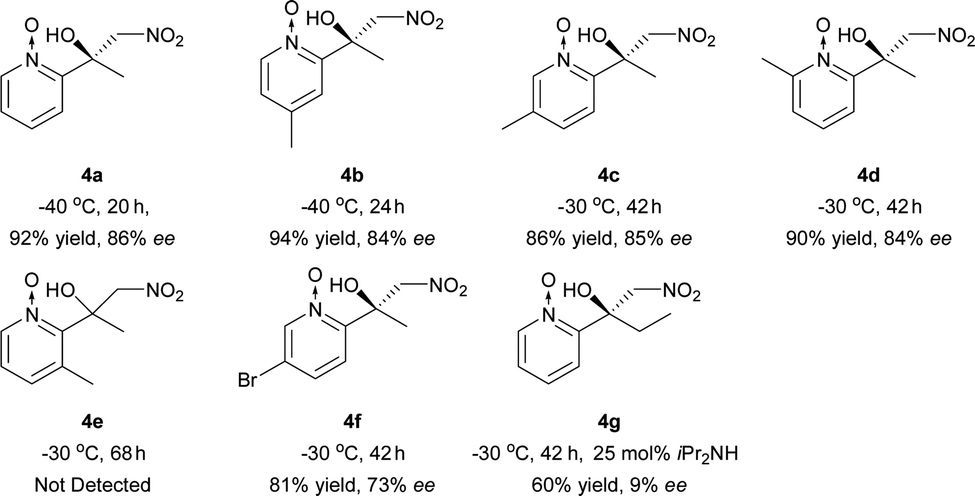
The Ni–L1 was also used in the asymmetric Henry reaction of 2-acylpyridine N-oxides (Table 6).17 High yields and good ee were obtained with methyl ketones (Table 6, 4a–4d, 4f). Whereas low ee were obtained with ethyl ketones (Table 6, 4g). The corresponding product of 3-methyl-2-acylpyridine N-oxide was not detected (Table 6, 4e).
|
|---|
| a Reactions were carried out with 2-acylpyridine N-oxides (0.2 mmol) with diisopropylamine (iPr2NH; 5 mol%) and 4 Å MS (30 mg) in a mixture of EtOH (0.8 mL) and CH3NO2 (0.2 mL) for 20–42 h. The catalyst was pre-prepared. EtOH: ethanol. |
 |
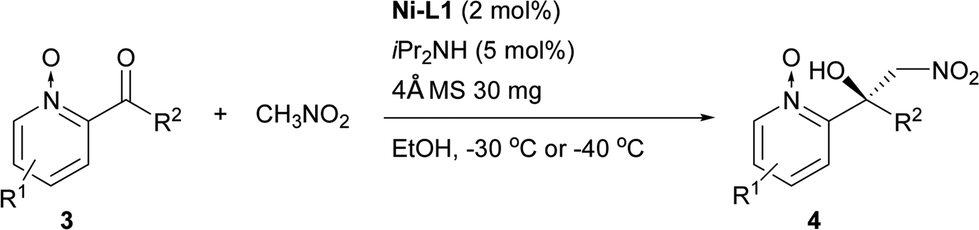
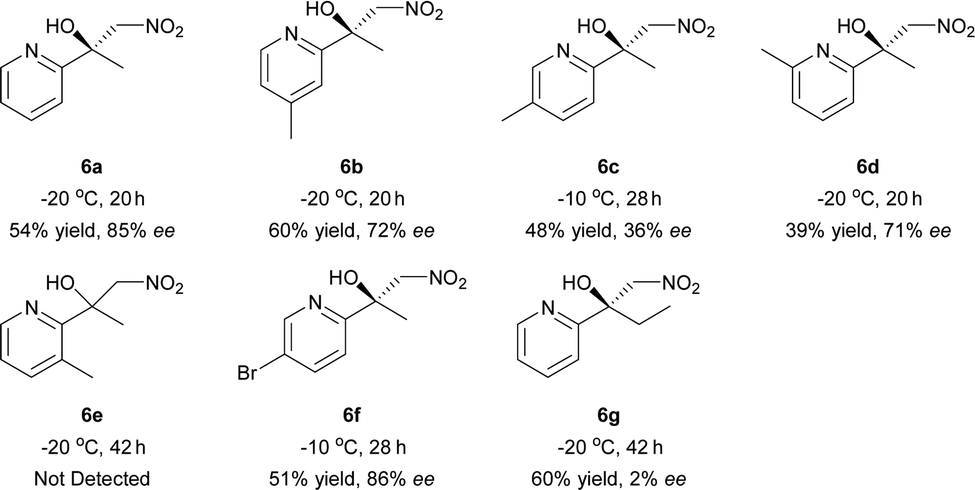
Inspired by the research of Tosaki et al.4 and Holmquist et al.10, the Henry reaction of 2-acylpyridines was also investigated (Table 7).17 In most cases, 50–60% yields and 70–86% ee were obtained with methyl ketones. Racemic products were obtained for ethyl ketones (Table 7, 6g). The Henry reaction of 3-methyl-2-acylpyridine did not take place at all. The results were similar to those obtained using 2-acylpyridine N-oxides, indicating that they had similar transition states.
|
|---|
| a Reactions were carried out with 2-acylpyridine (0.2 mmol) with iPr2NEt (10 mol%) and 4 Å MS (40 mg) in a mixture of THF (0.8 mL) and CH3NO2 (0.2 mL) for 20–42 h. The catalyst was pre-prepared. |
 |
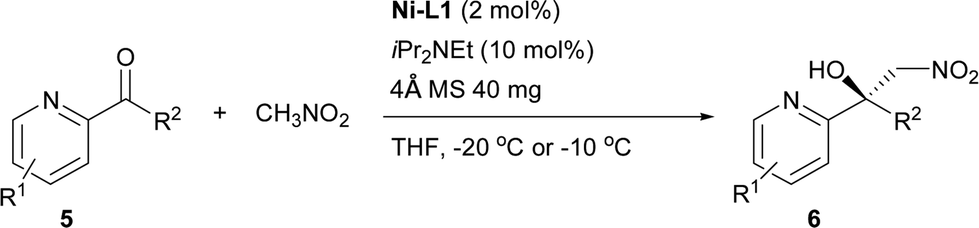
The proposed structure of Ni–L1 on the basis of the related structure of Fe–PyBisulidine complex,12a the geometry of L1 optimized using Chem3D at the MM2 level (Fig. 2) and the electrospray ionization-high resolution mass spectrometry (ESI-HRMS) analysis of the complex has previously been reported.11 To gain some insight into the active species, ESI-HRMS studies of the mixture of Ni–L1 and 3a were carried out. The spectrum displayed ions at m/z 1085.28625 and 1025.26555, which corresponded to C-I and C-II (Fig. 3). It was speculated that the complex C-I or C-II would be the active species. TS1–TS6 are proposed to rationalize the asymmetric induction. As illustrated in Fig. 4, the keto functionality is coordinated to Ni(II) in the more Lewis acidic equatorial position for maximal activation,18 whereas the nitronate generated by the amine is positioned by the hydrogen bonding.13g,13h
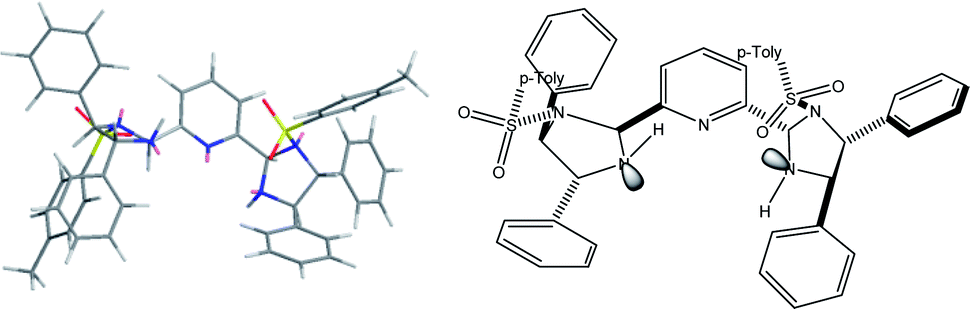 | ||
| Fig. 2 The geometry of L1 optimized using Chem3D (8.0) at the MM2 level. | ||
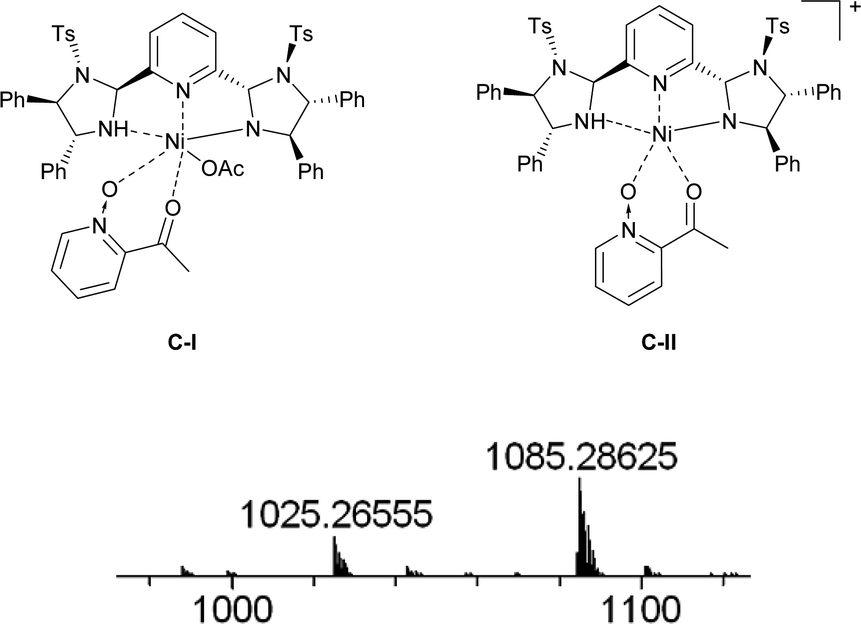 | ||
| Fig. 3 ESI-HRMS for C-I: m/z calc'd for C58H55N6NiO8S2+ [Ni(OAc)2 + ML1 + M3a − HOAc + H]+: 1085.28708, found: 1085.28625; ESI-HRMS for C-II: m/z calc'd for C56H51N6NiO6S2+ [Ni(OAc)2 + ML1 + M3a − HOAC − OAc]+: 1025.26595, found: 1025.26555. | ||
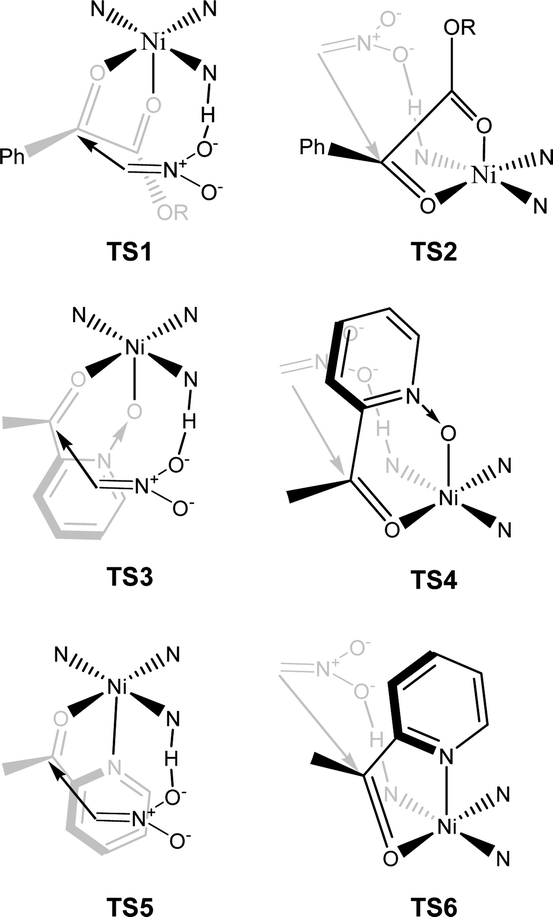 | ||
| Fig. 4 The proposed working model. | ||
Conclusions
A catalytic asymmetric Henry reaction of α-keto esters, 2-acylpyridines and 2-acylpyridine N-oxides, was developed using a Ni–PyBisulidine complex with a low catalyst loading (1–2 mol%). This is the first example of the direct asymmetric synthesis of tertiary nitro alcohols derived from 2-acylpyridines, which were simple methyl ketones. The catalytic system is tolerant of air and moisture. Further investigations into other versions of asymmetric catalysis are in progress.Experimental
General methods
Commercial reagents were used as purchased. High resolution mass spectra were recorded using a Bruker SolariX Fourier-transform ion cyclotron mass spectrometry (FT-ICR-MS) system. Nuclear magnetic resonance (NMR) spectra were recorded in the deuterated solvents [deuterated chloroform (CDCl3) or deuterated methanol (CD3OD)] as stated, using residual non-deuterated solvent as internal standard. The enantiomeric excess (ee) was determined using high-performance liquid chromatography (HPLC) analysis using the corresponding commercial chiral column as stated in the experimental procedures at 23 °C with an ultraviolet detector at 220 nm or 215 nm or 254 nm. Optical rotations were measured on a commercial polarimeter and are reported as follows: [α]TD (c = g per 100 mL solvent).General procedure for catalytic asymmetric reaction
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–15:1) to give the products. The absolute configuration of 2ac was determined using X-ray crystallographic analysis. The absolute configuration of 2ab was assigned by comparison with the sign of optical rotation value found in the literature.6b The absolute configuration of 2bc–2mc and 2aa was determined by analogy. The absolute configuration of 2nb was assigned by comparison with the sign of optical rotation values found in the literature.6a,6b,6e,6f,6i:1) to give the products. The absolute configuration of 4a–4d and 4f was assigned by comparison with the sign of optical rotation value found in the literature.10:1) to give the products. The absolute configuration of 6a was assigned by comparison with the sign of optical rotation value of its reduced product (7) found in the literature.10 The absolute configuration of 6b–6d and 6f was determined by analogy.:1) to give 12.3 mg (81%) of compound 7. [α]20D = +26.7 (c 0.21, MeOH) [lit.10 [α]20D = +33 (c 0.8, MeOH) in 86% ee]; 1H-NMR (400 MHz, CD3OD) δ 8.50 (d, 1H, J = 4.4), 7.81 (td, 1H, J = 7.7, 1.4), 7.68 (d, 1H, J = 8.0), 7.28–7.24 (m, 1H), 3.07 (d, 1H, J = 13.2), 2.89 (d, 1H, J = 13.2), 1.48 (s, 3H); 13C-NMR (100 MHz, CD3OD) δ 164.5(C), 147.9(CH), 137.1(CH), 121.9(CH), 120.0(CH), 74.6(C), 51.2(CH2), 25.5(CH3).
:1–15:1) to give the products. The absolute configuration of 2ac was determined using X-ray crystallographic analysis. The absolute configuration of 2ab was assigned by comparison with the sign of optical rotation value found in the literature.6b The absolute configuration of 2bc–2mc and 2aa was determined by analogy. The absolute configuration of 2nb was assigned by comparison with the sign of optical rotation values found in the literature.6a,6b,6e,6f,6i:1) to give the products. The absolute configuration of 4a–4d and 4f was assigned by comparison with the sign of optical rotation value found in the literature.10:1) to give the products. The absolute configuration of 6a was assigned by comparison with the sign of optical rotation value of its reduced product (7) found in the literature.10 The absolute configuration of 6b–6d and 6f was determined by analogy.:1) to give 12.3 mg (81%) of compound 7. [α]20D = +26.7 (c 0.21, MeOH) [lit.10 [α]20D = +33 (c 0.8, MeOH) in 86% ee]; 1H-NMR (400 MHz, CD3OD) δ 8.50 (d, 1H, J = 4.4), 7.81 (td, 1H, J = 7.7, 1.4), 7.68 (d, 1H, J = 8.0), 7.28–7.24 (m, 1H), 3.07 (d, 1H, J = 13.2), 2.89 (d, 1H, J = 13.2), 1.48 (s, 3H); 13C-NMR (100 MHz, CD3OD) δ 164.5(C), 147.9(CH), 137.1(CH), 121.9(CH), 120.0(CH), 74.6(C), 51.2(CH2), 25.5(CH3).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Authors are grateful for the generous financial support by the National Nature Science Foundation of China (21672031, 21372265), the Fundamental and Advanced Research Projects of Chongqing City (cstc2016jcyjA0250 and cstc2017jcyjAX0352), the Scientific and Technological Research Program of Chongqing Municipal Education Commission (KJ1702014).Notes and references
- (a) G. Rosini, in Comprehensive Organic Synthesis, ed. B. M. Trost, I. C. Fleming, H. Heathcock, Pergamon, New York, 1991, vol. 2, p. 321 Search PubMed; (b) M. Shibasaki and H. Gröer, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz, H. Yamamoto, Springer, Berlin, 1999, vol. 3, p. 1075 Search PubMed; (c) N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, Weinheim, Germany, 2001, ch. 3, p. 30 CrossRef.
- For reviews on the asymmetric Henry reaction. see: (a) F. A. Luzzio, Tetrahedron, 2001, 57, 915–945 CrossRef CAS; (b) C. Palomo, M. Oiarbide and A. Mielgo, Angew. Chem., Int. Ed., 2004, 43, 5442–5444 CrossRef CAS PubMed; (c) J. Boruwa, N. Gogoi, P. P. Saikia and N. C. Barua, Tetrahedron: Asymmetry, 2006, 17, 3315–3326 CrossRef CAS; (d) C. Palomo, M. Oiarbide and A. Laso, Eur. J. Org. Chem., 2007, 2561–2574 CrossRef CAS; (e) N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 4760–4772 CrossRef CAS PubMed; (f) S. E. Milner, T. S. Moody and A. R. Maguire, Eur. J. Org. Chem., 2012, 3059–3067 CrossRef CAS; (g) N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2013, 52, 223–234 CrossRef CAS PubMed.
- For reviews on the stereoselective preparation of quaternary stereogenic centers, see: (a) P. G. Cozzi, R. Hilgraf and N. Zimmerman, Eur. J. Org. Chem., 2007, 5969–5994 CrossRef CAS; (b) M. Bella and T. Gasperi, Synthesis, 2009, 1583–1614 CrossRef CAS; (c) O. Riant and J. Hannedouche, Org. Biomol. Chem., 2007, 5, 873–888 RSC; (d) Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis, ed. J. Christoffers, A. Baro, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed.
- S. Tosaki, K. Hara, V. Gnanadesikan, H. Morimoto, S. Harada, M. Sugita, N. Yamagiwa, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2006, 128, 11776–11777 CrossRef CAS PubMed.
- (a) F. Tur and J. M. Saá, Org. Lett., 2007, 9, 5079–5082 CrossRef CAS PubMed; (b) M. Bandini, R. Sinisi and A. Umani-Ronchi, Chem. Commun., 2008, 4360–4362 RSC; (c) J. M. Saá, F. Tur and J. González, Chirality, 2009, 21, 836–842 CrossRef PubMed; (d) H. Xu and C. Wolf, Chem. Commun., 2010, 46, 8026–8028 RSC; (e) C. Palacio and S. J. Connon, Org. Lett., 2011, 13, 1298–1301 CrossRef CAS PubMed; (f) A. Das, M. K. Choudhary, R. I. Kureshy, K. Jana, S. Verma, N. H. Khan, S. H. R. Abdi, H. C. Bajaj and B. Ganguly, Tetrahedron, 2015, 71, 5229 CrossRef CAS.
- (a) C. Christensen, K. Juhl, R. G. Hazell and K. A. Jørgensen, Chem. Commun., 2001, 2222–2223 RSC; (b) C. Christensen, K. Juhl, R. G. Hazell and K. A. Jørgensen, J. Org. Chem., 2002, 67, 4875–4881 CrossRef CAS PubMed; (c) S.-F. Lu, D.-M. Du, S.-W. Zhang and J. Xu, Tetrahedron: Asymmetry, 2004, 15, 3433–3441 CrossRef CAS; (d) B. M. Choudary, K. V. S. Ranganath, U. Pal, M. L. Kantam and B. Sreedhar, J. Am. Chem. Soc., 2005, 127, 13167–13171 CrossRef CAS PubMed; (e) D.-M. Du, S.-F. Lu, T. Fang and J. Xu, J. Org. Chem., 2005, 70, 3712–3715 CrossRef CAS PubMed; (f) H. Li, B. Wang and L. Deng, J. Am. Chem. Soc., 2006, 128, 732–733 CrossRef CAS PubMed; (g) B. Qin, X. Xiao, X. Liu, J. Huang, Y. Wen and X. Feng, J. Org. Chem., 2007, 72, 9323–9328 CrossRef CAS PubMed; (h) A. Bulut, A. Aslan and Ö. Dogan, J. Org. Chem., 2008, 73, 7373–7375 CrossRef CAS PubMed; (i) G. Blay, V. Hernández-Olmos and J. R. Pedro, Org. Biomol. Chem., 2008, 6, 468–476 RSC; (j) K. Takada, N. Takemura, K. Cho, Y. Sohtome and K. Nagasawa, Tetrahedron Lett., 2008, 49, 1623–1626 CrossRef CAS; (k) D. Uraguchi, T. Ito, S. Nakamura, S. Sakaki and T. Ooi, Chem. Lett., 2009, 38, 1052–1053 CrossRef CAS; (l) A. Cochi, T.-X. Métro, D. G. Pardo and J. Cossy, Org. Lett., 2010, 12, 3693–3695 CrossRef CAS PubMed; (m) H. Xu and C. Wolf, Synlett, 2010, 18, 2765–2770 Search PubMed; (n) Q. J. Yao, D. T. Khong, Q. Gao and Z. M. A. Judeh, Synthesis, 2014, 46, 1793–1801 CrossRef; (o) J. Großeheilmann, J. Bandomir and U. Kragl, Chem.–Eur. J., 2015, 21, 18957–18960 CrossRef PubMed.
- (a) P. S. Prathima, K. Srinivas, K. Balaswamy, R. Arundhathi, G. N. Reddy, B. Sridhar, M. M. Rao and P. R. Likhar, Tetrahedron: Asymmetry, 2011, 22, 2099–2103 CrossRef; (b) H. Xu and C. Wolf, Angew. Chem., Int. Ed., 2011, 50, 12249–12252 CrossRef CAS PubMed; (c) Y. Zhang, Z. J. Li, H. S. Xu, Y. Zhang and W. Wang, RSC Adv., 2011, 1, 389–392 RSC; (d) M.-Q. Li, J.-X. Zhang, X.-F. Huang, B. Wu and Z.-M. Liu, Eur. J. Org. Chem., 2011, 5237–5241 CrossRef CAS.
- (a) T. Mandal, S. Samanta and C.-G. Zhao, Org. Lett., 2007, 9, 943–945 CrossRef CAS PubMed; (b) X. Chen, J. Wang, Y. Zhu, D. Shang, B. Gao, X. Liu, X. Feng, Z. Su and C. Hu, Chem.–Eur. J., 2008, 14, 10896–10899 CrossRef CAS PubMed.
- G. Blay, V. Hernández-Olmos and J. R. Pedro, Chem.–Eur. J., 2011, 17, 3768–3773 CrossRef CAS PubMed.
- M. Holmquist, G. Blay, M. C. Muñoz and J. R. Pedro, Org. Lett., 2014, 16, 1204–1207 CrossRef CAS PubMed.
- Y. Zeng, P. Deng, S. Zhang, H. Yan, G. Liang, Y. Xiong and H. Zhou, Org. Biomol. Chem., 2016, 14, 9221–9224 CAS.
- For the pioneering work of PyBisulidine by Xiao, see (a) A. Gonzalez-de-Castro, C. M. Robertson and J. Xiao, J. Am. Chem. Soc., 2014, 136, 8350–8360 CrossRef CAS PubMed; (b) A. Gonzalez-de-Castro and J. Xiao, J. Am. Chem. Soc., 2015, 137, 8206–8218 CrossRef CAS PubMed.
- For selected examples of asymmetric catalysis using the PyBidines as the ligand, see: (a) T. Arai, A. Mishiro, N. Yokoyama, K. Suzuki and H. Sato, J. Am. Chem. Soc., 2010, 132, 5338–5339 CrossRef CAS PubMed; (b) T. Arai, A. Mishiro, E. Matsumura, A. Awata and M. Shirasugi, Chem.–Eur. J., 2012, 18, 11219–11222 CrossRef CAS PubMed; (c) T. Arai and Y. Ogino, Molecules, 2012, 17, 6170–6178 CrossRef CAS PubMed; (d) T. Arai, S. Kajikawa and E. Matsumura, Synlett, 2013, 24, 2045–2048 CrossRef CAS; (e) T. Arai, E. Matsumura and H. Masu, Org. Lett., 2014, 16, 2768–2771 CrossRef CAS PubMed; (f) T. Arai, K. Tsuchiya and E. Matsumura, Org. Lett., 2015, 17, 2416–2419 CrossRef CAS PubMed; (g) T. Arai, H. Ogawa, A. Awata, M. Sato, M. Watabe and M. Yamanaka, Angew. Chem., Int. Ed., 2015, 54, 1595–1599 CrossRef CAS PubMed; (h) T. Arai, T. Miyazaki, H. Ogawa and H. Masu, Org. Lett., 2016, 18, 5824–5827 CrossRef CAS PubMed; (i) T. Arai, A. Tsuchida, T. Miyazaki and A. Awata, Org. Lett., 2017, 19, 758–761 CrossRef CAS PubMed.
- For detailed discussion of the pre-prepared Ni–L1, see ref. 11 and its ESI.†.
- Crystallographic data for compound 2ac (CCDC 1564222) has been provided as a CIF file in the ESI.†.
- The Henry reaction of 1ac with nitromethane was tested under the optimized conditions..
- For the detailed optimization of the reaction, see ESI.†.
- (a) F. Meyer and H. Kozlowski, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty, T. J. Mever, Elsevier, 2003, vol. 6, ch. 3 Search PubMed; (b) D. A. Evans, D. Seidel, M. Rueping, H. W. Lam, J. T. Shaw and C. W. Downey, J. Am. Chem. Soc., 2003, 125, 12692–12693 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1564222. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra00552d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |