
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtective role of fenofibrate in sepsis-induced acute kidney injury in BALB/c mice
Zuowei Pei a,
Shuling Dengb,
Dengmei Xiec,
Mingyi Lvb,
Wenyan Guob,
Duping Liub,
Zhenzhen Zhengb and
Xiaofeng Long*b
a,
Shuling Dengb,
Dengmei Xiec,
Mingyi Lvb,
Wenyan Guob,
Duping Liub,
Zhenzhen Zhengb and
Xiaofeng Long*b
aDepartment of Cardiology, Affiliated Zhongshan Hospital of Dalian University, No. 6 Jiefang Street, Dalian, China
bDepartment of Intensive Care Units, Affiliated Zhongshan Hospital of Dalian University, No. 6 Jiefang Street, Dalian, China. E-mail: longxiaofeng88@sina.com; Fax: +86-0411-62893373; Tel: +86-0411-62893373
cDepartment of Clinical Pharmacy, Affiliated Zhongshan Hospital of Dalian University, No. 6 Jiefang Street, Dalian, China
First published on 10th August 2018
Abstract
Acute kidney injury (AKI) is a severe complication of sepsis, which largely contributes to the associated high mortality rate. Fenofibrate, a peroxisome proliferator activated receptor α (PPARα) agonist, has received considerable attention because of its effects related to renal damage-related energy metabolism and inflammation. The present study investigated the effects of fenofibrate on sepsis-associated AKI in BALB/c mice subjected to caecal ligation and puncture (CLP). Eight-week-old male BALB/c mice were divided into four groups: control group, fenofibrate group, caecal ligation and puncture (CLP) group, and fenofibrate + CLP group. CLP was performed after mice were gavaged with fenofibrate for 2 weeks. After 48 hours, we measured the histopathological alterations of the kidney tissue and plasma levels of serum creatinine (CRE), neutrophil gelatinase-associated lipocalin (NGAL), reactive oxygen species (ROS), ATP, and ADP. We evaluated PPARα and P53 protein levels as well as interleukin (IL)-1β, IL-6, and tumour necrosis factor-α mRNA levels. Our results showed that administering fenofibrate significantly reduced kidney histological alterations caused by CLP. Fenofibrate inhibited the plasma levels of ROS, CRE, NGAL, and increased the ATP/ADP ratio. Fenofibrate significantly inhibited elevations in P53, IL-1β, IL-6, and tumour necrosis factor-α expression. The results suggest that fenofibrate administration effectively modulates energy metabolism and may be a novel approach to treat sepsis-induced renal damage.
Introduction
Sepsis or septic shock is the predominant cause of acute kidney injury (AKI) in critically ill patients,1,2 but the available treatment options are limited. To improve the diagnosis and treatment of sepsis-associated AKI, we must first understand its pathogenesis. In recent years, the majority of studies on this type of injury have focused on haemodynamic factors after bacterial invasion. However, in many patients sepsis-associated-AKI occurs without overt signs of global renal hypoperfusion, and sepsis-associated-AKI has been described in the presence of normal or even increased renal blood flow.3 Therefore, elucidating the associations of sepsis, energy metabolism, and the production of proinflammatory cytokines has been a popular research topic. Sepsis-induced energy deficiency in organs constitutes another critical component of the pathophysiology of this disease. Studies have shown that inflammatory cytokines, such as tumour necrosis factor-α (TNF-α), interleukin (IL)-1β, and IL-6 play critical roles in the pathological process of kidney injury.4 Moreover, inhibition of the secretion of these inflammatory cytokines could attenuate the injury to kidney tissue. Thus, early anti-inflammatory therapy can improve renal function.PPARα has been shown to play a beneficial role in preventing various diseases by inhibiting the NFκB-induced expression of inflammatory mediators, including vascular cell adhesion molecule-1, IL-6, endothelin-1, and tissue factor in a broad range of cells, including endothelial cells, smooth muscle cells, and macrophages.5–7 The activation of PPARα by fibrates inhibits the IL-1-induced secretion of IL-6 in human aortic smooth muscle cells.8 In contrast, the aorta of PPARα-null mice exhibits an exacerbated response to lipopolysaccharide, revealing that the anti-inflammatory effect of fibrates on the vascular wall requires PPARα activation in vivo.9 The activation of PPARα was required for the L-carnitine-mediated protection of renal tubular cells.10 The PPARα agonist, fenofibrate, ameliorated diabetic nephropathy directly, which involves effects other than a systemic lipid-lowering effect, as evidenced by improvements in albuminuria, glomerular hypertrophy, and mesangial expansion in a type 2 diabetic model.11 However, whether fenofibrate has protective effects against caecal ligation and puncture (CLP)-induced AKI is not known. Therefore, in the present study, we investigated the protective effects and mechanisms underlying these effects of fenofibrate on CLP-induced AKI in mice.
Materials and methods
Animals
All animal studies were approved by the Animal Studies Committee of the affiliated Zhongshan Hospital of Dalian University. Male BALB/c mice were purchased from Dalian Medical University (Dalian, China). All mice were maintained under constant conditions (temperature, 23–25 °C; humidity, 40–60%; 12 hour light/dark cycle).Murine model of sepsis
To induce polymicrobial sepsis, we used an established murine model of CLP as previously described.12–14 The mice were an aesthetised with sodium pentobarbital (100 mg kg−1, intraperitoneal). The peritoneum was opened, and the bowel was exposed. Two-thirds of the caecum was tied off and punctured once with a 21-gauge needle. Gentle pressure was applied at the perforation sites to extrude a small amount of faeces, which was then returned to the peritoneal cavity. The laparotomy site was then stitched. Sham-operated mice underwent the same procedure, which included opening the peritoneum and exposing the bowel but did not include ligation and needle perforation of the caecum. At 8 weeks of age, forty male mice were randomly divided into the following four groups (n = 10/group): the control group, the fenofibrate (100 mg kg−1 d−1; Sigma-Aldrich, St. Louis, MO, USA) treatment group, the CLP group, and the CLP with fenofibrate treatment group.15 CLP was performed after mice were gavaged with fenofibrate for 2 weeks. Forty-eight hours after CLP, all mice that had survived were sacrificed; blood samples were obtained from the inferior vena cava and collected in serum tubes. The samples were then stored at −80 °C until use. Coronal sections of kidney tissues were fixed in 10% formalin and then embedded in paraffin for histological evaluation. The remainder of the kidney tissue was snap-frozen in liquid nitrogen for mRNA or immunohistochemical analysis. All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals. The study was approved by the ethical committee of the affiliated Zhongshan Hospital of Dalian University.Serum analysis
Serum concentrations of reactive oxygen species (ROS), creatinine (CRE), ATP, and ADP were measured using ELISA kits (Westang, Shanghai, China) according to the manufacture's instruction. The ELISA (Boster, Wuhan, China) was used for measuring serum concentration of NGAL as instructed by the manufacturer. Briefly, the standards and test samples were pipetted into the wells to allow binding of these proteins to the immobilized antibodies. After washing away any unbound substances, an enzyme linked polyclonal antibody specific for these proteins was added to the wells. Following removal of unbound antibody-enzyme reagent through washing, a substrate solution was added to the wells and color was developed. The optical density of each well was measured at the wave length of 510 nm or 550 nm on a microreader (51119000, Thermo Scientific Multiskan FC, USA). The concentration of ROS, CRE, ATP and ADP was accordingly calculated.Morphologic analysis
The kidney tissues were fixed with 10% buffered formalin solution for 30 minutes and then dehydrated in 75% ethanol overnight, followed by paraffin embedding. Serial sections (4 μm) were stained with hematoxylin and eosin (H&E) staining to assess the pathologic changes. Renal injury scores were determined by two researchers in a blinded fashion according to the extent of kidney injury, as previously described. Briefly, the scoring grading was mainly based on the hemorrhage, tubular cell necrosis, tubular dilatation, and cytoplasmic vacuole formation. The grading system was shown as the following scoring: 0 (normal kidney); 1 (0–5% injury, minimal damage); 2 (5–25% injury, mild damage); 3 (25–75% injury, moderate damage); and 4 (75–100% injury, severe damage).16,17Immunohistochemistry
Immunohistochemical analysis was performed using the Histone Simple Stain Kit (Nichirei, Tokyo, Japan), according to the manufacturer's instructions. Paraffin-embedded sections were deparaffinised with xylene and then rehydrated in a descending series of ethanol washes. The sections were treated for 15 minutes with 3% H2O2 in methanol to inactivate endogenous peroxidases and then incubated at room temperature for 1 hour with primary antibodies to PPARα (rabbit anti-PPARα antibody, 1:200; Proteintech, Wuhan, China) and P53 (rabbit anti-P53 antibody, 1:200; Proteintech). Tissue sections were observed with a microscope (Olympus, Tokyo, Japan).RNA isolation and real-time RT-PCR
Total RNA was isolated from kidney tissue using the ISOGEN (Nippon Gene, Tokyo, Japan), according to the manufacturer's protocol. Complementary DNA (cDNA) was synthesised from total RNA using a first-strand cDNA synthesis kit (SuperScript VILO cDNA Synthesis Kit; Life Technologies, Carlsbad, CA, USA), according to the manufacturer's protocol. Gene expression was analysed quantitatively by RT-PCR using fluorescent SYBR Green technology (Light Cycler; Roche Molecular Biochemicals). β-Actin cDNA was amplified and quantitated in each cDNA preparation in order to normalise the relative expression of the target genes. Primer sequences are listed in Table 1.| Gene | Primers |
|---|---|
| a TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; IL-1β, interleukin-1β. | |
| TNF-α | F: 5′-TCTCATGCACCACCATCAAGGACT-3′ |
| R: 5′-ACCACTCTCCCTTTGCAGAACTCA-3′ | |
| IL-6 | F: 5′-TACCAGTTGCCTTCTTGGGACTGA-3′ |
| R: 5′-TAAGCCTCCGACTTGTGAAGTGGT-3′ | |
| IL-1β | F: 5′-TGCCACCTTTTGACAGTGAT-3′ |
| R: 5′-TGTGCTGCTGCGAGATTTGA-3′ | |
| β-Actin | F: 5′-CGATGCCCTGAGGGTCTTT-3′ |
| R: 5′-TGGATGCCACAGGATTCCAT-3′ | |
Western blotting for kidney tissues
Proteins were extracted from kidney tissue using radio immunoprecipitation assay buffer (P0013B; Beyotime, Shanghai, China). Samples were electrophoresed on a 10% SDS-PAGE gel, and proteins were transferred to polyvinylidene fluoride membrane (Immobilon, Millipore, Billerica, MA, USA). Membranes were blocked in Tris-buffered saline with 0.1% Tween-20 (TBS-T) containing 5% skim milk and then incubated in primary antibody diluent (P0023A; Beyotime) and gently shaken overnight at 4 °C. Primary antibodies against PPARα (rabbit anti-PPARα antibody, 1:1000; Proteintech), P53 (rabbit anti-P53 antibody, 1:1000; Proteintech), and anti-β-actin (1:1000; Cell Signaling Technology) were utilised. Membranes were then incubated with secondary antibody (anti-rabbit Ig-G, 1:1000; Cell Signaling Technology for 1 hour). This analysis was carried out independently three times. Protein levels are expressed as protein/β-actin ratios to minimise loading differences. The relative signal intensity was quantified using NIH ImageJ software.Statistical analysis
All data are presented as the mean ± SEM. Statistical analysis was performed using SPSS software version 23.0 (SPSS Inc., Chicago, IL, USA). Inter-group variation was measured by one-way ANOVA and subsequent Tukey's test. The minimal level for significance was P < 0.05.Results
Metabolic characterization
The metabolic characteristics of BALB/c mice from the four groups exposed to different treatments are summarized in Fig. 1. Kidney/body weights did not differ among the four groups. The CLP mice group showed markedly increased CRE, NGAL, and ROS levels, but these levels significantly decreased in the Fen + CLP group. The level of ATP/ADP significantly increased in the Fen + CLP group compared to those in the CLP group. Compared to the CLP mice group, there are the significant difference in the control, Fen, and Fen + CLP mice groups. There was no difference among the control, Fen, and Fen + CLP groups. | ||
| Fig. 1 Metabolic data from the four groups of BALB/cm ice after different treatments. Kidney/body weights and ROS, ATP/ADP, CRE, and NGAL levels of four groups after different treatments are presented. Data are means ± SEM; n = 6–7 per group. *P < 0.05, **P < 0.01 vs. CLP group. | ||
Fenofibrate reduced histopathological damage in kidney tissue
To evaluate the histopathological damage in kidney tissue, H&E staining was performed (Fig. 2). The H&E staining results showed that the kidneys from the CLP group mice displayed hemorrhage, inflammatory infiltration, detachment and swelling of the tubular epithelial cells, interstitial edema, tubular cell casts and dilatation, and necrosis (Fig. 2(A)). The renal injury scoring (CLP versus Fen + CLP group, 3.417 ± 0.3817 versus 2.583 ± 0.3371, P < 0.05, Fig. 2(B)) showed that fenofibrate treatment could significantly decrease the renal injury of septic BALB/c mice, which was consistent with the H&E results. | ||
| Fig. 2 Histopathological damage in kidney tissue from the four groups of BALB/c mice subjected to different treatments. (A) Histological analysis of H&E-stained kidney samples demonstrated a markedly reducing inflammatory infiltration in the kidney tissue of the Fen + CLP group of mice compared to that observed in the CLP group of mice. (B) Renal injury score. Scale bar = 100 μm. Arrows indicate inflammatory infiltration. Data are means ± SEM; n = 6–7 per group. *P < 0.05, **P < 0.01 vs. CLP group. | ||
Fenofibrate increased PPARα expression in kidney tissue
To evaluate PPARα expression in kidney tissue, PPARα immunostaining was performed (Fig. 3A). The Fen + CLP group exhibited markedly increased PPARα expression in kidney tissue compared to that observed in the CLP group. Immunoblotting was performed to detect PPARα protein (Fig. 3B). We found that PPARα protein expression was significantly increased in the Fen + CLP group compared to that in the CLP group (Fig. 3C). These results indicate that fenofibrate increased PPARα expression in the CLP group of mice. | ||
| Fig. 3 PPARα expression in kidney tissue from the four groups of BALB/cm ice after different treatments. Representative immunohistochemistry for PPARαin kidney tissue. Scale bar = 100 μm. Arrows indicate positively stained cells. (B) Immunoblotting analysis for PPARα expression in kidney tissue. (C) Bar graph showing quantification of PPARα protein expression. Data are expressed as the means ± SEM; n = 3–4 in each group. *P < 0.05 vs. CLP group. | ||
Fenofibrate reduced P53 expression in kidney tissue
To evaluate P53 expression in kidney tissue, immunohistochemical analysis of P53 was performed (Fig. 4A). The Fen + CLP group exhibited markedly reduced staining of P53 in kidney tissue compared to that observed in the CLP group. Immunoblotting was performed to assess P53 protein levels (Fig. 4B). We found that P53 protein expression was significantly suppressed in the Fen + CLP group compared to that in the CLP group (Fig. 4C). These results indicate that fenofibrate reduced P53 in the CLP group of mice.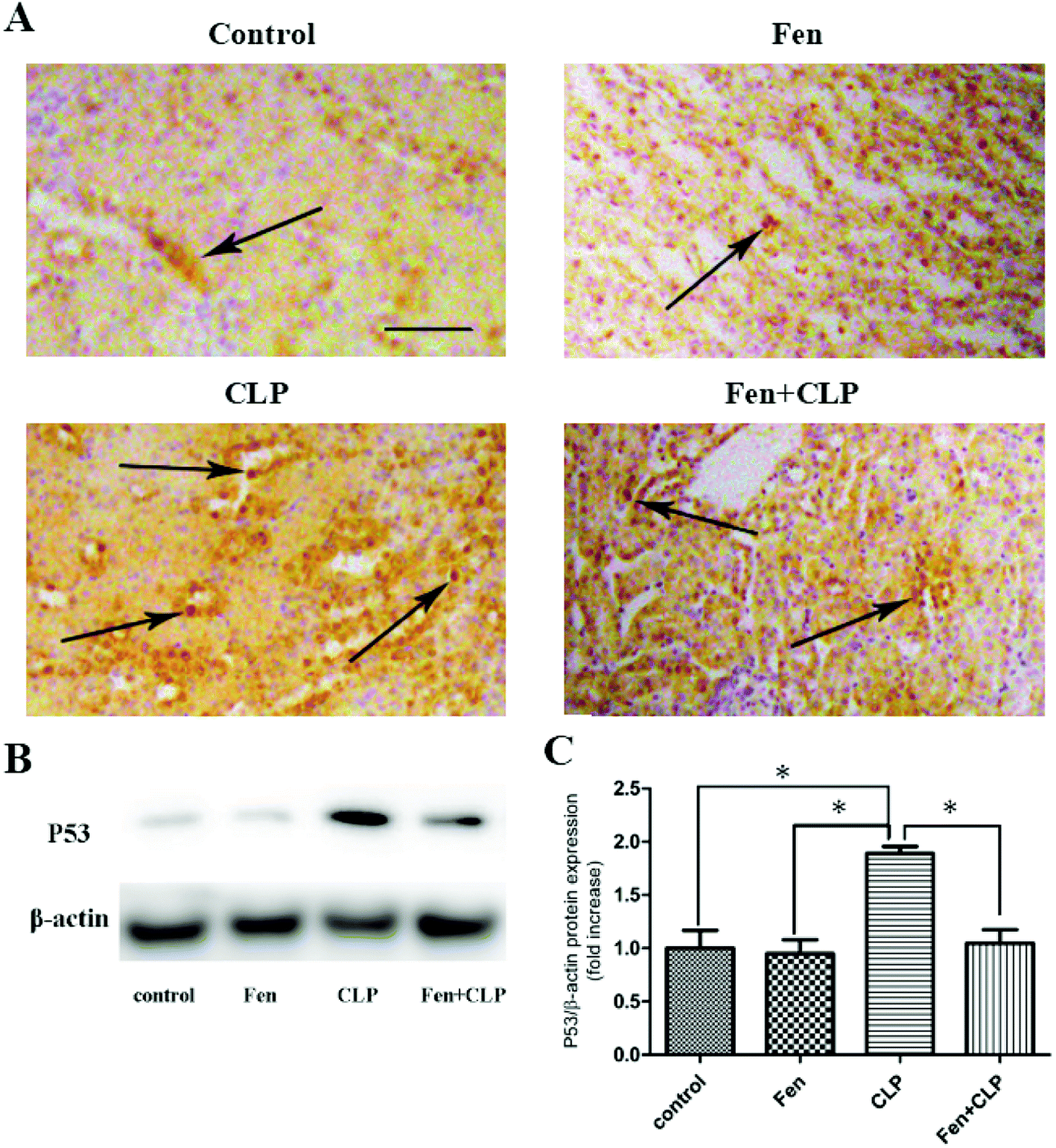 | ||
| Fig. 4 P53 expression in kidney tissue from the four groups of BALB/cm ice after different treatments. Representative immunohistochemistry for P53 in kidney tissue; scale bar = 100 μm. Arrows indicate positively stained cells. (B) Immunoblotting analysis for P53 expression in kidney tissue. (C) Bar graph showing quantification of P53 protein expression. Data are expressed as the means ± SEM; n = 3–4 in each group. *P < 0.05 vs. CLP group. | ||
Fenofibrate reduced interleukin (IL)-6, IL-1β, and tumour necrosis factor (TNF)-α gene expression in kidney tissues of the CLP group of mice
To examine the involvement of pro-inflammatory cytokines in the gene expression in kidney tissue from mice in the four experimental groups, the expression of genes encoding IL-6, IL-1β, and TNF-α was measured using real-time PCR (Fig. 5). IL-6, IL-1β, and TNF-α genes were upregulated in the CLP group of mice; however, this upregulation was attenuated in the Fen + CLP group of mice.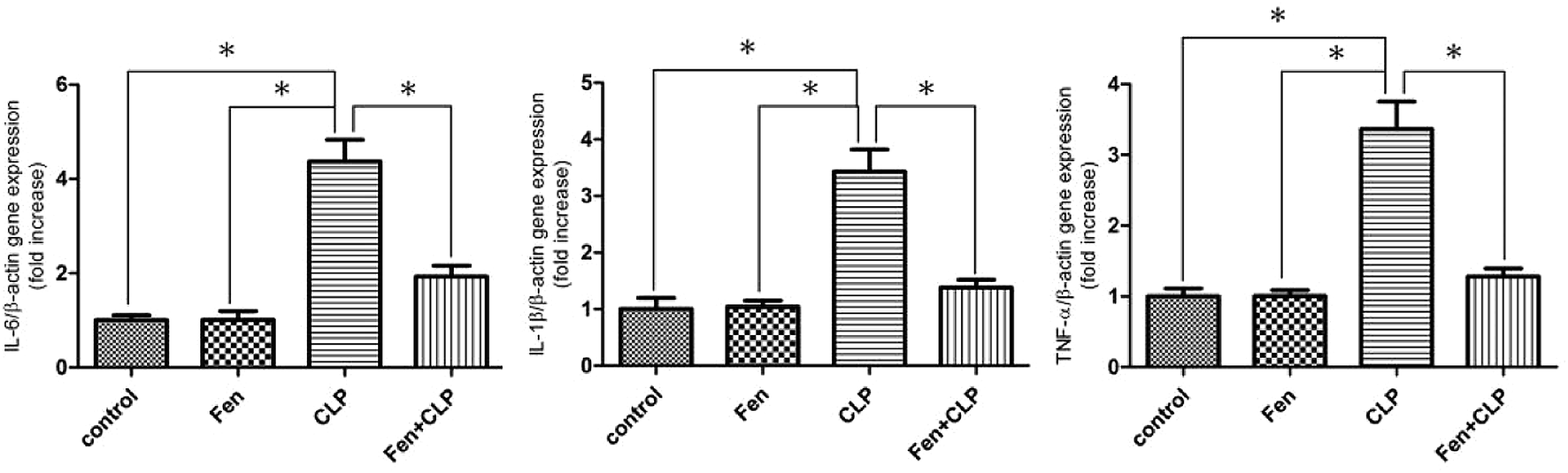 | ||
| Fig. 5 Expression of pro-inflammatory genes in kidney tissue from the four groups of BALB/c mice after different treatments. Relative mRNA expression of IL-6, IL-1β, and TNF-α in kidney tissue of the four groups after different treatments. Data are expressed as the means ± SEM; n = 6 in each group. *P < 0.05 vs. CLP group. | ||
Survival rate
By observing for 48 hours, we compared the survival rate of four groups using Kaplan–Meier method and the log-rank test. There were no death in control and Fen groups. Four of the 10 mice in CLP group were dead, while two out of the 10 mice gavaged with fenofibrate were dead post-CLP. In general, the survival rate in the Fen + CLP group was significantly higher than that of CLP group (Fig. 6). | ||
| Fig. 6 Survival analysis of four groups. Each point in the figure represents the mean survival rate at each time point. The survival rate is compared by Kaplan–Meier method and the log-rank test (n = 10/group, *P < 0.05 vs. CLP group). | ||
Discussion
This study shows that fenofibrate has a protective effect against kidney damage since fenofibrate treatment increased ATP/ADP ratio and PPARα expression and decreased the levels of ROS, P53, and pro-inflammatory cytokines.Previous studies have demonstrated that mitochondrial dysfunction contribute to septic AKI, such as the depletion of ATP, the overproduction of ROS.18 Yang RL et al. have reported that ischemic injury of the kidney result from ATP depletion.19 They found that imbalanced energy metabolism, rather than tubular cell apoptosis, may be the initiator of renal dysfunction during lipopolysaccharide-induced septic shock.19 Meanwhile, the ATP level can inhibit cell apoptosis and minimize organ dysfunction.20 We obtained the similar result. According to the metabolic characteristics of the mice, we found that fenofibrate treatment increased the ATP/ADP ratio compared to that observed in the CLP group of mice. The reduction of the ATP/ADP ratio means a low ATP synthesis efficiency.21 Several studies have shown that relatively high levels of ROS cause redox imbalance, induce cell apoptosis or necrosis, and deteriorate the inflammation response in physiological and pathological conditions.22–24 We found that fenofibrate treatment decreased the level of ROS compared to that observed in the CLP group of mice. In addition, CRE and NGAL are the sensitive indicators of kidney damage. In our study, fenofibrate treatment decreased the levels of CRE and NGAL compared to that observed in the CLP group of mice. As a result, increased histopathological damage led to the release of additional inflammatory molecules in the CLP group than in the Fen + CLP group. Taken together, serum and histological results confirmed that renal damage occurred in the CLP group, but this damage was significantly suppressed in the Fen + CLP group. Above results demonstrated the protective effects of fenofibrate, and the observation was further confirmed by survival rate. The survival rate results demonstrated the protective effects of fenofibrate as well. We found the survival rate in the Fen + CLP group was significantly higher than that of CLP group.
Sepsis-induced energetic deficiency in organs constitutes another critical component of the pathophysiology of the disease. PPARα is a ligand-activated nuclear receptor transcription factor. This transcription factor sits at the interface between inflammatory and metabolic pathways and regulates aspects of each.25 Endogenous PPAR ligands include specific fatty acids and their derivatives. Many exogenous PPAR agonists are used in clinical practice. The fibrate class of drugs, including fenofibrate, activates PPARα. PPARα is widely expressed in the body (especially in the heart, kidney, and liver); it exerts a general anti-inflammatory influence over the immune response and also regulate senergy metabolism.
Several studies have indicated that PPARα activation leads to an increase in fatty acid catabolism and ATP production as well as a decrease in the levels of cytotoxic fatty acid peroxidation products, promotion of cell viability, and inhibition of renal epithelium cell death.26–28 In our study, we analysed PPARα expression in kidney tissue by immunohistochemical analysis and immunoblotting. We observed that PPARα expression was significantly decreased in the CLP group; however, the PPARα level was markedly increased in the Fen + CLP group compared with that in the CLP group.
P53 is a stress-induced transcription factor that can be activated by a number of adverse stimuli, including DNA damage, hypoxia, and ROS. Increased expression of P53 protein under these conditions causes growth arrest or apoptosis.29,30 In vitro studies have shown that fenofibrate activates AMPK in various cells, such as myocytes, retinal endothelial cells, and human umbilical vein endothelial cells.31–33 AMPK signalling also inhibits the inflammatory response, which is mediated by several transcription factors that are the downstream targets of AMPK, such as P53.34 Our results showed that the Fen + CLP group exhibited markedly reduced P53 expression in kidney tissue compared to that observed in the CLP group, and P53 protein expression was significantly suppressed in the Fen + CLP group compared to the CLP group. These results indicate that fenofibrate reduced P53 in the CLP group of mice.
Pro-inflammatory genes (TNF-α, IL-1β, and IL-6) have been reported to be expressed at high levels and contribute to renal damage in sepsis. The present study showed that TNF-α, IL-1β, and IL-6 gene expression was reduced in the Fen + CLP group compared to the expression in the CLP group.
Limitations
It should be noted that this study has used only one dose fenofibrate (100 mg kg−1 d−1). In some previous studies, we found some researchers study the effect of fenofibrate (100 mg kg−1 d−1) pretreatment on preventing the development of diabetic nephropathy and on acute lung injury induced by intestinal ischemia/reperfusion in mice.35,36 We directly chose the dose of 100 mg kg−1 d−1 according to the previous study. However, fenofibrate have the potential risk of inducing rhabdomyolysis and acute renal failure.37,38 Lacking of study of dose-dependence and potential toxicities of fenofibrate in our experiment. It would be better if we perform dose ranging experiments rather than use one dose of fenofibrate. This area deserves further preclinical and clinical investigations.Conclusions
In conclusion, our study established that fenofibrate can help mitigate septic renal damage as shown by the upregulation of PPARα and ATP/ADP as well as the suppression of ROS and P53. These findings provide new insights into the role of fenofibrate in sepsis-induced AKI and raise the possibility of a novel therapeutic intervention to treat AKI.Author contributions
Xiaofeng Long designed this study; Zuowei Pei and Wenyan Guo helped in performing experiments; Mingyi Lv and Dengmei Xie analyzed data and interpreted the results of experiments; Zhenzhen Zheng and Shuling Deng prepared figures; Zuowei Pei drafted the manuscript; Duping Liu helped to revising of manuscript. All authors read and approved the final manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was finally supported by the Natural Science Foundation of Liaoning Province, China (No. 201602023).References
- A. Romanovsky, C. Morgan and S. M. Bagshaw, Pathophysiology and management of septic acute kidney injury, Pediatr. Nephrol., 2014, 29, 1–12 CrossRef PubMed
.
- J. Chvojka, R. Sykora, A. Krouzecky, J. Radej, V. Varnerova and T. Karvunidis, et al., Renal haemodynamic, microcirculatory, metabolic and histopathological responses to peritonitis-induced septic shock in pigs, Crit Care, 2008, 12 DOI:10.1186/cc7164
- L. Wan, S. M. Bagshaw, C. Langenberg, C. Langenberg, T. Saotome and C. May, et al., Pathophysiology of septic acute kidney injury: what do we really know?, Crit. Care Med., 2008, 36, 198–203 CrossRef PubMed
- G. Ramesh and W. B. Reeves, TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity, J. Clin. Invest., 2002, 110, 835–842 CrossRef PubMed
- N. Marx, G. K. Sukhova, T. Collins, P. Libby and J. Plutzky, PPARalpha activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells, Circulation, 1999, 99, 3125–3131 CrossRef PubMed
- P. Delerive, K. De Bosscher, S. Besnard, W. Vanden Berghe, J. M. Peters and F. J. Gonzalez, et al., Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1, J. Biol. Chem., 1999, 274, 32048–32054 CrossRef PubMed
- B. P. Neve, D. Corseaux, G. Chinetti, C. Zawadzki, J. C. Fruchart and P. Duriez, et al., PPAR alpha agonists inhibit tissue factor expression in human monocytes and macrophages, Circulation, 2001, 103, 207–212 CrossRef PubMed
- B. Staels, W. Koenig, A. Habib, R. Merval, M. Lebret and I. P. Torra, et al., Activation of human aortic smooth-muscle cells is inhibited by PPAR alpha but not by PPARgamma activators, Nature, 1998, 393, 790–793 CrossRef PubMed
- P. Delerive, P. Gervois, J. C. Fruchart and B. Staels, Induction of Ikappa Balpha expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-alpha activators, J. Biol. Chem., 2000, 275, 36703–36707 CrossRef PubMed
- Y. M. Sue, H. C. Chou, C. C. Chang, N. J. Yang, Y. Chou and S. H. Juan, L-carnitine protects against carboplatin-mediated renal injury: AMPK- and PPARα- dependent inactivation of NFAT3, PLoS One, 2014, 9 DOI:10.1371/journal.pone.0104079
- C. W. Park, Y. Zhang, X. Zhang, J. Wu, L. Chen and D. R. Cha, et al., PPAR alpha agonist fenofibrate improves diabetic nephropathy in db/db mice, Kidney Int., 2006, 69, 1511–1517 CrossRef PubMed
- D. Rittirsch, M. S. Huber-Laang, M. A. Flierl and P. A. Ward, Immunodesign of experimental sepsis by cecal ligation and puncture, Nat. Protoc., 2009, 4, 31–36 CrossRef PubMed
- H. Wen, Sepsis induced by cecal ligation and puncture, Methods Mol. Biol., 2013, 1031, 117–124 CrossRef PubMed
- B. C. Su, H. N. Huang, T. W. Lin, C. D. Hsiao and J. Y. Chen, Epinecidin-1 protects mice from LPS-induced endotoxemia and cecal ligation and puncture-induced polymicrobial sepsis, Biochim. Biophys. Acta, 2017, 1863, 3028–3037 CrossRef PubMed
- I. A. Bukhari, A. A. Almotrefi, O. Y. Mohamed, A. A. Al-Masri and S. A. Sheikh, Protective effect of fenofibrate against ischemia-/reperfusion-induced cardiac arrhythmias in isolated rat hearts, Fundam. Clin. Pharmacol., 2018, 32, 141–146 CrossRef PubMed
- X. Hong, X. Zhao, G. Wang, Z. Zhang, H. Pei and Z. Liu, Luteolin Treatment Protects Against Renal Ischemia-Reperfusion Injury in Rats, Mediators Inflammation, 2017, 2017 DOI:10.1155/2017/9783893
- C. Shingu, H. Koga, S. Hagiwara, S. Matsumoto, K. Goto, I. Yokoi and T. Noguchi, Hydrogen-rich saline solution attenuates renal ischemia-reperfusion injury, J. Anesth., 2010, 24, 569–574 CrossRef PubMed
- N. B. Madungwe, Y. Feng, M. Lie, N. Tombo, L. Liu, F. Kaya and J. C. Bopassa, Mitochondrial Inner Membrane Protein (Mitofilin) Knockdown Induces Cell Death by Apoptosis Via an AIF-PARP-Dependent Mechanism and Cell Cycle Arrest, Am. J. Physiol.: Cell Physiol., 2018, 315, 28–43 CrossRef PubMed
- R. L. Yang, X. T. Wang, D. W. Liu and S. B. Liu, Energy and oxygen metabolism disorder during septic acute kidney injury, Kidney Blood Pressure Res., 2014, 39, 240–251 CrossRef PubMed
- A. V. Birk, S. Liu, Y. Soong, W. Mills, P. Singh and J. D. Warren, et al., The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin, J. Am. Soc. Nephrol., 2013, 24, 1250–1261 CrossRef PubMed
- E. N. Maldonado and J. J. Lemasters, ATP/ADP ratio, the missed connection between mitochondria and the Warburg effect, Mitochondrion, 2014, 19, 78–84 CrossRef PubMed
- M. L. Circu and T. Y. Aw, Reactive oxygen species, cellular redox systems, and apoptosis, Free Radical Biol. Med., 2010, 48, 749–762 CrossRef PubMed
- D. Wu, N. Luo, L. Wang, Z. Zhao, H. Bu and G. Xu, et al. Hydrogen sulfide ameliorates chronic renal failure in rats by inhibiting apoptosis and inflammation through ROS/MAPK and NF-κB signaling pathways, Sci. Rep., 2017, 7 DOI:10.1038/s41598-017-00557-2
- H. Wu, K. Jiang, N. Yin, X. Ma, G. Zhao and C. Qiu, et al., Thymol mitigates lipopolysaccharide-induced endometritis by regulating the TLR4- and ROS-mediated NF-κB signaling pathways, Oncotarget, 2017, 8, 20042–20055 Search PubMed
- W. Wahli and L. Michalik, PPARs at the crossroads of lipid signaling and inflammation, Trends Endocrinol. Metab., 2012, 23, 351–363 CrossRef PubMed
- F. Ouali, F. Djouadi, C. Merlet-Benichou and J. Bastin, Dietary lipids regulate beta-oxidation enzyme gene expression in the developing rat kidney, Am. J. Physiol., 1998, 275, 777–784 Search PubMed
- Y. Luo, Q. He, G. Kuang, Q. Jiang and J. Yang, PPAR-alpha and PPAR-beta expression changes in the hippocampus of rats undergoing global cerebral ischemia/reperfusion due to PPAR-gamma status, Behav. Brain Funct., 2014, 10 DOI:10.1186/1744-9081-10-21
- M. Pawlak, P. Lefebvre and B. Staels, Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease, J. Hepatol., 2015, 62, 720–733 CrossRef PubMed
- S. W. Lowe, E. M. Schmitt, S. W. Smith, B. A. Osborne and T. Jacks, p53 is required for radiation-induced apoptosis in mouse thymocytes, Nature, 1993, 362, 847–849 CrossRef PubMed
- K. Polyak, Y. Xia, J. L. Zweier, K. W. Kinzler and B. Vogelstein, A model for p53-induced apoptosis, Nature, 1997, 389, 300–305 CrossRef PubMed
- W. L. Chen, Y. L. Chen, Y. M. Chiang, S. G. Wang and H. M. Lee, Fenofibrate lowers lipid accumulation in myotubes by modulating the PPARalpha/AMPK/FoxO1/ATGL pathway, Biochem. Pharmacol., 2012, 84, 522–531 CrossRef PubMed
- J. Kim, J. H. Ahn, J. H. Kim, Y. S. Yu, H. S. Kim and J. Ha, et al., Fenofibrate regulates retinal endothelial cell survival through the AMPK signal transduction pathway, Exp. Eye Res., 2007, 84, 886–893 CrossRef PubMed
- H. Murakami, R. Murakami, F. Kambe, X. Cao, R. Takahashi and T. Asai, et al., Fenofibrate activates AMPK and increases eNOS phosphorylation in HUVEC, Biochem. Biophys. Res. Commun., 2006, 341, 973–978 CrossRef PubMed
- K. H. Vousden and K. M. Ryan, p53 and metabolism, Nat. Rev. Cancer, 2009, 9, 691–700 CrossRef PubMed
- Y. Cheng, J. Zhang, W. Guo, F. Li, W. Sun and J. Chen, et al., Up-regulation of Nrf2 is involved in FGF21-mediated fenofibrate protection against type 1 diabetic nephropathy, Free Radical Biol. Med., 2016, 93, 94–109 CrossRef PubMed
- Q. Zhu, G. He, J. Wang, Y. Wang and W. Chen, Protective effects of fenofibrate against acute lung injury induced by intestinal ischemia/reperfusion in mice, Sci. Rep., 2016, 6 DOI:10.1038/SREP22044
- M. Tahmaz, B. Kumbasar, K. Ergen, U. Ure, G. Karatemiz and R. Kazancioglu, Acute renal failure secondary to fenofibrate monotherapy-induced rhabdomyolysis, Renal Failure, 2007, 29, 927–930 CrossRef PubMed
- J. Wu, Y. Song, H. Li and J. Chen, Rhabdomyolysis associated with fibrate therapy: review of 76 published cases and a new case report, Eur. J. Clin. Pharmacol., 2009, 65, 1169–1174 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2018 |