DOI:
10.1039/C8RA00385H
(Paper)
RSC Adv., 2018,
8, 9762-9769
Preparation and characterization of an electrospun colon-specific delivery system for salmon calcitonin†
Received
13th January 2018
, Accepted 3rd March 2018
First published on 8th March 2018
Abstract
A novel electrospun colon-specific delivery system for salmon calcitonin (SCT) was developed to improve its stability and bioavailability. Firstly, the pectin-coated SCT liposomes were prepared by film dispersion method and then a liposomes/sodium alginate/polyvinyl alcohol fiber mat was fabricated by electrospining. Scanning electron microscopy analysis indicated that the obtained nanofibers were uniform and smooth with an average diameter of about 350 nm. The release of SCT in different simulated digestive fluids was studied and corresponding release kinetics models were built. It was found that the fiber mat containing pectin-coated SCT liposomes had better stability and colon-specific properties compared with that containing uncoated SCT liposomes and the release of SCT in the colon followed the case II transport mechanism. In addition, there is no significant change in the bioactivity of released SCT measured by ELISA. This study shows that the electrospun colon-specific fiber mat is a potential delivery system for bioactive peptides.
1 Introduction
Calcitonin is an active peptide composed of thirty-two amino acids, which plays an important role in maintaining calcium balance and inhibiting osteoclast activity, effectively reducing serum calcium and promoting bone absorption of calcium.1 Compared with other sources, salmon calcitonin (SCT) has been widely used in clinic for its advantages of high activity, long action time and less side effects.2 At present, SCT has been used clinically in the form of intramuscular and subcutaneous injections and nasal spray formulations.3 Considering the long-term nature of the treatment, nasal administration is more convenient and easier to accept by patients than frequent injection. However, current nasal spray formulations irritate nasal mucosa and cause side effects such as rhinitis, rhinorrhea, and allergic rhinitis.4 What's more, the bioavailability of SCT through nasal spray is much lower than injection, i.e., only approximately 3% for clinically used sprays.5 Therefore, it is necessary to develop a new type of SCT transmission system to avoid the above problems.
The instability and low bioavailability of SCT when exposed to upper gastrointestinal tract (GIT) conditions significantly compromised the envisioned benefits and thus limiting its application.6 Therefore, controlled release of which at the right place is a key approach to improve its effectiveness and bioavailability.6 Presently, different delivery systems have been used to improve oral bioavailability of SCT. Among them, the oral colon-specific delivery approach has attracted increasing attention, since this kind of delivery system could take advantage of the potentially favorable characteristics of the colon environment, offering obvious advantages over administration to the small intestine.7,8 Especially, the polysaccharides-based microflora-activated colonic delivery system, such as chitosan, alginate, and cellulose etc., have gained significant attention owing to the abundant colonic microflora population and their corresponding enzyme activities on degradation of polysaccharides that occurred only in colon.9
In recent years, some new forms, such as microspheres, liposomes and nanoparticles, have been used to encapsulate SCT to enhance its stability and bioavailability.10–12 However, they can't effectively improve the oral bioavailability of SCT. Liposomes are generally used in drug delivery and are designed to reduce toxicity, increase efficacy, enhance targeting and improve stability of drug.13–15 However, as an oral delivery system, liposomes have poor stability, so it is necessary to improve the stability of liposomes by other methods. Electrospinning is a simple and effective method of direct and continuous preparation of nanofibers and a mild method of operation as well.16,17 In the past few years, electrospinning has been well established for encapsulating bioactive compounds.18–20 It can be even used to develop a colon-targeted delivery system to achieve slow and sustained release of protein.21
In order to improve its stability and bioavailability, a novel colon-specific delivery system for SCT was developed by electrospinning in this study. Pectin is an anionic polymer which is easily degraded by microbial enzymes in the colon, thus it was coated on the surface of liposomes to further enhance their stability. Firstly, the uncoated and pectin-coated SCT liposomes were prepared by film dispersion method. Then, the liposomes were added into the mixed polymer solution containing sodium alginate (SA) and polyvinyl alcohol (PVA) and the mixture was electrospun to obtain fiber mat. The morphology of fiber mat and the release behavior of SCT in simulated digestive fluids were investigated, respectively. To understand the colon-specific release mechanism, the release kinetics of SCT was investigated by establishing corresponding mathematics model. Finally, the bioactivity of released SCT from fiber mat was determined by SCT ELISA kit.
2 Materials and methods
2.1. Materials
Low-viscosity SA (from brown algae) and pectinase were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Pectin, pepsin and trypsin were obtained from Aladdin Biological Technology Co., Ltd (Shanghai, China). PVA (Mw: 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–124000) was purchased from Tianma Fine Chemical Factory (Guangzhou, China). SCT was purchased from Dalian Meilun Biological Technology Co., Ltd (Dalian, China). Lecithin (from soybean) was purchased from Shanghai Source Poly Biological Technology Co., Ltd (Shanghai, China). Cholesterol was purchased from Shanghai Biological Technology Co., Ltd (Shanghai, China). Ether and methanol were all analytical grade and were used as received. SCT ELISA kit was purchased from Shanghai Chang Jin Biological Technology Co., Ltd (Shanghai, China). Deionized water was used to prepare all the solutions.
000–124000) was purchased from Tianma Fine Chemical Factory (Guangzhou, China). SCT was purchased from Dalian Meilun Biological Technology Co., Ltd (Dalian, China). Lecithin (from soybean) was purchased from Shanghai Source Poly Biological Technology Co., Ltd (Shanghai, China). Cholesterol was purchased from Shanghai Biological Technology Co., Ltd (Shanghai, China). Ether and methanol were all analytical grade and were used as received. SCT ELISA kit was purchased from Shanghai Chang Jin Biological Technology Co., Ltd (Shanghai, China). Deionized water was used to prepare all the solutions.
2.2. Preparation of SCT liposomes and pectin-coated SCT liposomes
SCT liposomes were prepared with lecithin, cholesterol and SCT as materials by film dispersion method. Briefly, a certain amount of lecithin and cholesterol were added to a 100 mL round bottom flask and dissolved in 9 mL mixed solvent of chloroform and methanol (8:1, v/v). Then lipid film was produced by evaporating the solvents using a rotary evaporator operate at 35 °C and 70 rpm. The resulting lipid film was hydrated with 5 mL SCT phosphate solution in 70 °C for 10 s followed by eluting for 5 min and this process repeated three times. The obtained solution was incubated at 10 °C for 1 h and then was subjected to ultrasound in ice bath for 5 min to produce liposomes. After that, the resulting liposome solution was through the 0.22 μm membrane to generate narrowly distributed SCT liposome solution. The pectin solution was prepared by adding a certain amount of which into 100 mL distilled water and then stirred at room temperature for 2 h. Finally, the SCT liposome solution was gradually added to the pectin solution (1:1, v/v) and then stirred for 2 h at room temperature to obtain pectin-coated SCT liposome solution.
2.3. Characterization of the liposomes
2.3.1 Size and zeta potential measurement. The particle size and zeta potential of the liposomes were measured using a Nano ZS Zetasizer instrument (Malvern Instruments, Worcestershire, UK). All measurements were carried out at 25 °C, and all the experiments were performed in triplicate.
2.3.2 TEM analysis. The morphology of a single liposome was observed by TEM (JEOL, Japan). The liposome solution was diluted with distilled water and then dripped onto the copper grid. The sample was negatively stained with phosphotungstic acid and dried at room temperature, and then was examined at 80 kV.
2.4. Encapsulation efficiency of SCT
2.4.1 HPLC method. The content of SCT was determined by Agilent 1200 high performance liquid chromatography (HPLC). A ZORBAX Eclipse XDB-C18 column (4.6 mm × 250 mm) was used as the stationary phase. The mobile phase consisted of mobile phase A (an aqueous solution containing 0.1% trifluoroacetic acid) and mobile phase B (an aqueous solution containing 0.1% trifluoroacetic acid:acetonitrile (3:2, v/v)). The conditions for gradient elution were as follows: 45–70% B for 20 min, 70–45% B from 20 min to 22 min and 45% from 22 min to 23 min. The flow rate of the mobile phase was 1.0 mL min−1 and the injection volume was 20 μL. The detector wavelength was set at 210 nm for quantification of SCT. The calibration curve constructed over the concentration range of 50–150 μg mL−1. Six different concentrations of standard SCT were measured for three times. The regression equation of the calibration curve is y = 12419x − 116907 (where y is the peak area and x is the concentration of SCT in μg mL−1, R2 = 0.9981, n = 6).
2.4.2 Determination of encapsulation efficiency. The liposomes were separated from the resulting solution by ultra-filtered centrifugation at 10000 rpm (25 °C, 15 min). The amount of SCT in the upper liposomes (SCTf) and the total amount of SCT (SCTt) were measured by the above-mentioned HPLC method. The SCT encapsulation efficiency (EE) was calculated using the following equation:
2.5. Electrospinning
A mixed solution was prepared by adding SA solution (dissolved in distilled water) to the PVA solution (dissolved in distilled water) in a appropriate mass ratio. Then the appropriate amount of uncoated or pectin-coated SCT liposome solutions were added into the above mixture, respectively, and mixed evenly to form spinning solutions. The electrospinning was performed using a power supply (ES50P-5W/DAM, Gamma, USA) and a syringe pump (NE-300, New Era Pump Systems Inc., USA). A homemade spray device (a 5 mL plastic syringe and a metal needle of 21 gauge) was adopted to give a constant flow rate. The distance between needle tip and collector (a grounded collecting plate covered by aluminium foil) was varied from 11–15 cm. The electrospinning was carried out at a temperature of 25 °C and a humidity of around 35–40%. The obtained fiber mat was dried in a vacuum at room temperature for 12 h before analysis.
2.6. Morphology of electrospun nanofibers
The morphology of nanofibers was observed with scanning electron microscopy (SEM, Hitachi, Japan) at accelerating voltage of 10 kV. The sample on aluminum foil was coated with Pt using a sputter coater (K550, Emitech, UK) under vacuum before observation. The diameter distribution of fibers was analyzed and calculated by SEM.
2.7. In vitro release test
The in vitro release of SCT from fiber mat containing uncoated or pectin-coated liposomes was carried out under different simulated digestive fluids using a dissolution rate test apparatus (RCZ-8B, TiandaTianfa Co., Ltd., Tianjin, China) (100 rpm, 37 °C) according to the USP23 method. Simulated gastric fluid containing 10 mg mL−1 pepsin (SGF, pH 1.2) was prepared by 0.1 mol L−1 HCl, simulated intestinal fluid containing 10 mg mL−1 trypsin (SIF, pH 6.8) and simulated colonic fluid containing pectinase (SCF, pH 7.4) were prepared by phosphate buffer solution.21 The release percentage (Q%) of SCT from the fiber mat was calculated by using the following equation:
(where Mt is the amount of cumulative release of SCT at time t and M0 is the total amount of SCT loaded in fiber mat).
The release behavior of SCT was investigated by a continuous simulation GIT. First, the fiber mat was immersed in SGF for 2 h, then transferred to SIF for 4 h, and then put into SCF for 16 h, and 1 mL of medium was sampled and substituted for the same volume of fresh medium. The release amount of SCT was determined and calculated according to the above method.
2.8. Release mechanism study
The release data of SCT in SGF, SIF and SCF were individually analyzed using First-order, Higuchi, Weibull and Ritger–Peppas model (eqn (1), (2), (3) and (4)) to describe the release mechanism:| | |
First-order: Q = 1 − exp(kt)
| (1) |
| | |
Weibull: Q = 1 − exp(−atb)
| (3) |
| | |
Ritger–Peppas: Q = ktn
| (4) |
where Q is the accumulated fraction of SCT release in time t; k is the release rate constant; a is the scale parameter; b is the shape parameter; n is the release exponent.
2.9. Bioactivity examination of released SCT
The bioactivity of released SCT was determined by SCT ELISA kit.22 The kit includes a set of SCT standards and the calibration curve was prepared by the method supplied by the kit. Firstly, the SCT sample was added into the 48-well microtiter plate (Costar 3590 High Binding, Corning, New York, USA) with 50 μL per well, and 100 μL of HRP-conjugate reagent was added to each well and then incubated at 37 °C for 1 h. The plate was washed before adding chromogen solution A (50 μL per well), chromogen solution B (50 μL per well) and incubating at 37 °C for 15 min. The reaction was stopped by the addition of stop solution (50 μL per well). The optical density (OD) was detected at 450 nm by using a Cytation™ 5 imaging reader (BioTek Instruments Co. Ltd. Vermont, USA).
2.10. Statistics
The statistical analysis was performed using one way analysis of variance (ANOVA). Differences were considered statistically significant at p < 0.05. All data were carried out in three parallel experiments and presented as the mean ± standard deviation.
3 Results and discussion
3.1. Preparation and characterization of liposomes
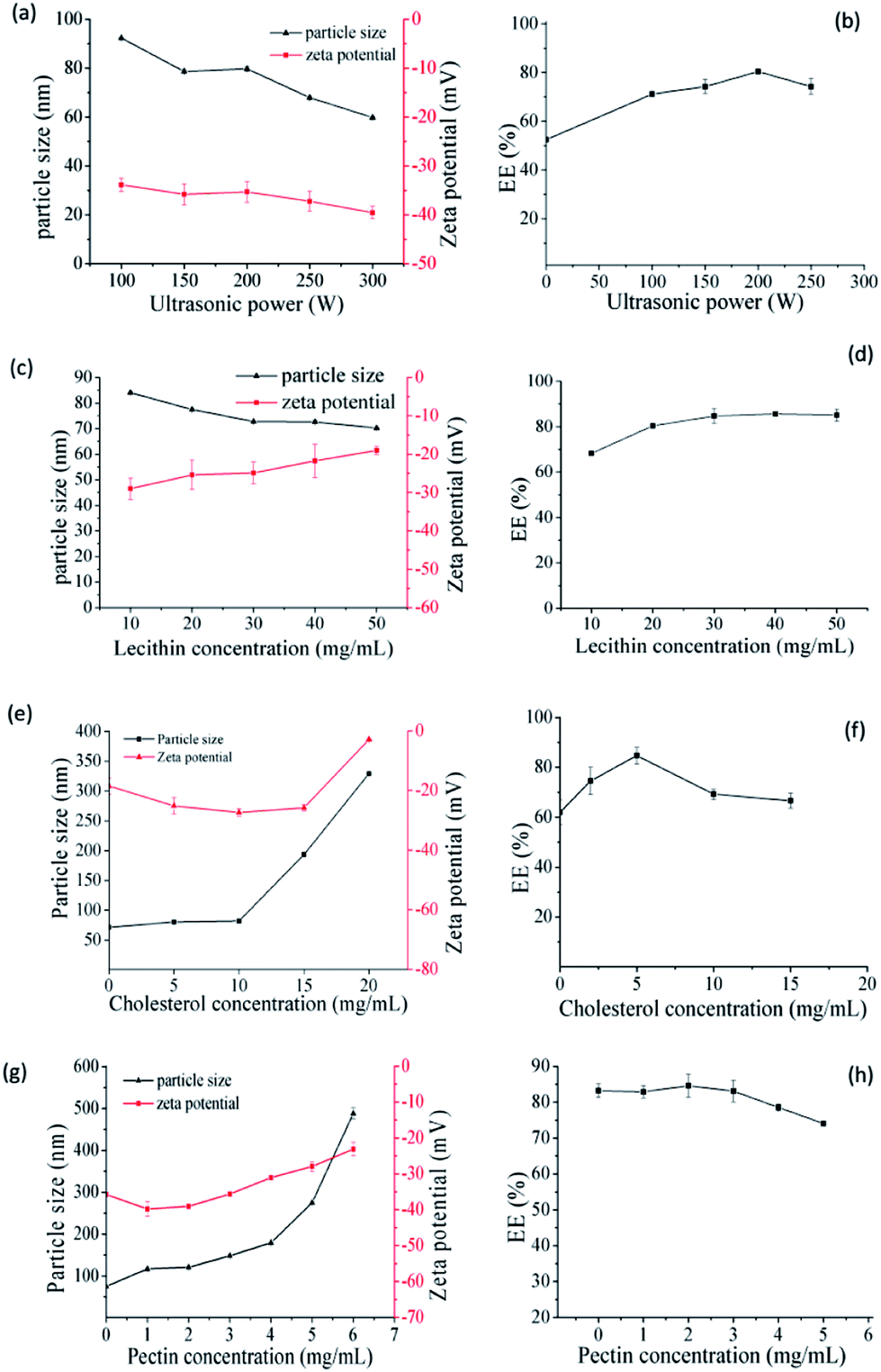
Firstly, the SCT liposomes were prepared respectively and some parameters were investigated to obtain the liposomes with low size and high EE (Fig. 1). As shown in Fig. 1a, ultrasound had obvious effect on decreasing the particle size of liposomes and the trend of particle size and zeta potential decreased with the increase of ultrasonic power. It was due to liposomes could form a more stable double layer structure thus enhancing their stability under ultrasonic conditions.23,24 While for EE, it increased with the increase of ultrasonic power and reached the maximum at 200 W, while further increase in the ultrasonic power reduced its value. The possible reason was that more SCT were absorbed into the film with the increase of ultrasonic power, however, further increase in the ultrasonic power after saturation would lead to part leakage of SCT from liposome, thereby reducing the EE of liposomes.25 As can be seen in Fig. 1c and d, the particle size declined while the zeta potential increased with the increase of lecithin concentration. It was because the mass ratio of cholesterol to lecithin decreased with the increase of lecithin concentration, which led to the instability of double layer structure of liposomes.26,27 For the EE of SCT, it increased gradually with the increase of lecithin concentration to 30 mg mL−1 and there was no obvious change with further increase of lecithin concentration. It was because that increasing lecithin concentration was beneficial to improve the EE of liposomes. However, when the SCT was completely absorbed into the lipid film to form liposomes, further increase in the lecithin concentration could not enhance its EE anymore.28 The suitable lecithin concentration was 30 mg mL−1. As shown in Fig. 1e and f, the particle size increased with increasing cholesterol concentration. While for zeta potential, it decreased with the increase of cholesterol concentration and reached the minimum at 10 mg mL−1. As to EE, it increased with the increase of cholesterol concentration and reached the maximum at 5 mg mL−1, further increase in the cholesterol concentration above 5 mg mL−1 decreased its value. Generally, cholesterol can be embedded into the phospholipid molecule thus influencing the fluidity of lipid film. Addition of appropriate amount of cholesterol could improve the stability of the liposome. However, excessive cholesterols would destroy the balance between the hydrophobic and hydrophilic molecules of the lipid film thus resulting in unstable liposomes.29,30 The results showed that the optimal conditions for preparation of SCT liposomes were as follows: lecithin concentration = 30 mg mL−1, cholesterol concentration = 5 mg mL−1 and ultrasonic power = 200 W.
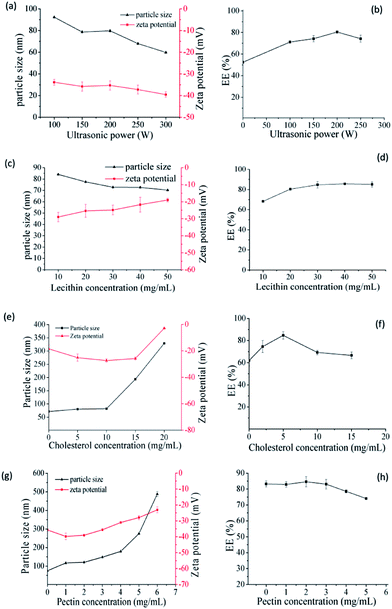 |
| | Fig. 1 Effect of ultrasonic power on the size, zeta potential (a) and EE (b) of SCT liposomes (preparation conditions: lecithin concentration = 20 mg mL−1, cholesterol concentration = 5 mg mL−1); effect of lecithin on the size, zeta potential (c) and EE (d) of SCT liposomes (preparation conditions: ultrasonic power = 200 W, cholesterol concentration = 5 mg mL−1); effect of cholesterol on the size, zeta potential (e) and EE (f) of SCT liposomes (preparation conditions: ultrasonic power = 200 W, lecithin concentration = 30 mg mL−1); effect of pectin on the size, zeta potential (g) and EE (h) of pectin-coated SCT liposomes (preparation conditions: ultrasonic = 200 W, lecithin concentration = 30 mg mL−1, cholesterol concentration = 5 mg mL−1). | |
Since pectin is easily degraded by microbial enzymes in the colon and is often selected as a material to achieve the drug's colon targeting release, it was chosen to be coated on the above SCT liposomes. The SCT liposome solution prepared under the optimal conditions was added into different concentration of pectin solution with volumetric ratio being 1:1, thereby obtaining pectin-coated SCT liposomes. Fig. 1g showed that the particle size of pectin-coated SCT liposomes increased gradually with the increase of pectin concentration. As for zeta potential, it declined with the increase of pectin concentration up to 2 mg mL−1 due to the influence of negatively charged hydroxyl ends of pectin molecules.31 But it went up gradually when the pectin concentration was above 2 mg mL−1. The increase of particle size under low pectin concentration may be attributed to the adsorption of pectin molecule onto the surface of liposomes thus increasing their thickness.32 This was in consistent with the result reported by Thirawong et al.10 However, further increase in the pectin concentration caused the accumulation and precipitation of partial liposomes thus resulting in the increase of zeta potential and particle size. As can be seen in Fig. 1h, there was no obvious change in the EE when the pectin concentration was below 3 mg mL−1. However, the EE declined with further increase in the pectin concentration above 3 mg mL−1 due to the instability of pectin-coated SCT liposomes. The suitable pectin concentration was 2 mg mL−1.
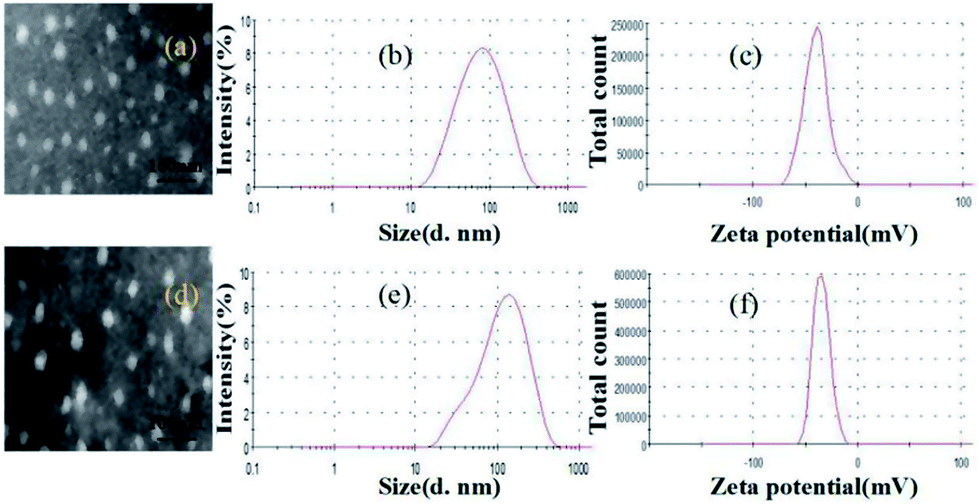
Fig. 2 showed the characteristics of uncoated and pectin-coated SCT liposomes prepared under the optimized conditions. TEM images indicated that both liposomes were spherical in shape, and their average diameters were around 20 nm and 40 nm, respectively (Fig. 2a and d). It revealed that pectin was successfully coated on the surface of liposomes. The results of dynamic light scattering analysis showed that both liposomes had typical size distribution profiles with a mean diameter of 74.98 nm in a narrow size distribution (PDI = 0.27) for uncoated SCT liposomes (Fig. 2b) and 120.56 nm in a narrow size distribution (PDI = 0.29) for pectin-coated SCT liposomes (Fig. 2e). Generally, a zeta potential of ±30.00 mV is required as a minimum for a physical stable nanosuspension stabilized by electrostatic repulsion.33 Fig. 2c and f showed the zeta potential of the uncoated SCT liposomes and pectin-coated SCT liposomes were about −35.75 mV and −39.05 mV, respectively.
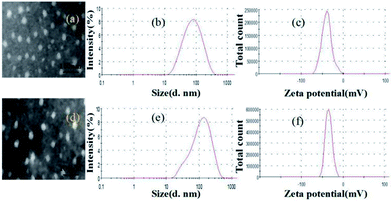 |
| | Fig. 2 Characteristics of uncoated SCT liposomes: (a) TEM image; (b) size distribution; (c) zeta potential distribution; characteristics of pectin-coated SCT liposomes: (d) TEM image; (e) size distribution; (f) zeta potential distribution. | |
3.2. Electrospinning and morphology of nanofibers
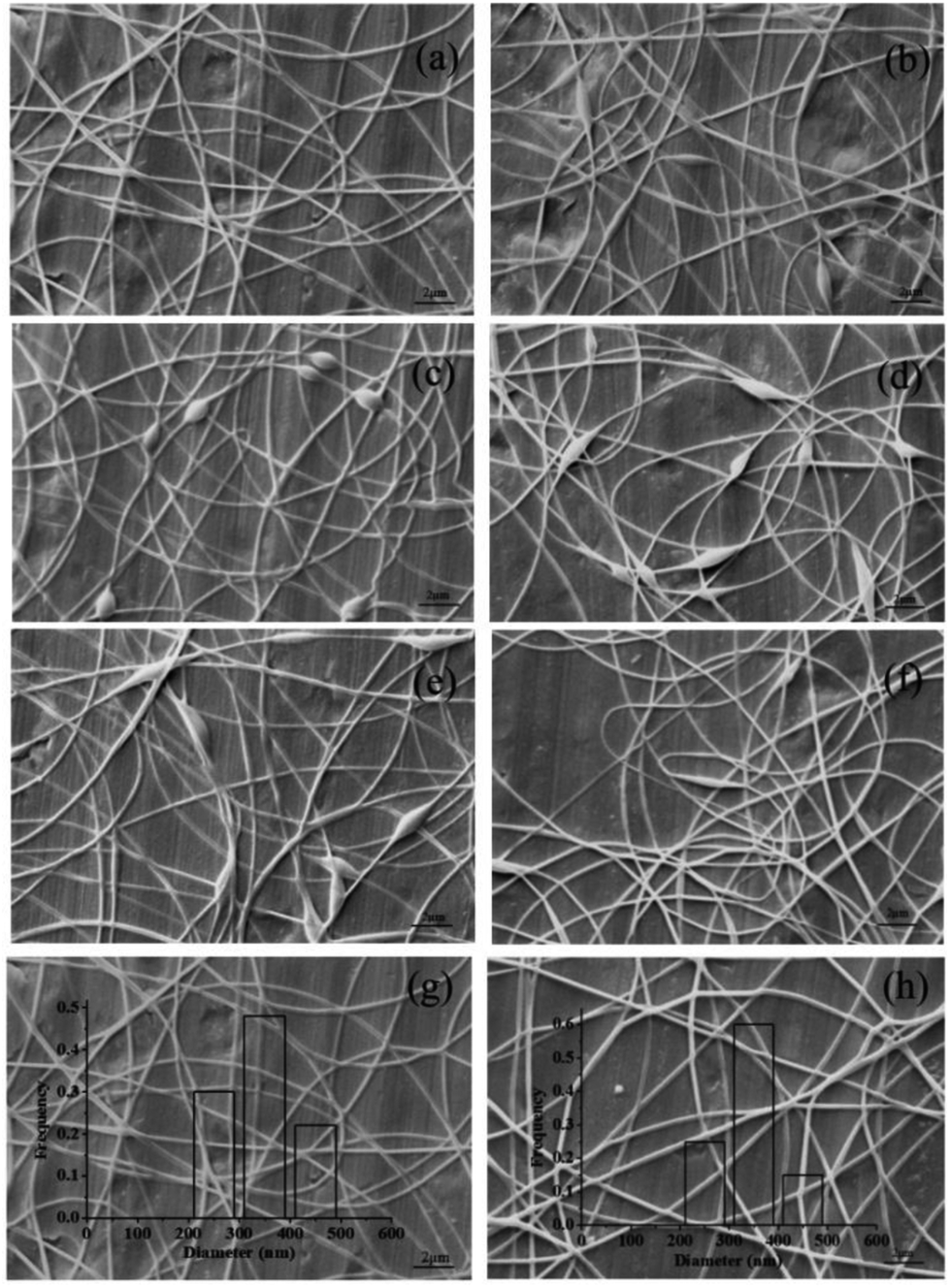
3.2.1 Effect of spinning solution composition on nanofibers. The obtained pectin-coated SCT liposomes were added into a mixed solution of SA and PVA for electrospinning. SA could protect SCT from gastric acid attack, however, the spinnability of which is poor and it was necessary to add appropriate PVA into SA solution to obtain nanofibers with good morphology.34 Fig. 3 showed the effect of spinning solution composition on the morphology of nanofibers. As shown in the SEM images of Fig. 3a–c, with the increase of PVA concentration from 60–100 g L−1, the formed nanofibers had less beads and the smooth nanofibers were obtained when PVA concentration was 100 g L−1 (Fig. 3c). It was attributed to the realization of the balance between the viscosity and conductivity in the higher viscosity of PVA solution.35 Fig. 3d and e indicated that it was not helpful for generation of smooth nanofibers at lower or higher SA concentration and the suitable SA concentration was regarded as 20 g L−1 (Fig. 3c). Similarly, lower or higher mass ratio of PVA to SA in the mixed solution was not beneficial to form the nanofibers with good morphology and more beaded fibers were presented in the mat (Fig. 3g and f). Therefore, the suitable composition of electrospinning solution was 100 g L−1 PVA:20 g L−1 SA = 8:2 (w/w). SEM image indicated that the resulting nanofibers were uniform and smooth with an average diameter about 360 nm (Fig. 3h). FTIR analysis showed that there was a certain interaction between liposomes, SA and PVA (Fig. S.1†).
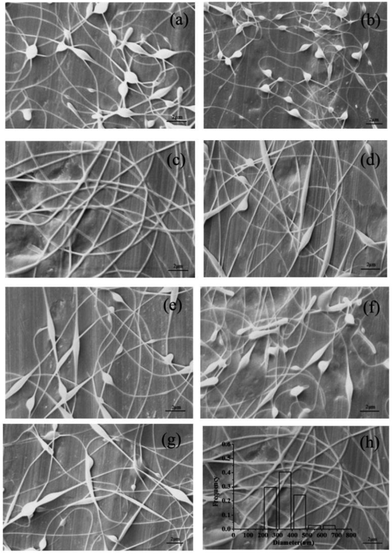 |
| | Fig. 3 SEM images of the electrospun nanofibers containing pectin-coated SCT liposomes under the following conditions: (a) 60 g L−1 PVA:20 g L−1 SA = 8:2 (w/w); (b) 80 g L−1 PVA:20 g L−1 SA = 8:2 (w/w); (c) 100 g L−1 PVA:20 g L−1 SA = 8:2 (w/w); (d) 100 g L−1 PVA:10 g L−1 SA = 8:2 (w/w); (e) 100 g L−1 PVA:30 g L−1 SA = 8:2 (w/w); (f) 100 g L−1 PVA:20 g L−1 SA = 7:3 (w/w); (g) 100 g L−1 PVA:20 g L−1 SA = 9:1 (w/w); and characterization of the optimized nanofibers: (h) SEM image. | |
3.2.2 Effect of electrospinning parameters on nanofibers. Electrospinning parameters also have important influence on the morphology of nanofibers. Hence, the impacts of electrospinning parameters, including distance, voltage and flow rate, were investigated and the results were shown in Fig. 4. The SEM images indicated that some beaded fibers in the mat appeared with the decrease of distance between the needle tip and the collector (Fig. 4a–c). This was because the decrease of the distance could increase the electric field intensity and shorten the flight time of the jet thus resulting in insufficient stretch and bead formation.36 The flow rate determined the velocity of jet flow and a suitable flow rate for obtaining uniform and smooth fibers was found to be 0.3 mL h−1 (Fig. 4a). Higher flow rate was not conducive to solvent evaporation and easy to form fibers with more beads (Fig. 4d). Lower flow rate was beneficial to solvent evaporation, but it will reduce spinning efficiency (data not shown).16 Fig. 4e showed that when the voltage was reduced, the weaker electric force could not overcome the surface tension, resulting in the formation of beaded nanofibers. However, higher voltage led to shorter flight time of the jet thus generating more beads in the fibers (Fig. 4f). The optimal electrospinning process parameters were: distance = 15 cm, voltage = 16 kV and flow rate = 0.3 mL h−1. SEM image showed that the nanfibers prepared under the optimal conditions were uniform and smooth with an average diameter about 350 nm (Fig. 4g). For comparison, the nanofibers containing uncoated SCT liposomes were also prepared. SEM image indicated that the resulting nanofibers had smooth surface and the diameter distribution was in the range of 210–470 nm (Fig. 4h). TGA analysis indicated that the obtained fiber mat containing pectin-coated SCT liposomes or uncoated SCT liposomes had good thermal stability (Fig. S.2†).
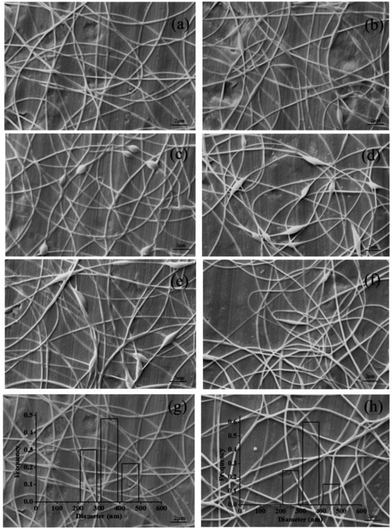 |
| | Fig. 4 SEM images of the electrospun nanofibers containing pectin coated liposomes under the following conditions: (a) 15 cm, 16 kV, 0.3 mL h−1; (b) 13 cm, 16 kV, 0.3 mL h−1; (c) 11 cm, 16 kV, 0.3 mL h−1; (d) 15 cm, 16 kV, 0.5 mL h−1; (e) 15 cm, 13 kV, 0.3 mL h−1; (f) 15 cm, 19 kV, 0.3 mL h−1; and SEM images of the optimized nanofibers containing pectin-coated SCT liposomes (g) and uncoated SCT liposomes (h). | |
3.3. In vitro release behaviour
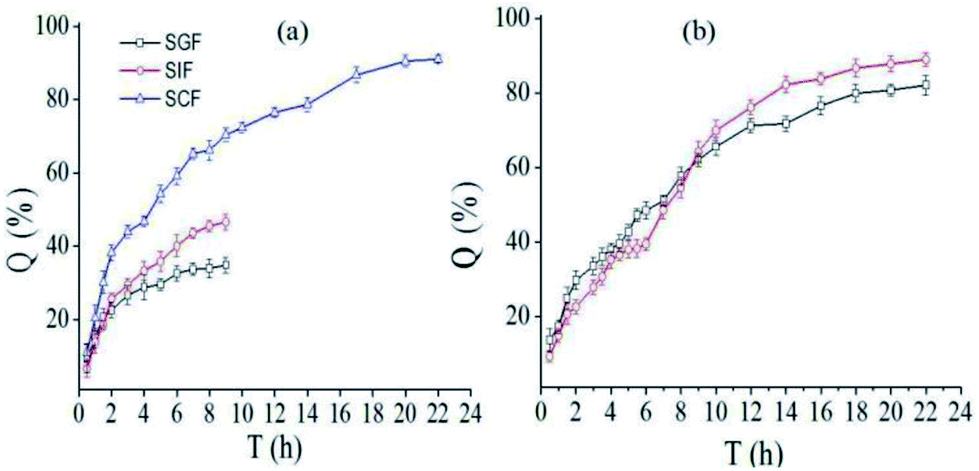
Generally, food stayed in stomach, intestinal and colon for about 2, 4 and 10 h, respectively. Fig. 5a showed the release profile of SCT from nanofibers containing pectin-coated SCT liposomes in different simulated medium. It was found that around 34.9% SCT was released in SGF and approximately 46.6% SCT was released in SIF during 9 h. While around 91.1% SCT was released in SCF during 22 h. The release profile of SCT in continuous simulated GIT (SGF 2 h, SIF 4 h and SCF 16 h) was showed in Fig. 5b. The amount of released SCT from the fiber mat containing pectin-coated liposomes were 22.6% in SGF, 16.8% in SIF, and 49.6% in SCF, respectively. The corresponding values of released SCT from the fiber mat containing uncoated liposomes were 29.7% in SGF, 19.8% in SIF, and 33.6% in SCF, respectively. The results demonstrated that the fiber mat containing pectin-coated liposomes had better stability and colon-specific property in GIF.
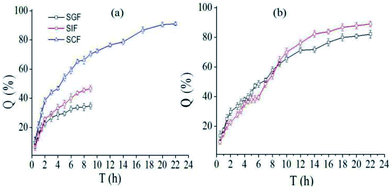 |
| | Fig. 5 In vitro release profile of SCT from electrospun fiber mat: (a) the release profile of SCT from fiber mat containing pectin-coated SCT liposomes in different simulated medium; (b) total release of SCT from fiber mat containing uncoated (□) or pectin-coated SCT liposomes (○) in continuous simulated GIT (SGF: 2 h, SIF: 4 h, SGF: 16 h). | |
3.4. Release mechanism
To design matrix optimally delivered to a target position, it is necessary to clarify the release mechanism of bioactive compound from the matrix in the GIT. In this work, four release models were used to clarify the release mechanism of SCT from fiber mat containing pectin-coated SCT liposomes (Fig. 5a), and the release mechanism is concluded by the kinetic parameters that characterize the models. The fitted curves were showed in Fig. 6 and the most suitable model was presented by the value of correlation coefficient (R2) closer to 1. In SGF, Weibull model (ln(ln[1/(1 − Q)]) = 0.4859lnt − 1.8104, a = 1.8104, b = 0.4859, R2 = 0.94852) was the best to describe the release behavior of SCT. While in SIF, Higuchi models (Q = 0.17148t1/2 − 0.02276, R2 = 0.9765) was more suitable to describe the release profile. The results indicated that the release of SCT in SGF and SIF followed the Fickian diffusion mechanism.37 In SCF, higher values of R2 were obtained for Weibull model (ln(ln[1/(1 − Q)]) = 0.7541lnt − 1.4515, a = 1.4515, b = 0.7541, R2 = 0.98886), the value of b was in the intermediate values (0.75–1), suggesting that the release of SCT followed the case II transport with axial and radial release from cylinders, in which the swelling and erosion of polymer matrix was dominant.38–40
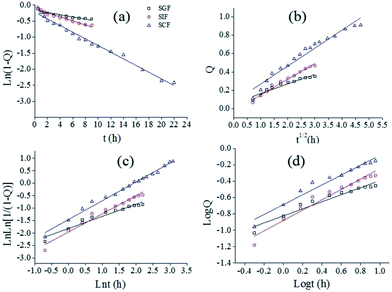 |
| | Fig. 6 Fitting curves of released SCT with different model in different medium (a) first-order model (b) Higuchi model (c) Weibull model (d) Ritger–Peppas model. | |
3.5. Bioactivity of released SCT
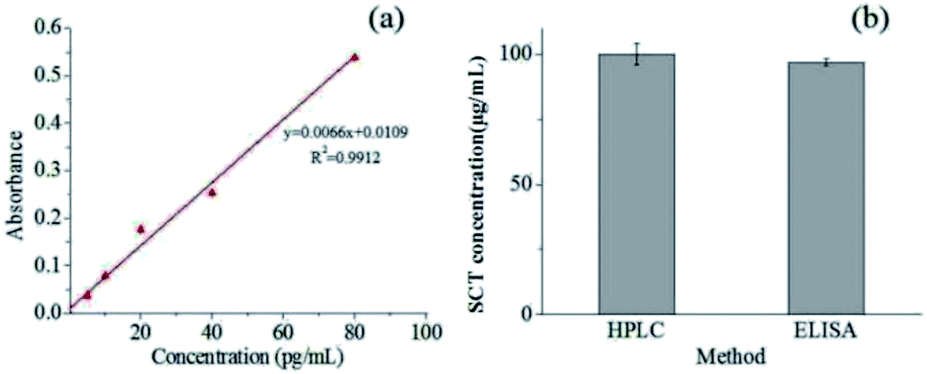
In order to determine the effects of the liposomes preparation and electrospinning process on the SCT bioactivity, SCT ELISA kit was applied to examine the bioactivity of the released SCT from the fiber mat containing pectin-coating liposomes. The calibration curve of SCT was showed in Fig. 7a and the regression equation of the calibration curve is y = 0.0066x + 0.0109 (where y is the absorbance and x is the concentration of SCT in pg mL−1, R2 = 0.9912, n = 6). The fiber mat containing pectin-coated SCT liposomes was immersed in SCF for 24 h, then 2 mL of medium was sampled for bioactivity analysis. Fig. 7b showed that the SCT concentration in the sample was 100.2 ± 4.0 μg mL−1 measured by HPLC, while the corresponding value was 97.0 ± 1.5 μg mL−1 detected by SCT ELISA kit. The results indicate that there is no significant change in the bioactivity of released SCT (p > 0.05).
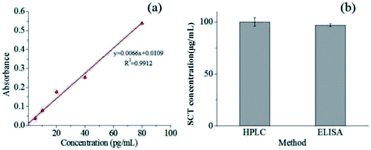 |
| | Fig. 7 (a) Calibration curve of SCT measured by SCT ELISA kit; (b) concentration of SCT obtained by HPLC and ELISA examination. | |
4 Conclusions
A novel colon-specific delivery system for bioactive SCT was successfully developed by electrospinning. Addition of pectin as coating layer can efficiently enhance the stability of liposomes in GIF, and the resulting electrospun fiber mat containing the pectin-coated liposomes had better colon-specific property. The nanofibers fabricated under the optimal electrospinning conditions were uniform and smooth with an average diameter about 350 nm. The release of SCT in colon followed the case II transport mechanism and the swelling and erosion of polymer matrix were dominant. The process of liposome preparation and electrospinning had little influence on the bioactivity of SCT. This study indicates that the electrospun colon-specific fiber mat is a promising delivery system for bioactive peptide.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 31671852), the Natural Science Foundation of Guangdong Province (No. 2017A030313148) and Special Funds for the Cultivation of Guangdong College Students' Scientific and Technological Innovation (No. pdjh2017b0036).
References
- M. Azria, D. H. Copp and J. M. Zanelli, Calcif. Tissue Int., 1995, 57, 405–408 CrossRef CAS PubMed.
- C. R. Schneyer, Md. Med. J., 1991, 40, 469–473 CAS.
- M. Torres-Lugo and N. A. Peppas, Biomaterials, 2000, 21, 1191–1196 CrossRef CAS PubMed.
- M. Ugwoke, N. Verbeke and R. Kinget, J. Pharm. Pharmacol., 2001, 53, 3–21 CrossRef CAS PubMed.
- W. A. Lee, R. D. Ennis, J. P. Longenecker and P. Bengtsson, Pharm. Res., 1994, 11, 747–750 CrossRef CAS.
- Z. L. Wan, J. Guo and X. Q. Yang, Food Funct., 2015, 6, 2876–2889 CAS.
- A. Maroni, L. Zema, M. D. Del Curto, A. Foppoli and A. Gazzaniga, Adv. Drug Delivery Rev., 2012, 64, 540–556 CrossRef CAS PubMed.
- F. J. Varum, E. L. Mcconnell, J. J. Sousa, F. Veiga and A. W. Basit, Crit. Rev. Ther. Drug Carrier Syst., 2008, 25, 207–258 CrossRef CAS PubMed.
- T. Cerchiara, A. Abruzzo, C. Parolin, B. Vitali, F. Bigucci, M. C. Gallucci, F. P. Nicoletta and B. Luppi, Carbohydr. Polym., 2016, 143, 124–130 CrossRef CAS PubMed.
- N. Thirawong, J. Thongborisute, H. Takeuchi and P. Sriamornsak, J. Controlled Release, 2008, 125, 236–245 CrossRef CAS PubMed.
- K. H. Song, S. J. Chung and C. K. Shim, J. Controlled Release, 2005, 106, 298–308 CrossRef CAS PubMed.
- Y. S. Youn, J. Y. Jung, S. H. Oh, S. D. Yoo and K. C. Lee, J. Controlled Release, 2006, 114, 334–342 CrossRef CAS PubMed.
- T. Lian and R. J. Y. Ho, J. Pharm. Sci., 2001, 90, 667–680 CrossRef CAS PubMed.
- L. Lin, Y. J. Dai and H. Y. Cui, Carbohydr. Polym., 2017, 178, 131–140 CrossRef CAS PubMed.
- H. Y. Cui, Y. Lu and L. Lin, Carbohydr. Polym., 2017, 177, 156–164 CrossRef CAS PubMed.
- P. Wen, D. H. Zhu, H. Wu, M. H. Zong, Y. R. Jing and S. Y. Han, Food Control, 2016, 59, 366–376 CrossRef CAS.
- H. Y. Cui, J. Wu, C. Z. Li and L. Lin, LWT–Food Sci. Technol., 2017, 81, 233–242 CrossRef CAS.
- H. Yang, P. Wen, K. Feng, M. H. Zong, W. Y. Lou and H. Wu, RSC Adv., 2017, 7, 14939–14946 RSC.
- H. Y. Cui, M. Bai, M. M. A. Rashed and L. Lin, Int. J. Food Microbiol., 2018, 266, 69–78 CrossRef CAS PubMed.
- H. Y. Cui, M. Bai and L. Lin, Carbohydr. Polym., 2018, 179, 360–369 CrossRef CAS PubMed.
- P. Wen, Y. Wen, X. Huang, M. H. Zong and H. Wu, J. Agric. Food Chem., 2017, 65, 4789–4796 Search PubMed.
- C. Tas, S. Mansoor, H. Kalluri, V. G. Zarnitsyn, S. Choi, A. K. Banga and M. R. Prausnitz, Int. J. Pharm., 2012, 423, 257–263 CrossRef CAS PubMed.
- C. Schwarz, W. Mehnert and R. H. Müller, Eur. J. Pharm. Biopharm., 2000, 50, 161–177 CrossRef.
- B. Heurtault, P. Saulnier, B. Pech, J. E. Proust and J. P. Benoit, Biomaterials, 2003, 24, 4283–4300 CrossRef CAS PubMed.
- A. S. Zidan and H. Aldawsari, Drug Des., Dev. Ther., 2015, 9, 3885–3898 CrossRef PubMed.
- W. W. Sulkowski, D. Pentak, K. Nowak and A. Sułkowska, J. Mol. Struct., 2005, 744–747, 737–747 CrossRef CAS.
- K. Aramaki, Y. Watanabe, J. Takahashi, Y. Tsuji, A. Ogata and Y. Konno, Colloids Surf., A, 2016, 506, 732–738 CrossRef CAS.
- N. E. Kateb, L. Cynober, J. C. Chaumeil and G. Dumortier, J. Microencapsulation, 2008, 25, 399–413 CrossRef PubMed.
- M. Alexander, L. A. Acero, Y. Fang and M. Corredig, LWT–Food Sci. Technol., 2012, 47, 427–436 CrossRef CAS.
- E. A. Smith, W. Wang and P. K. Dea, Chem. Phys. Lipids, 2012, 165, 151–159 CrossRef CAS PubMed.
- S. Nguyen, S. J. Alund, M. Hiorth, A. L. Kjøniksen and G. Smistad, Colloids Surf., B, 2011, 88, 664–673 CrossRef CAS PubMed.
- J. Zhang and S. Wang, Int. J. Pharm., 2009, 372, 66–75 CrossRef CAS PubMed.
- R. H. Müller, C. Jacobs and O. Kayser, Adv. Drug Delivery Rev., 2001, 47, 3–19 CrossRef.
- C. D. Saquing, C. Tang, B. Monian, C. A. Bonino, J. L. Manasco, E. Alsberg and S. A. Khan, Ind. Eng. Chem. Res., 2013, 52, 8692–8704 CrossRef CAS.
- K. Feng, P. Wen, H. Yang, N. Li, W. Y. Lou, M. H. Zong and H. Wu, RSC Adv., 2017, 7, 1572–1580 RSC.
- X. Zong, K. Kim, D. Fang, S. Ran, B. S. Hsiao and B. Chu, Polymer, 2002, 43, 4403–4412 CrossRef CAS.
- M. Fathi and J. Varshosaz, J. Funct. Foods, 2013, 5, 1382–1391 CrossRef CAS.
- P. C. Ferrari, G. F. Oliveira, F. C. S. Chibebe and R. C. Evangelista, Carbohydr. Polym., 2009, 78, 557–563 CrossRef CAS.
- X. Wang, D. G. Yu, X. Y. Li, S. W. Bligh and G. R. Williams, Int. J. Pharm., 2015, 490, 384–390 CrossRef CAS PubMed.
- K. Kosmidis, E. Rinaki, P. Argyrakis and P. Macheras, Int. J. Pharm., 2003, 254, 183–188 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00385h |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab