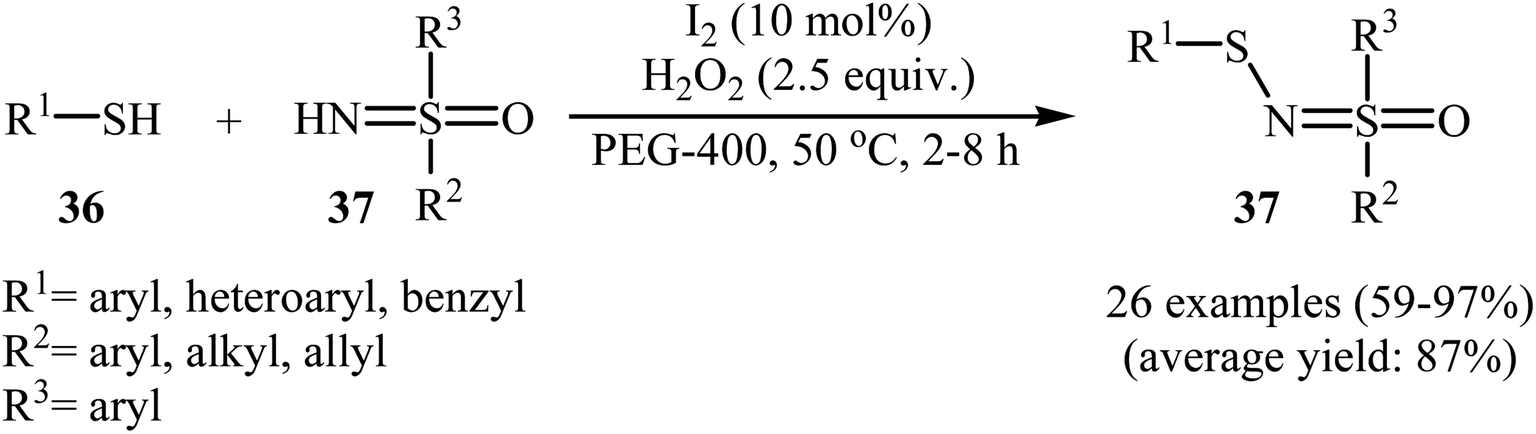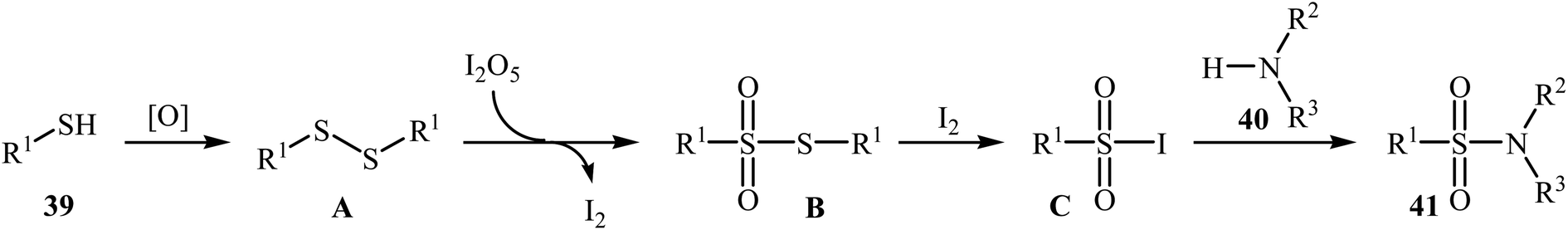DOI:
10.1039/C8RA00356D
(Review Article)
RSC Adv., 2018,
8, 18456-18469
Recent advances in sulfur–nitrogen bond formation via cross-dehydrogenative coupling reactions
Received
12th January 2018
, Accepted 12th April 2018
First published on 21st May 2018
Abstract
This focus-review surveys literature methods for the construction of sulfur–nitrogen bonds through cross-dehydrogenative coupling reactions between thiols and N–H compounds with a particular emphasis on the mechanistic aspects of the reactions. The literature has been surveyed until the end of 2017.
 Fatemeh Alsadat Hosseini Nasab | Fatemeh Alsadat Hosseini Nasab was born in Sirjan, Kerman, Iran, in 1980. She received her B.S. degree in Pure Chemistry from University of Shahid Bahonar, Kerman, Iran, and her M.S. degree in Organic Chemistry from Shahid Bahonar University, Kerman, Iran, in 2006 under the supervision of Dr M. Eslami. Now she is working at Hormozgan University of Bandar Abbas, as an Instructor. Her research interests include organic synthesis and new nanochemistry. |
 Leila Zare Fekri | Leila Zare Fekri was born in Rasht, Iran, in 1980. She received her B.Sc. degree in Pure Chemistry from Guilan University, Rasht, Iran, and her M.Sc. degree in Organic Chemistry from Guilan University, Rasht, Iran, in 2006 under the supervision of Prof. N. Mahmoodi. She received her Ph.D. degree in 2010 under the supervision N. Mahmoodi in Guilan University, Rasht, Iran. Now she is working at Payame Noor University of Guilan as Assistant Professor. Her research interests include organic synthesis, green chemistry, one-pot reaction, new nanochemistry and calculated chemistry. |
 Aazam Monfared | Aazam Monfared was born in Tehran, Iran, in 1965. She received her B.S. degree in Pure Chemistry from University of Shahid Beheshti, Tehran, Iran, and her M.S. degree in Organic Chemistry from Shahid Beheshti University, Tehran, Iran, in 1991 under the supervision of Prof. A. Rustaiyan. She received her Ph.D. degree in 1999 under the supervision of Prof. A. Rustaiyan in Shahid Beheshti University, Tehran, Iran. Now, she is working at Payame Noor University of Tehran as Associate Professor. Her research interests include organic synthesis, phytochemistry, drug synthesis, nano chemistry, methodologies and theoretical chemistry. |
 Akram Hosseinian | Akram Hosseinian was born in Ahar, Iran, in 1973. She received her B.S. degree in Pure Chemistry from University of Tehran, Iran, and her M.S. degree in Inorganic Chemistry from Tarbiat Modares University, Tehran, Iran, in 2000 under the supervision of Prof. A. R. Mahjoub. She completed her Ph.D. degree in 2007 under the supervision of Prof. A. R. Mahjoub. Now she is working at University of Tehran as Associate Professor. Her research interests include inorganic and organic synthesis, new methodologies in nano material synthesis. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in Pure Chemistry from University of Tabriz, Tabriz, Iran, and his M.S. degree in Organic Chemistry from Tehran University, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his Ph.D. degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as full Professor of Organic Chemistry. His research interests include theoretical organic chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
1. Introduction
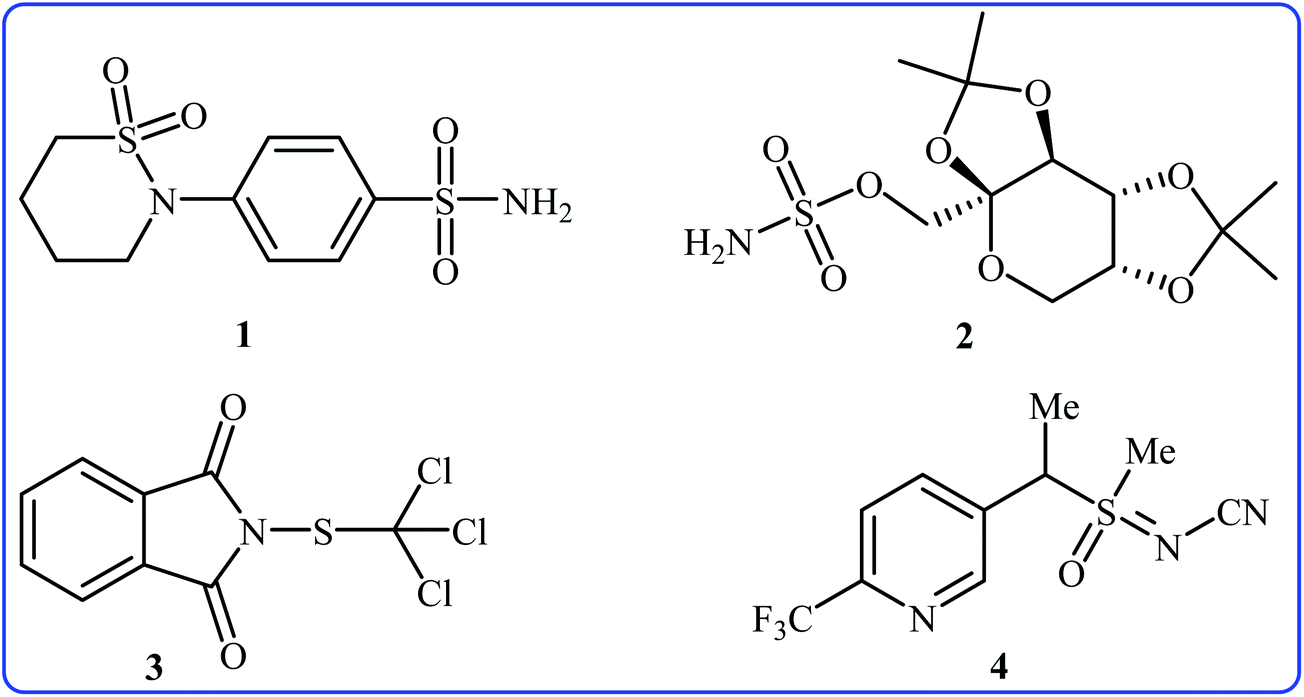
Needless to say, organosulfur compounds play a very important role in various fields such as organic synthesis, materials science, agriculture, and medicinal chemistry.1–4 Among them, compounds bearing sulfur–nitrogen bonds are important building blocks in a variety of biologically active substances. For instance, sultiame 1 (Fig. 1) with brand name Ospolot is a synthetic sulfa-based drug marketed worldwide for the treatment of epilepsy.5,6 The drug works by reducing the activity of carbonic anhydrase, an enzyme responsible for catalyzing the interconversion of carbon dioxide and water to carbonic acid by using a metal hydroxide nucleophilic mechanism.7 Topiramate 2 with trade name Topamax is a promising anticonvulsant medicine available in a number of countries worldwide. The drug is also used for migraine prevention in adults.8 Folpet 3 is a fungicide used for broad-spectrum control of fungal pathogens.9 Sulfoxaflor 4 is an insecticide with a sulfoximine motif, this compound is very active against a wide range of sap-feeding insect pests and widely used around the world.10 Thus, construction of biologically and synthetically important sulfur–nitrogen bonds is one of the essential transformations in organic synthesis. Conventional methods to achieve such linkages involve cross-coupling of S-halo compounds and N–H compounds in the presence of strong bases11 or metal-catalyzed cross-coupling reactions between N-halogenated compounds and thiols/disulfides.12 However, the limited availability of commercial starting materials, the formation of undesired by-products and/or harsh reaction conditions are the main drawbacks of these methods. Therefore, the search for novel straightforward and eco-friendly green approaches to the synthesis of sulfur–nitrogen bond-containing compounds remains a challenge.
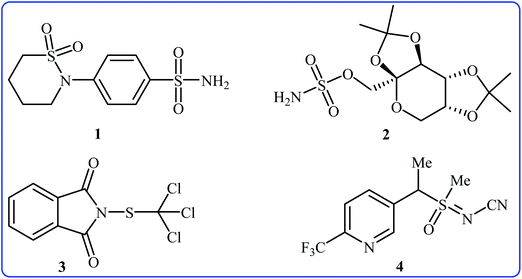 |
| | Fig. 1 Selected examples of biologically active sulfur–nitrogen bond-containing organosulfur compounds. | |
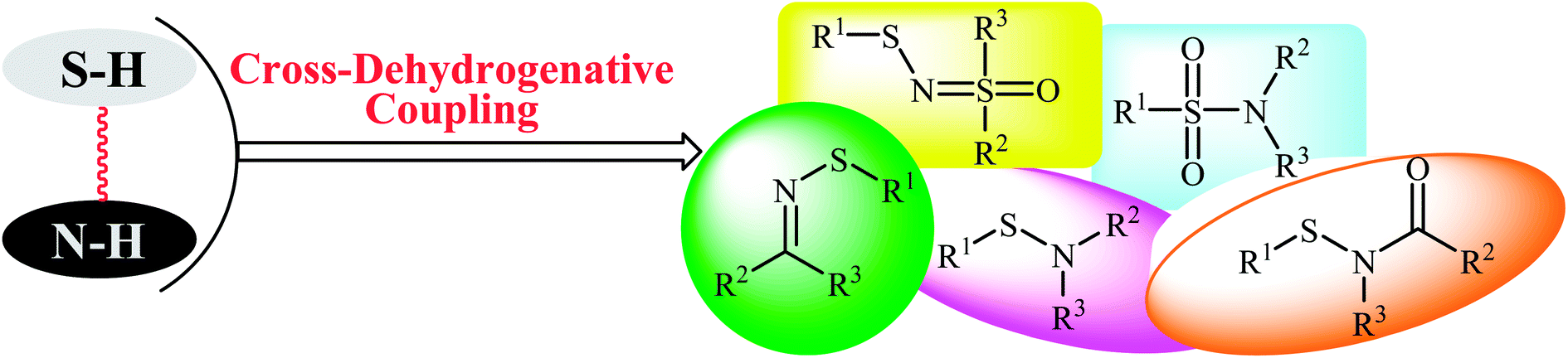
Metal-catalyzed and metal-free cross-coupling reactions have been shown to be one of the most powerful methods for the construction of C–C, C–X, and X–X′ bonds (X, X′ = heteroatom).13–15 Cross-dehydrogenative coupling (often called oxidative coupling) reactions are green and sustainable alternatives to traditional coupling procedures which rely on the use of two unmodified coupling partners.15,16 Indeed, in these reactions the relatively inactive C(X)–H bonds can be treated as a functional group, similar to the traditionally used C(X)–(pseudo)halide bonds. Along this line, recently, oxidative coupling of thiols and N–H compounds has gained a lot of attention because of the atom-economic and waste-minimization characters. The most obvious benefit of this route for construction of sulfur–nitrogen bond is that there is no need for preparation and isolation of halogenated reagents, thus improving pot, atom, and step economy. An overview of this emerging area is presented in this mini-review with a particular emphasis on the mechanistic aspects of the reactions. It is noted that the reactions were classified according to the type of products (Fig. 2). Literature has been surveyed until the end of 2017.
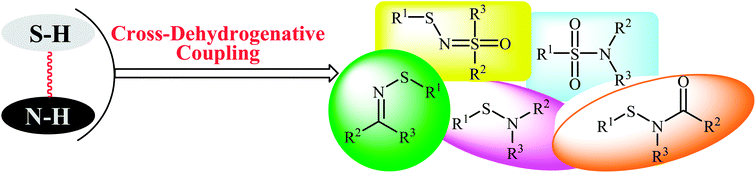 |
| | Fig. 2 Synthesis of a variety of sulfur–nitrogen bond-containing organosulfur compounds via cross-dehydrogenative coupling reactions. | |
2. Synthesis of sulfenamides
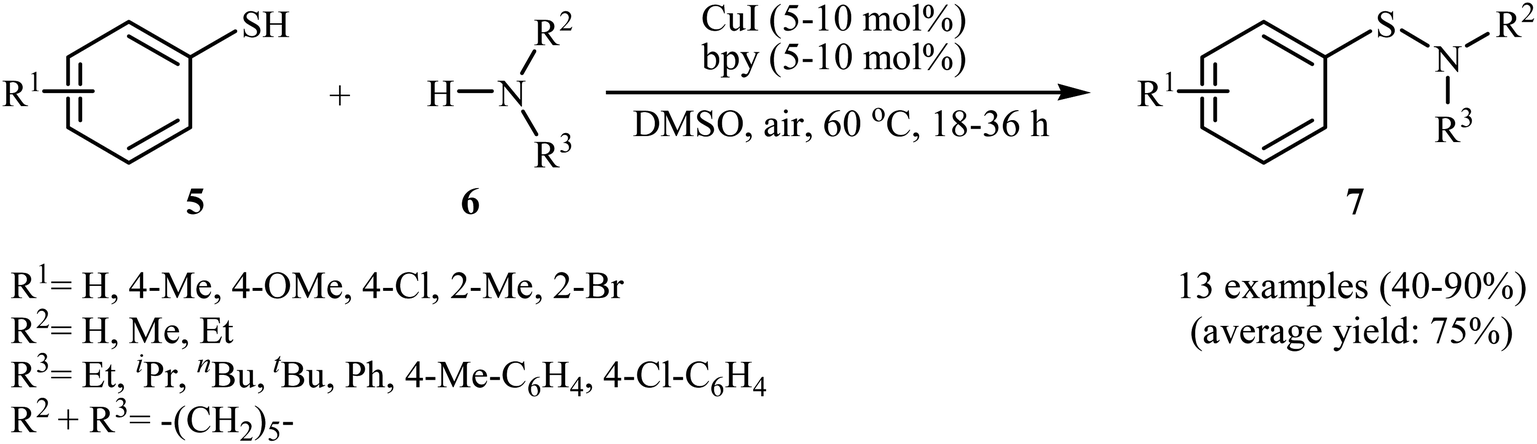
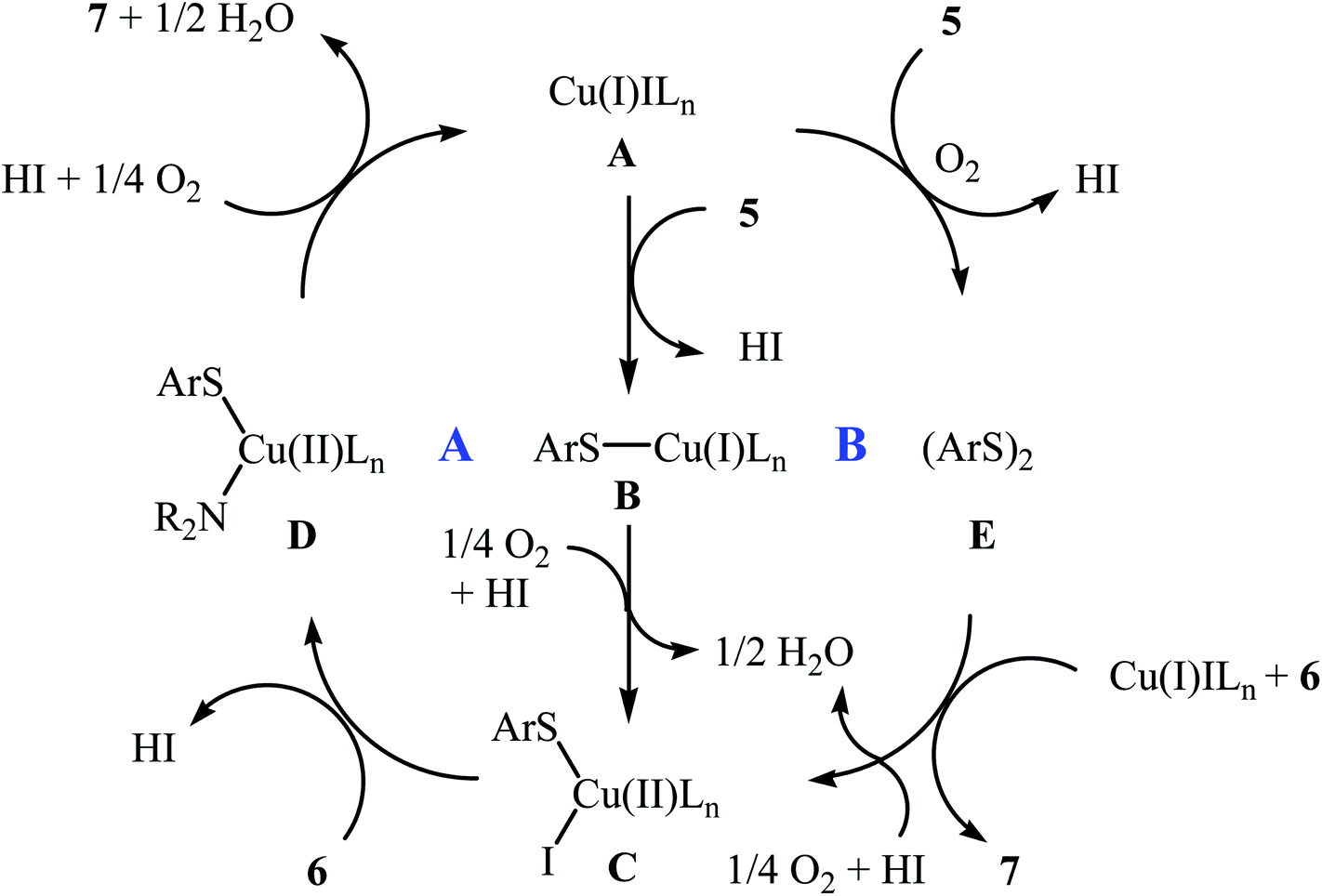
In 2010, N. Taniguchi reported the first example of Cu(I)-catalyzed cross-dehydrogenative coupling of aryl thiols 5 with amines 6 in the presence of Cu(I)/bpy (2,2′-bipyridine) combination as a catalytic system in air as the terminal oxidant, which allows for preparation of a broad range of sulfenamides 7 in moderate to excellent yields (Scheme 1).17a The reaction is noteworthy in that both primary and secondary amines are tolerated. The results demonstrated that aliphatic amines generally gave higher yields than arylamines. It should be mentioned that the electronic character of the substituents in aryl thiols had a strong effect on the facility of reaction. While electron-donating, mild electron-withdrawing and unsubstituted thiophenol gave the desired products in good yields, the strongly electron-withdrawing nitro-substituted thiophenol failed to afford the product. Interestingly, when the reaction was carried out under the oxygen atmosphere, the corresponding sulfonamides were obtained in moderate to high yields. Furthermore, the author observed that using PdCl2/CuI binary catalyst, the reaction of aryl thiols with primary amines in air selectively afforded sulfinamides derivatives. The author proposed mechanism for the formation of sulfenamides is shown in Scheme 2. A possible process for the formation of sulfenamides 7 should involve the formation of intermediate B via oxidative addition of starting aryl thiol 5 with copper catalyst A, followed by oxidation of this intermediate with air oxygen to give the intermediate C, which after reaction with amine 6 delivers the intermediate D. Finally, reductive elimination of intermediate D affords the desired product 7 (cycle A, Scheme 2). In another possibility, oxidative homocoupling of thiol 5 can happen to give the disulfide E, which after reaction with amine 6 affords sulfenamide 7 with the generation of intermediate C (cycle B, Scheme 2).17
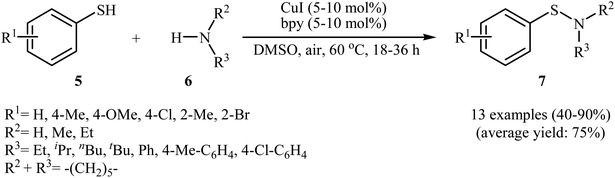 |
| | Scheme 1 Cu(I)-catalyzed S–N coupling of aryl thiols 5 with amines 6. | |
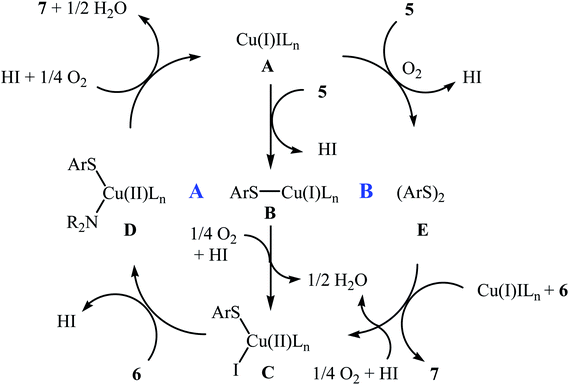 |
| | Scheme 2 Mechanistic proposal for the reaction in Scheme 1. | |
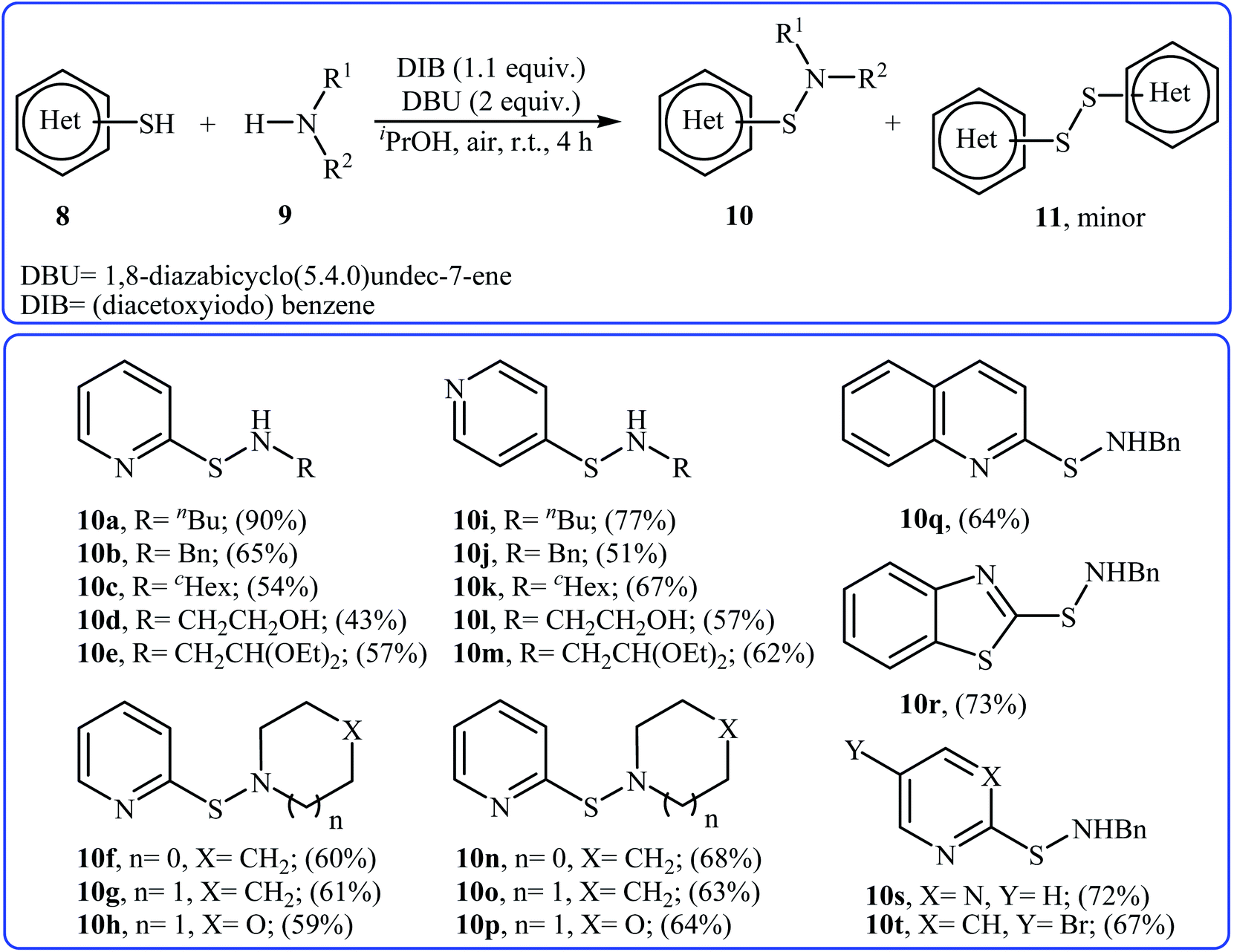
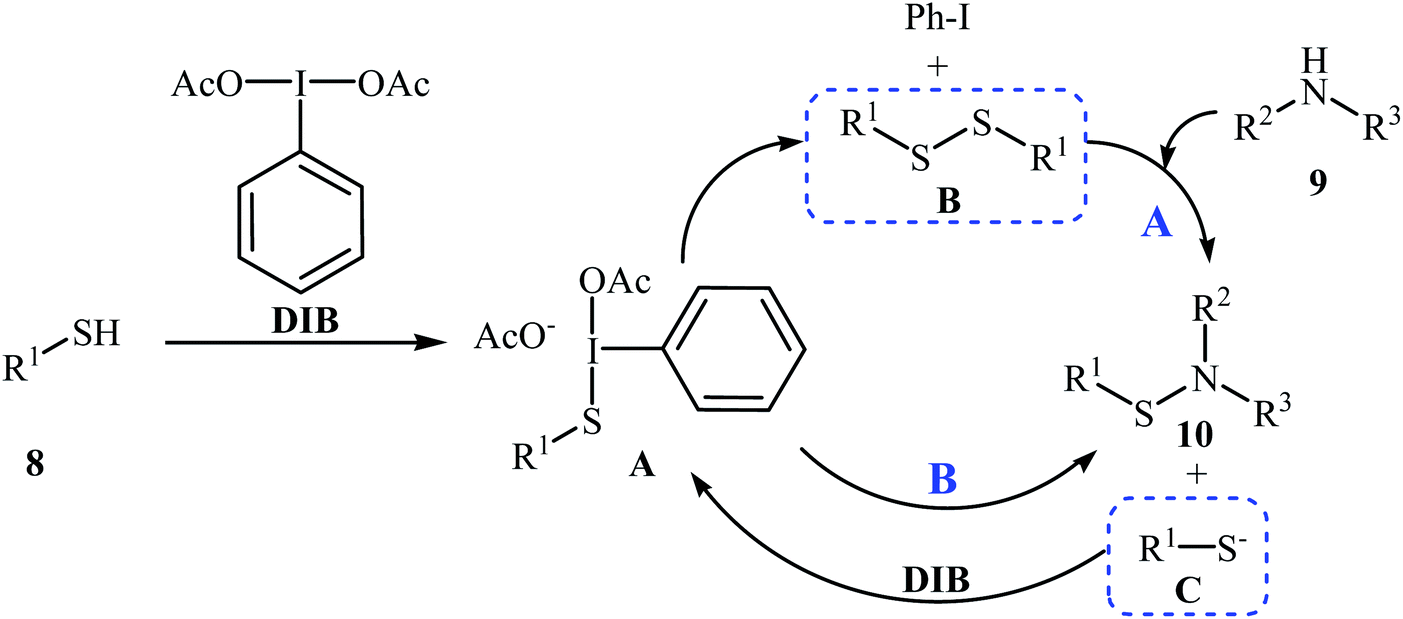
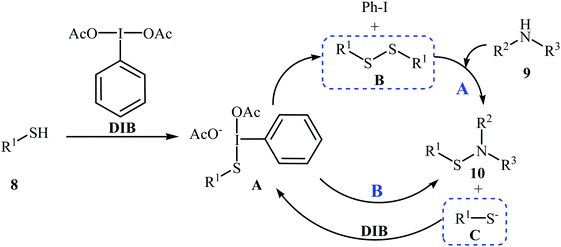
Four years later, the group of Wacharasindhu studied the possibility of synthesizing heterocyclic sulfenamides 10 through hypervalent iodine-catalyzed cross-dehydrogenative coupling of N-heterocyclic thiols 8 with amines 9.18 Thus, the careful analysis of the reaction optimization results showed that the optimum condition for this cross-coupling reaction was the addition of DBU (2 equiv.) and amine (2 equiv.) at 0 °C, to a solution of DIB (1.1 equiv.) and thiol (1.0 equiv.) in iPrOH. The optimized protocol tolerated a variety of N-heterocyclic thiols, such as 2-mercaptopyridine, 4-mercaptopyridine, 2-mercaptopyrimidine, 2-mercaptobenzothiazol, and 2-mercaptoquinoline, and provided heterocyclic sulfenamides 10 in moderate to high yields with a mixture of corresponding disulfides 11 (Scheme 3). In this study, the authors reported some limitations in their methodology, since considerable difficulties were found when they attempted to react the sterically hindered tert-butylamine and the less nucleophilic aniline. In these cases, they observed only disulfide products with no formation of sulfenamide product. The mechanism shown in Scheme 4 was proposed for the formation of sulfonamides 10.
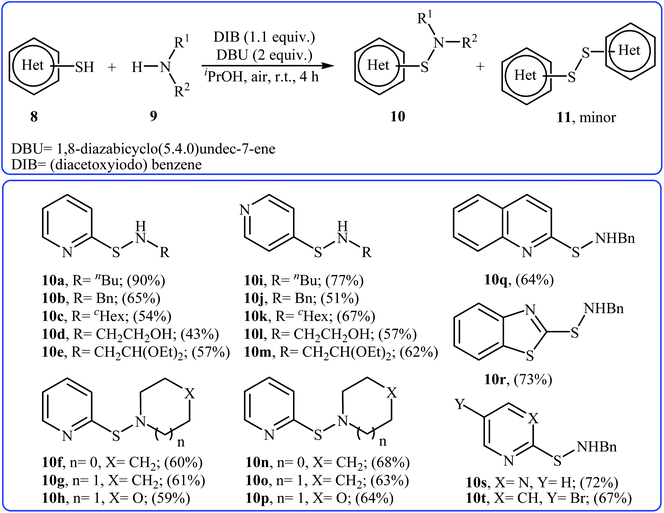 |
| | Scheme 3 Synthesis of sulfenamides 10 through hypervalent iodine-catalyzed cross-dehydrogenative coupling of N-heterocyclic thiols 8 with amines 9. | |
 |
| | Scheme 4 Mechanistic proposal for the formation of 10. | |
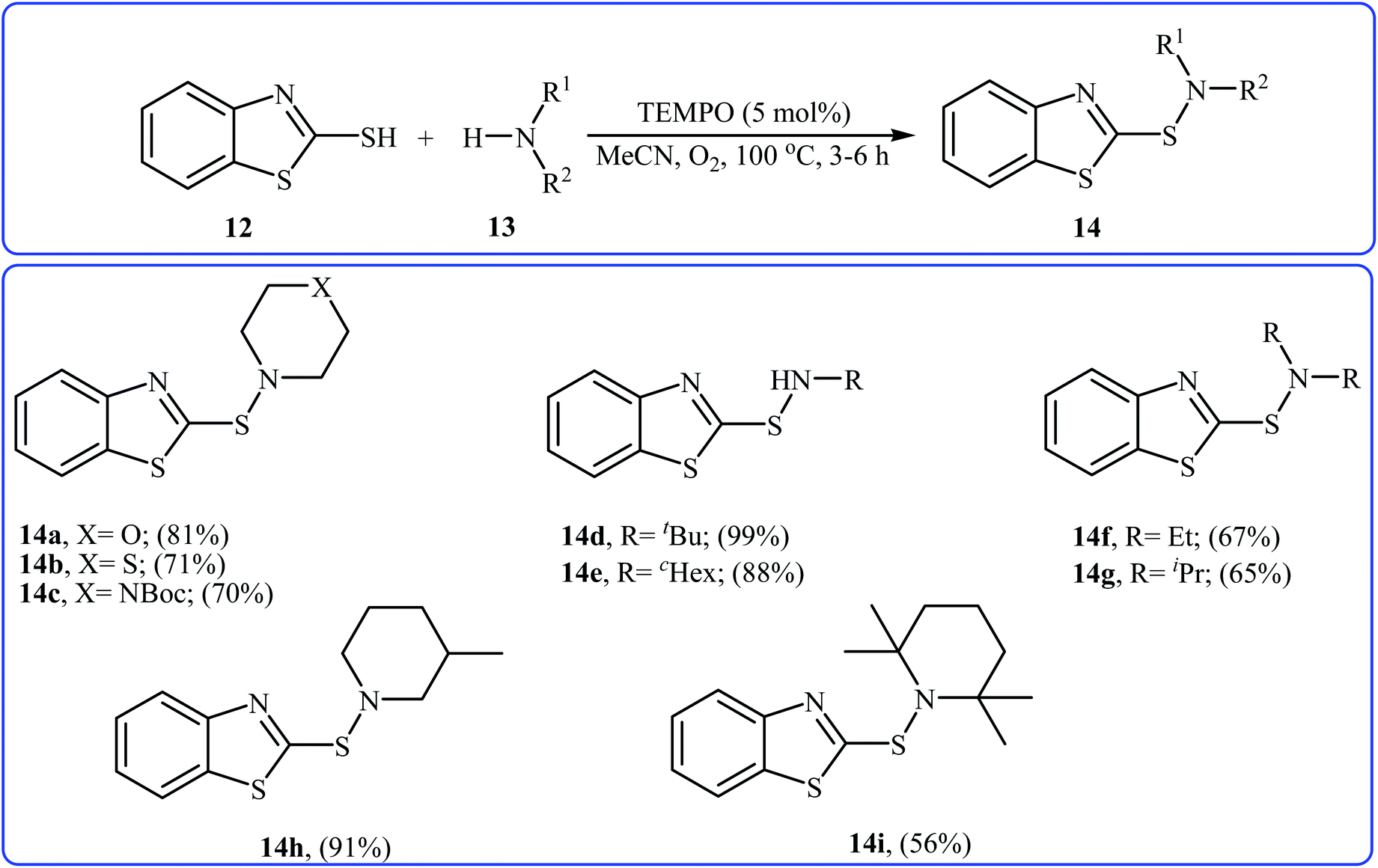
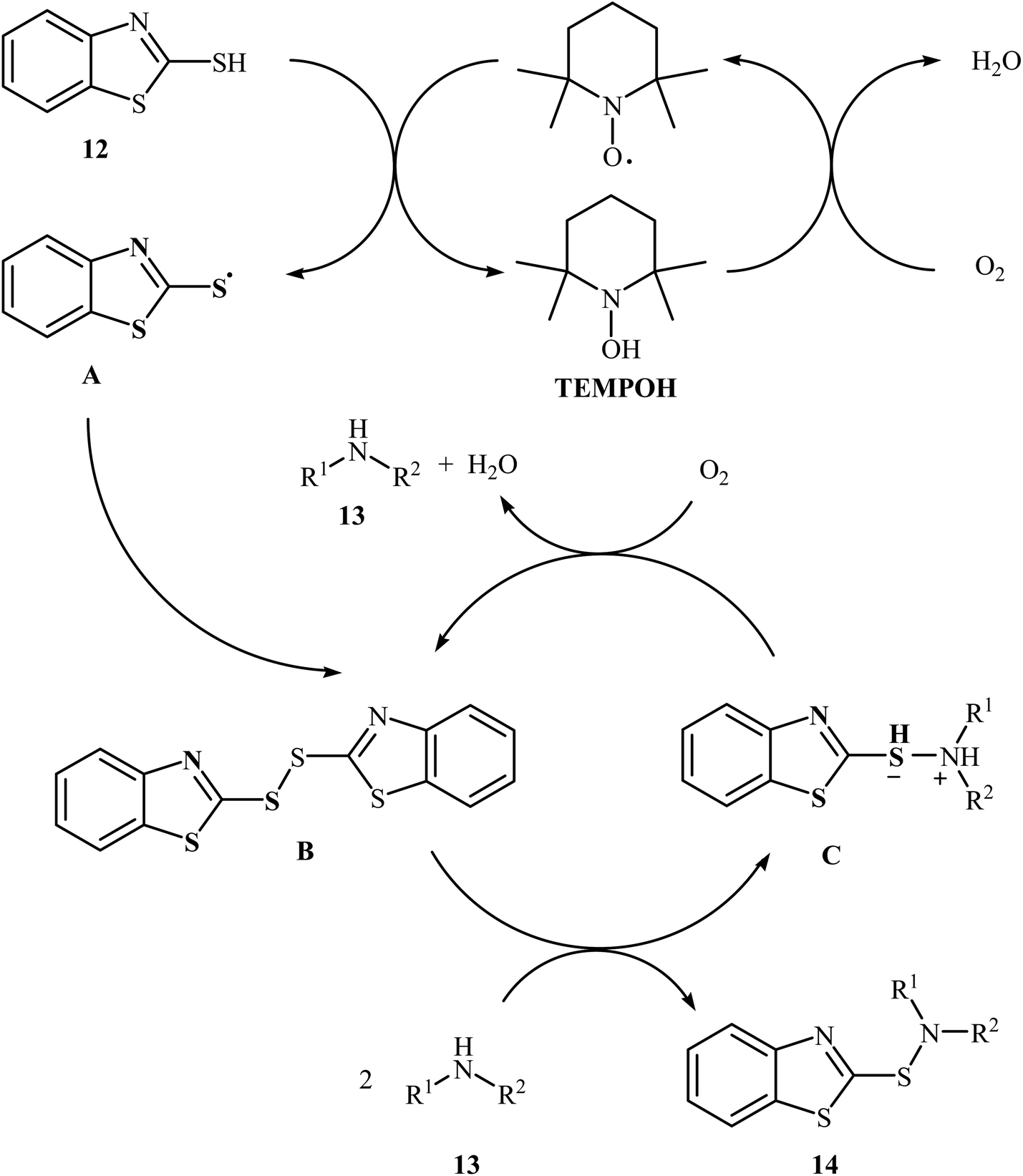
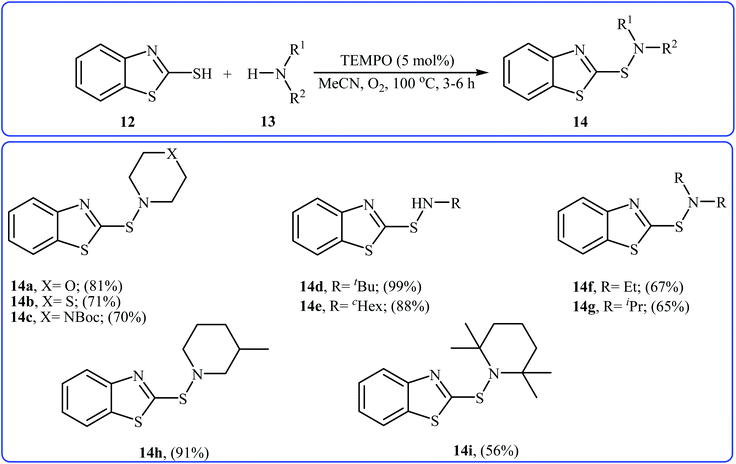
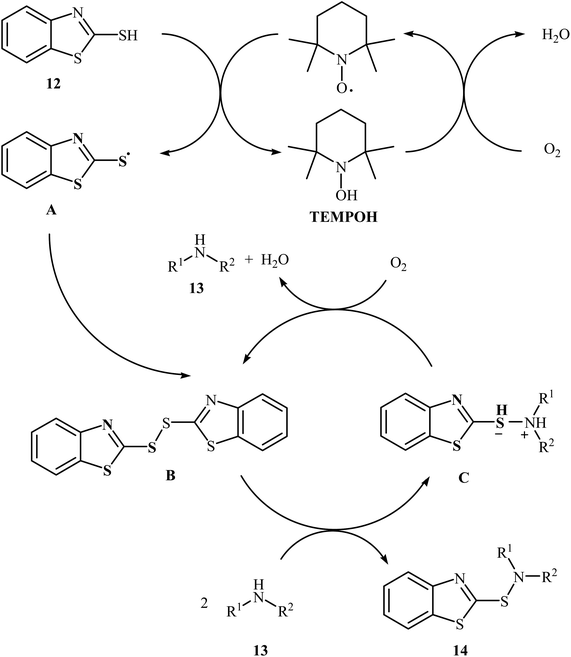
Very recently, Yuan's research team developed an efficient synthesis of 2-benzothiazole-sulfenamides 14 via direct aerobic oxidative amination of 2-mercaptobenzothiazole 12 under metal-free conditions.19 In this transformation, 2,2,6,6-(tetramethylpiperidin-1-yl)oxyl (TEMPO) was utilized as the catalyst and O2 as the oxidant without any co-oxidant. Solvent has a dramatic role in the reaction and among the various solvents such as MeOH, EtOH, DCM, DCE, DMF, MeCN, EtOAc, toluene, acetone, chlorobenzene, ethylbenzene; MeCN was the most effective solvent. Various aliphatic amines 13 were reacted well under the optimized reaction conditions to produce the target sulfonamides 14 in good to almost quantitative yields (Scheme 5). A plausible mechanism was proposed for this transformation (Scheme 6). It consists of the following key steps: (i) hydrogen-transfer reaction between a thiol 12 and TEMPO forms the thiyl radical A and TEMPOH; (ii) oxidation of TEMPOH by O2 regenerates TEMPO; (iii) coupling of two thiyl radicals A gives disulfide B; and (iv) reaction of disulfide B and amine 13 affords the expected sulfonamide 14 and mercaptide ion C, which can be oxidized in situ to regenerate the disulfide B.
 |
| | Scheme 5 Yuan's synthesis of 2-benzothiazole-sulfenamides 14. | |
 |
| | Scheme 6 Mechanism that accounts for the formation of 2-benzothiazole-sulfenamides 14. | |
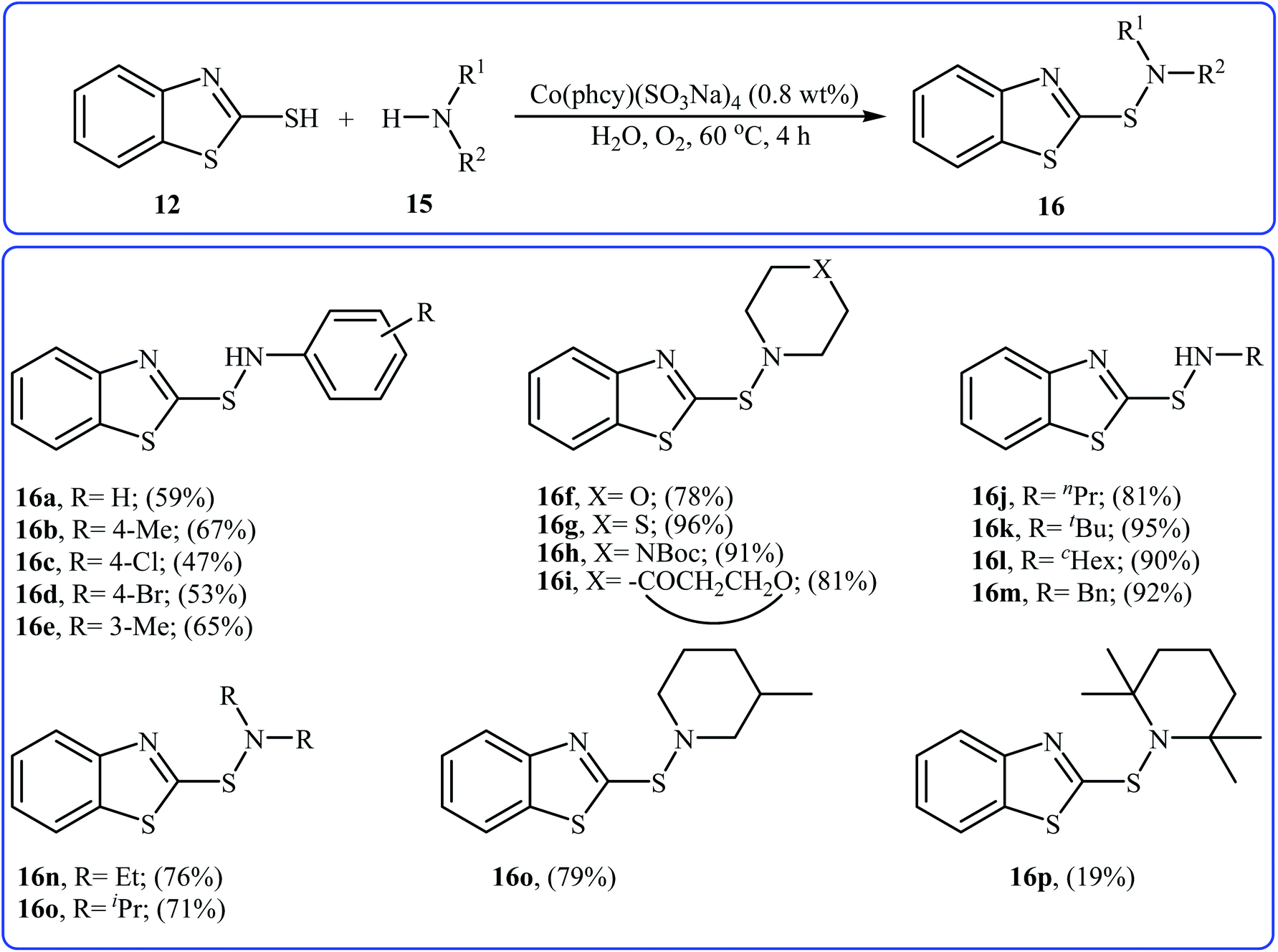
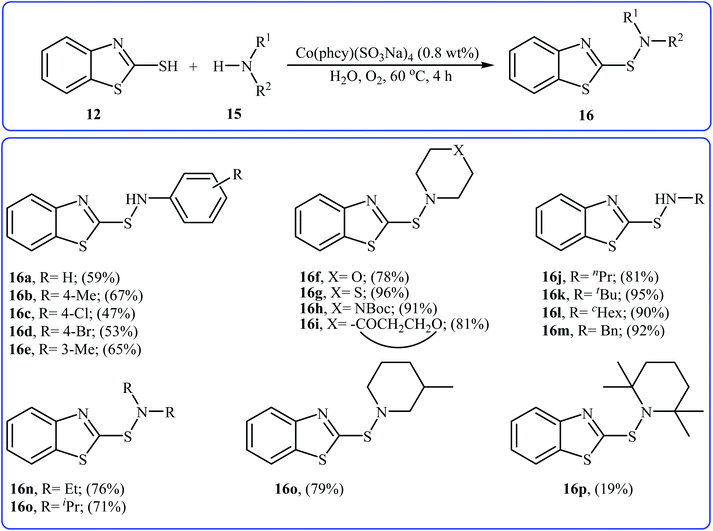
With the aim of designing a greener procedure to sulfenamides via aerobic oxidative coupling of thiols with amines, the same research group was able to demonstrate that a range of 2-benzothiazole-sulfenamides 16 could be obtained from the reaction of 2-mercaptobenzothiazole 12 with various aromatic and aliphatic amines 15 in the water media, employing Co(phcy)(SO3Na)4 as a catalyst under O2 atmosphere (Scheme 7).20 Beside good yields, broad substrate scope, scale-up ability, and easy work-up were the advantages, mentioned for this green synthetic approach. According to the author proposed mechanism, the reaction proceeds along the similar mechanistic pathway that described in Scheme 6. The authors also nicely showed the application of this environment-friendly catalytic system for the oxidative homocoupling of a variety of aromatic and aliphatic thiols to the construction of corresponding disulfides in excellent to almost quantitative yields.
 |
| | Scheme 7 Co(phcy)(SO3Na)4 catalyzed synthesis of 2-benzothiazole-sulfenamides 16 reported by Yuan.20 | |
3. Synthesis of N-acylsulfenamides
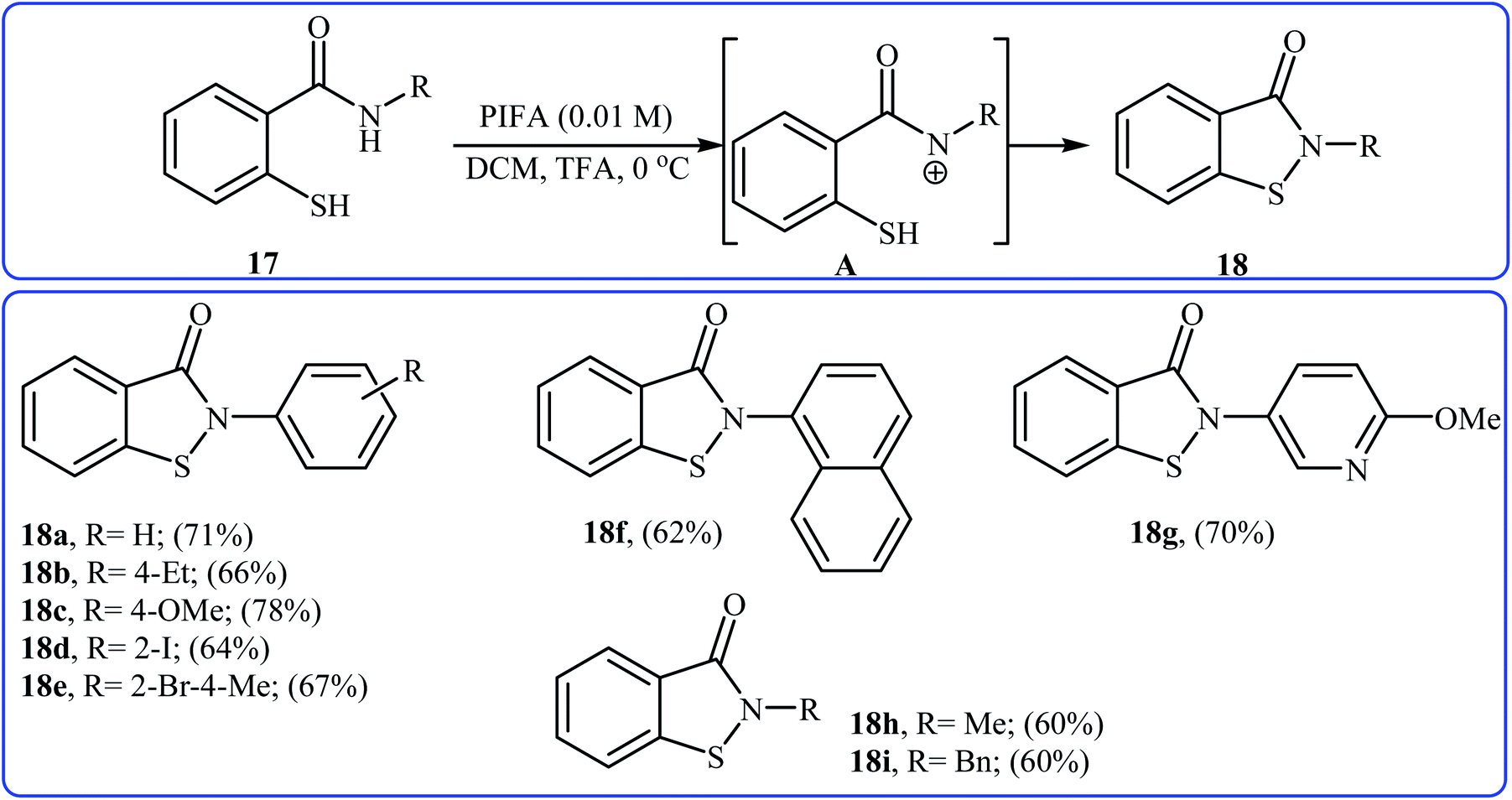
In 2006, Tellitu and Domínguez along with co-workers synthesized a library of biologically important N-substituted benzo[d]isothiazol-3(2H)-one derivatives 18 via the hypervalent iodine reagent [phenyliodine(III)-bis(trifluoroacetate)] (PIFA) mediated intramolecular oxidative cyclization of corresponding N-substituted 2-mercapto-benzamides 17 employing TFA as an additive in DCM at 0 °C.21 As shown in Scheme 8 the reaction showed good functional group tolerance, including bromo, iodo, and methoxy functionalities and gave corresponding cyclic products in moderate to good yields. However, N-allyl substituted 2-mercapto-benzamide failed to participate in this cyclization. It is noted that starting 2-mercapto-benzamides can be easily synthesized from the reaction of commercially available methyl thiosalicylate with primary amines. According to the authors proposed mechanism, the key step of the reaction involves the formation of a reactive N-acylnitrenium ion intermediate A. Shortly afterwards, the group of Cosford employed this interesting synthetic approach to access a series of novel drug-like benzoisothiazolones with significant inhibitory activities against phosphomannose isomerase (PMI), an enzyme that converts mannose-6-phosphate (Man-6-P) into fructose-6-phosphate (Fru-6-P).22
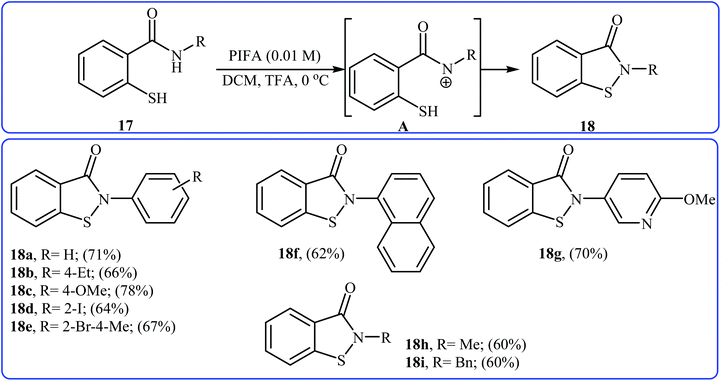 |
| | Scheme 8 Tellitu's synthesis of benzo[d]isothiazol-3(2H)-one derivatives 18. | |
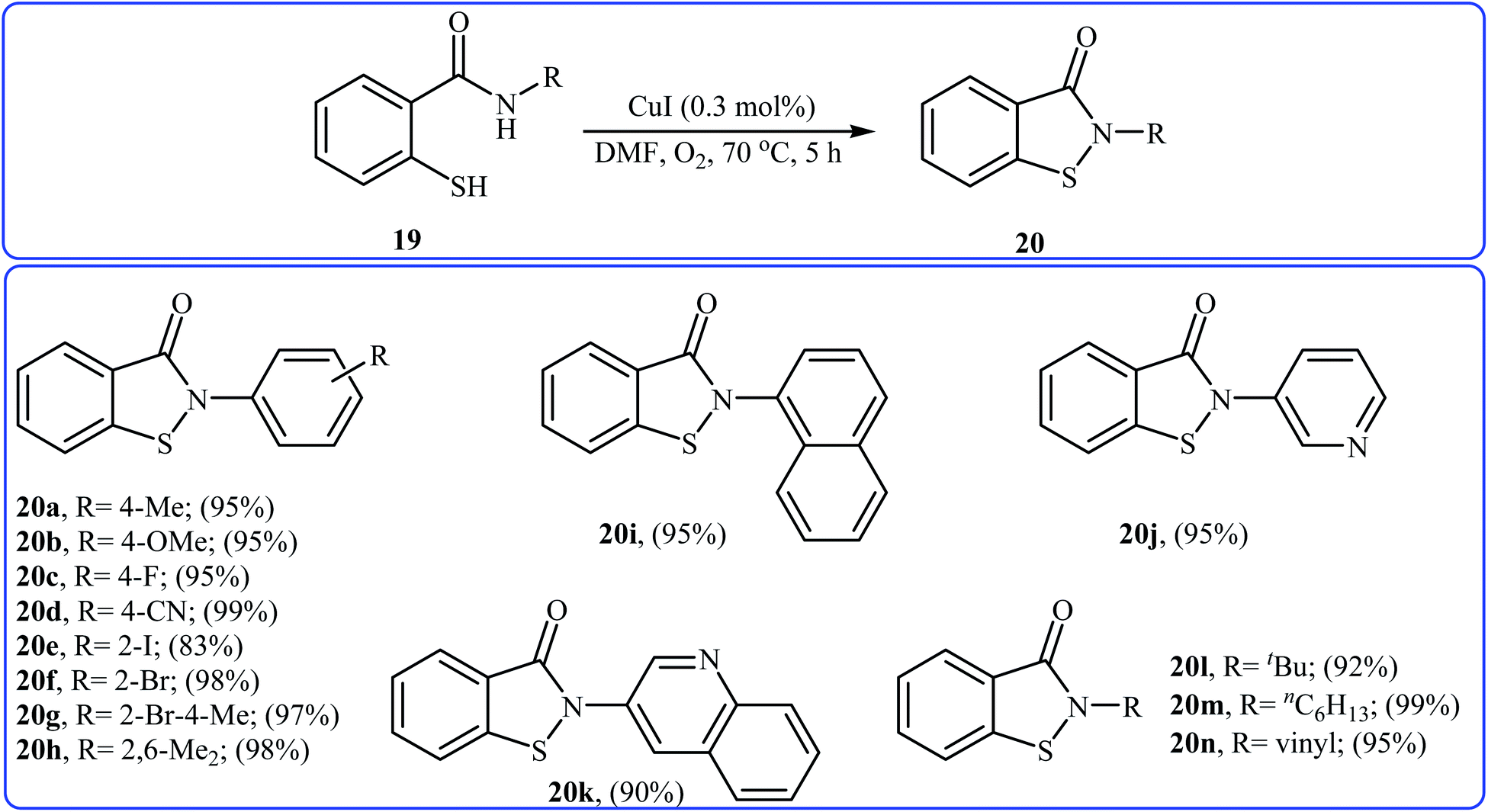
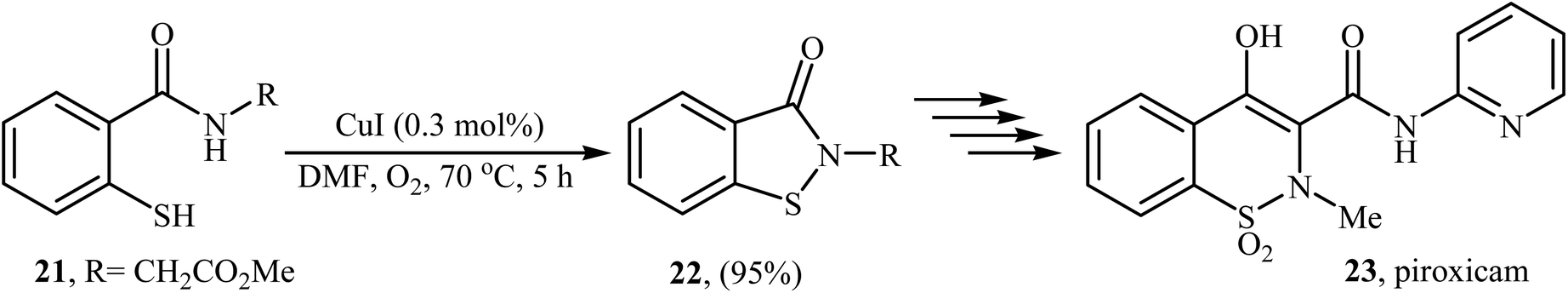
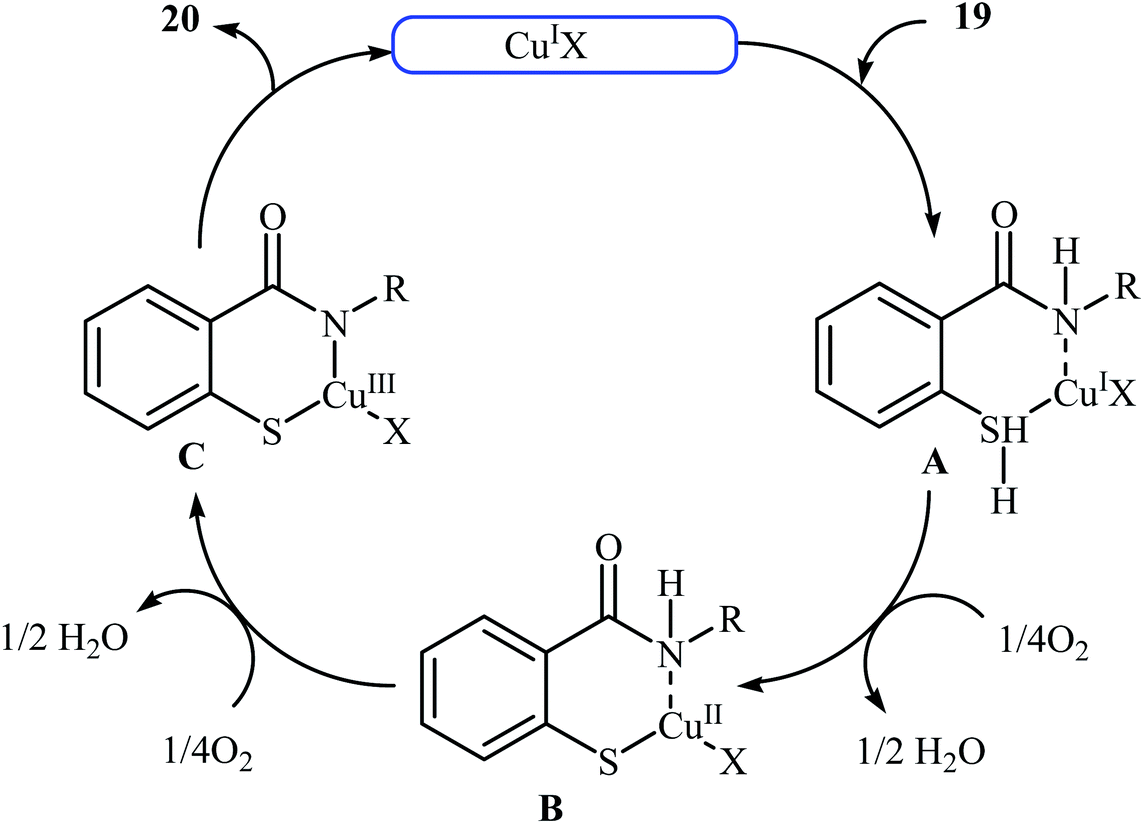
Inspired by these works, Wang and co-workers also showed that N-substituted benzoisothiazolones 20 were successfully formed from corresponding 2-mercapto-benzamides 19 in a simple process employing commercially available and inexpensive CuI as a catalyst and oxygen as an environmentally benign oxidant, in DMF as solvent and at 70 °C.23 This method afforded the desired benzoisothiazolones in high to quantitative yields with various alkyl, aryl, and allyl substituents on nitrogen atom (Scheme 9). The process can also be scaled up to provide multigram quantities of the target compound without obvious loss in the yield or outcome. The authors nicely applied this methodology to the high yielding synthesis of piroxicam 23, a nonsteroidal anti-inflammatory drug, precursor (Scheme 10). The following mechanism has been proposed to explain the formation of compounds 20 (Scheme 11). The complex A is first formed by the coordination of 2-mercapto-benzamide 19 to a copper atom. A subsequent oxidative formation of a Cu–S bond via the elimination of H2O provides the intermediate B, which then undergoes oxidative formation of a Cu–N bond via the elimination of H2O2 to form intermediate C that, after reductive elimination affords the final product 20. In their subsequent study, they reinvestigated the same reaction by using KBr as a green catalyst under air condition. A diverse range of benzo[d]isothiazol-3(2H)-ones (14 examples) in very impressive yields (up to 95%) with good functional group tolerance were obtained.24
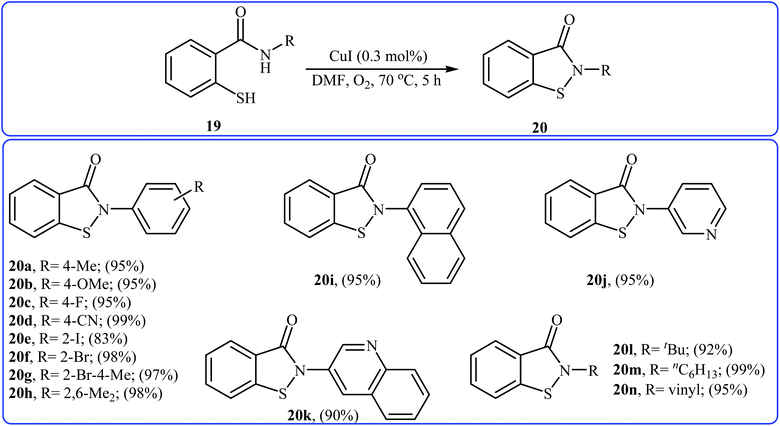 |
| | Scheme 9 Cu(I)-catalyzed synthesis of benzoisothiazolones 20 through intramolecular S–N cross-dehydrogenative coupling of 2-mercapto-benzamides 19. | |
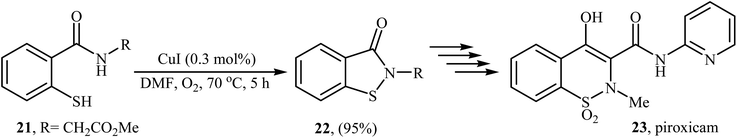 |
| | Scheme 10 Wang's synthesis of piroxicam 23. | |
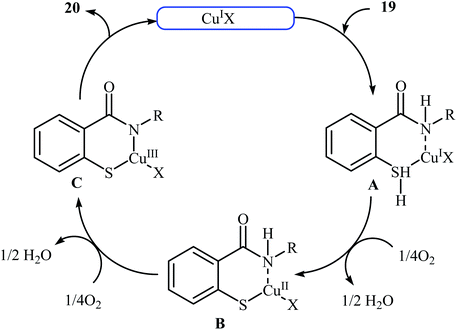 |
| | Scheme 11 Mechanistic proposal for the reactions in Scheme 9. | |
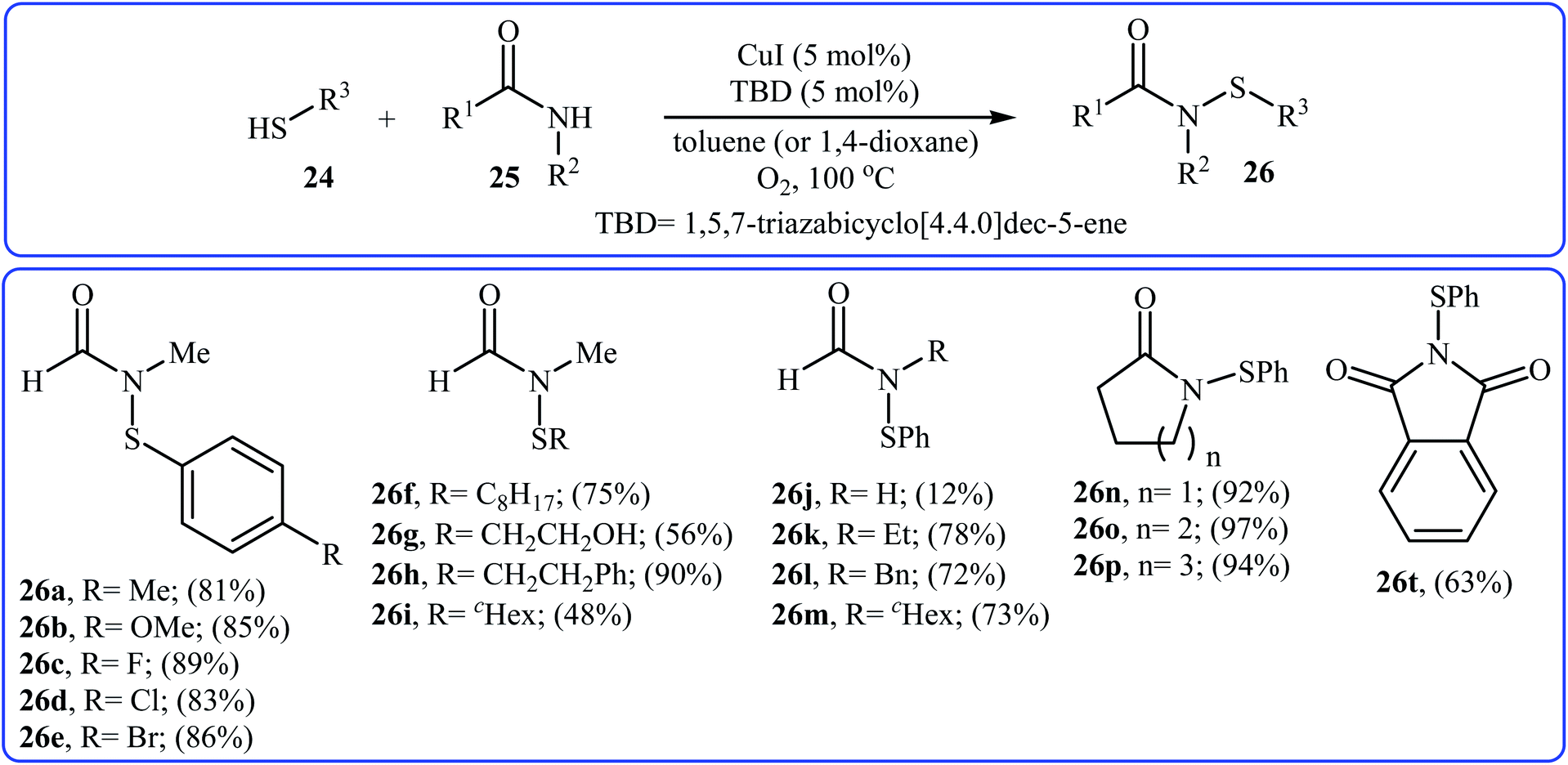
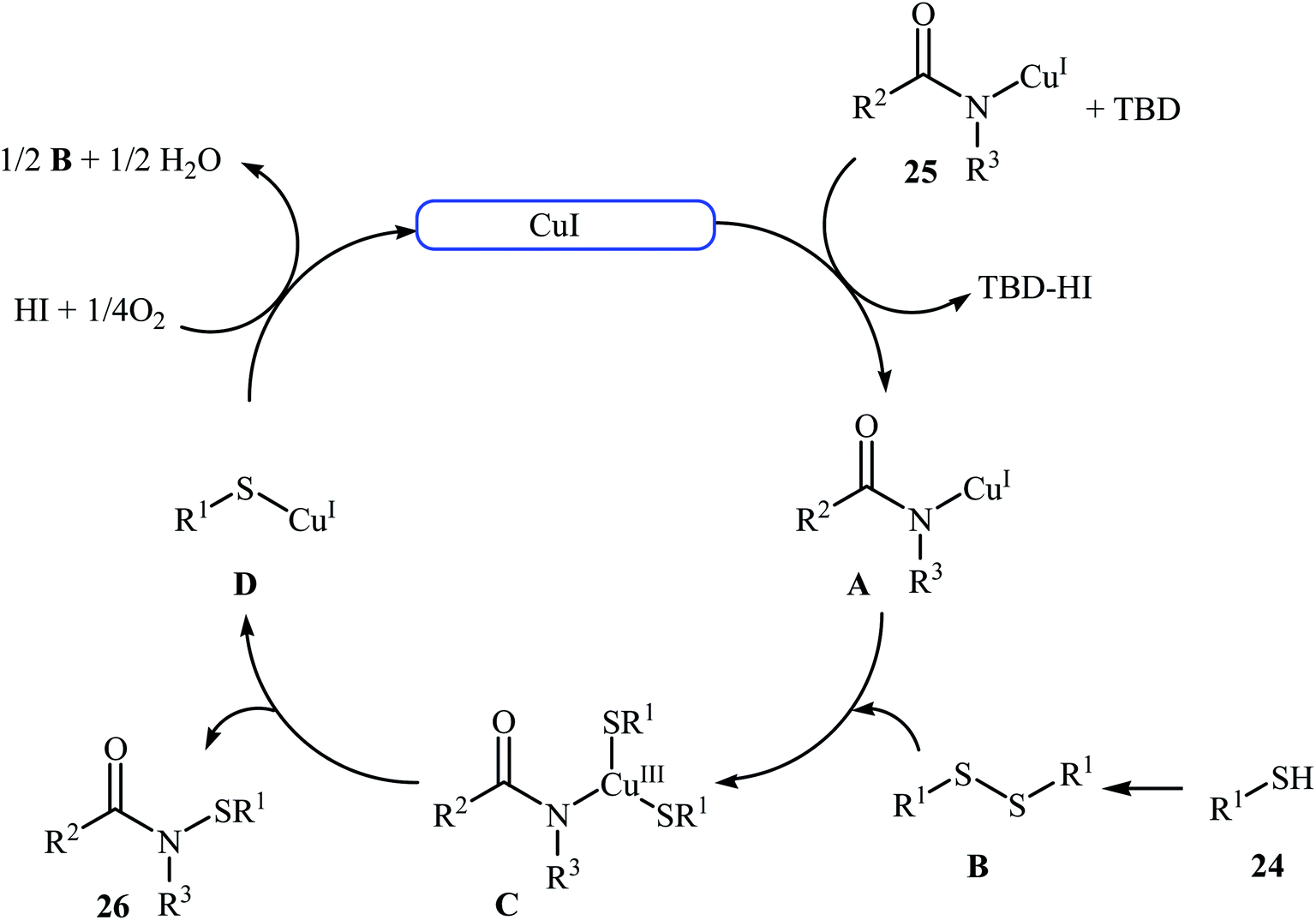
The first copper-catalyzed intermolecular cross-dehydrogenative coupling reaction between thiols and amides was reported by Jang's group in 2015.25 In this study, various copper salts (e.g., CuI, CuBr, CuOAc, and CuBr2) and base (e.g., K2CO3, tBuOK, TBD, and DBU) were examined and the system CuI/TBD was found to be optimal for this transformation. Various thiols 24 and secondary amides 25 were reacted well under the optimized reaction conditions to produce the corresponding N-carbonyl sulfonamides 26 in moderate to excellent yields (Scheme 12). However, this protocol for N-sulfenylation of primary amides was considerably less efficient. It is noted that this CuI-catalyzed reaction is equally efficient for both the aromatic and aliphatic thiols. A wide range of sensitive functional groups including ester, fluoro, chloro, bromo, alkoxy, and hyrdoxy are tolerated by the reaction conditions employed. This procedure offers scope for further manipulation of products. Mechanistic investigations revealed that the reaction starts with the generation of amide–CuI complex A via coordination of amide 25 to a copper ion, and then the reaction of intermediate A with in situ generated disulfide B from homocoupling of corresponding thiol 24 furnishes copper complex C. Subsequently, intermediate C undergoes reductive elimination to provide the expected sulfenamide 26 and generates PhS–CuI complex D. Finally, under aerobic oxidation conditions, PhS–CuI complex D converts into a catalytically more reactive CuI complex and disulfide B (Scheme 13).
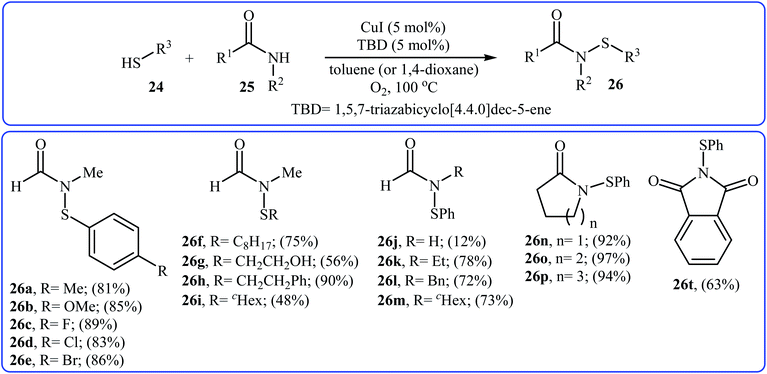 |
| | Scheme 12 Cu(I)-catalyzed synthesis of N-formyl/acylsulfenamides 26 reported by Jang.25 | |
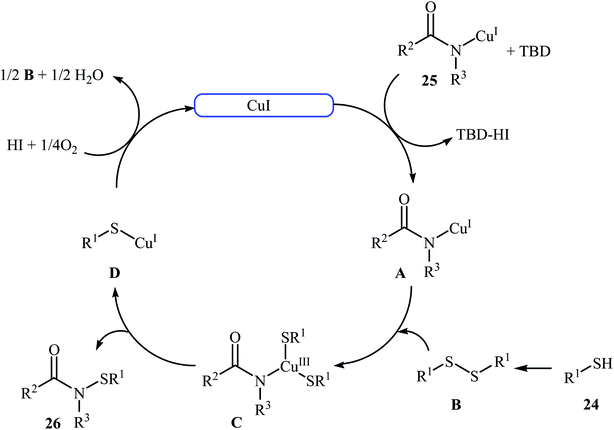 |
| | Scheme 13 Mechanistic proposal for the formation of 26. | |
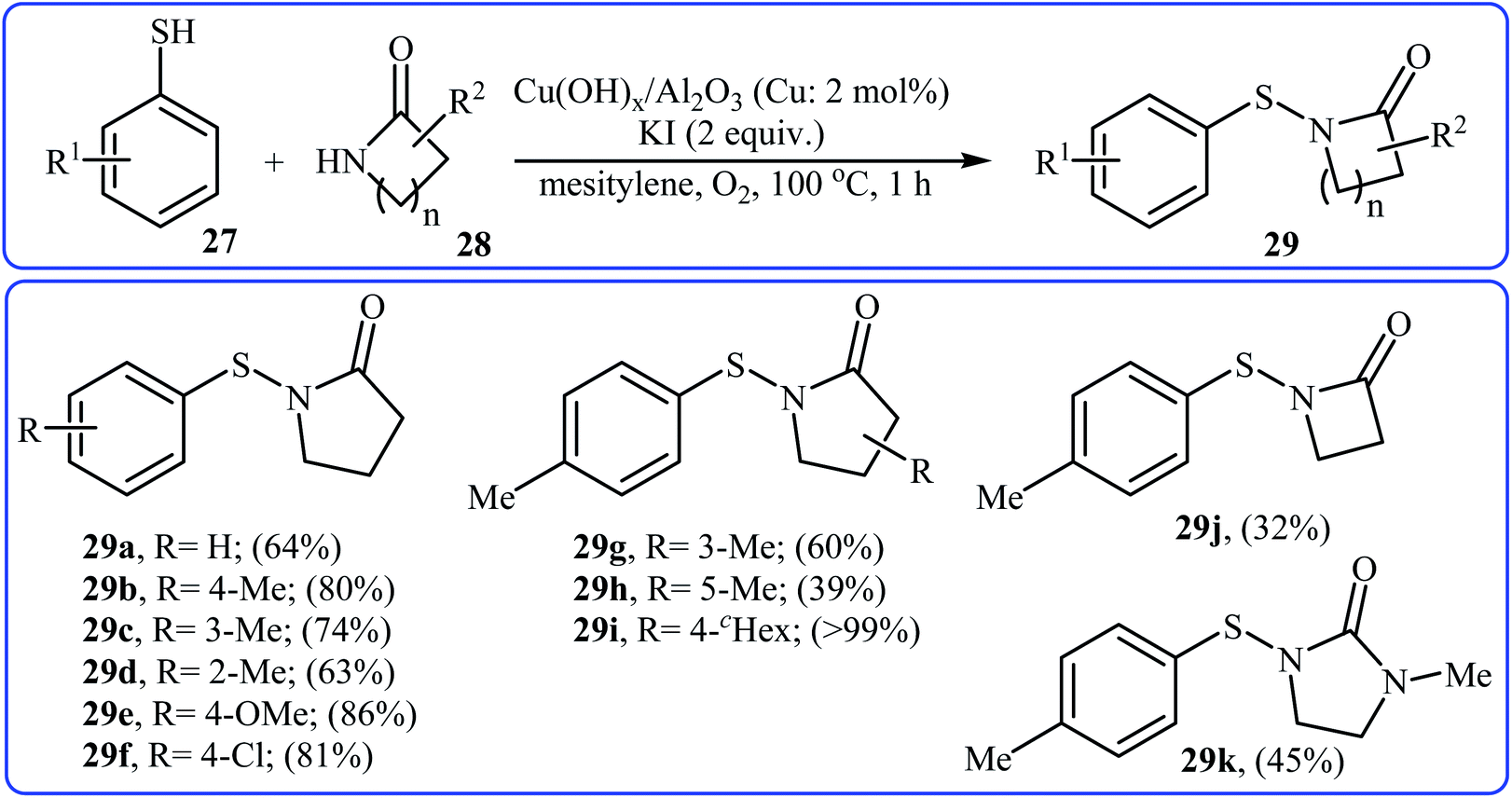
Recently, an efficient method for the synthesis of N-acylsulfenamides 29 via Cu-catalyzed cross-coupling of aryl thiols 27 and lactams 28 has been developed by Sakagami and co-workers (Scheme 14). This intermolecular thiolation was achieved in mesitylene using a supported copper hydroxide catalyst Cu(OH)x/Al2O3 (Cu: 2 mol%) at 100 °C for 1 h under an oxygen atmosphere to afford N-acylsulfenamides 3 in moderate to excellent yields.26
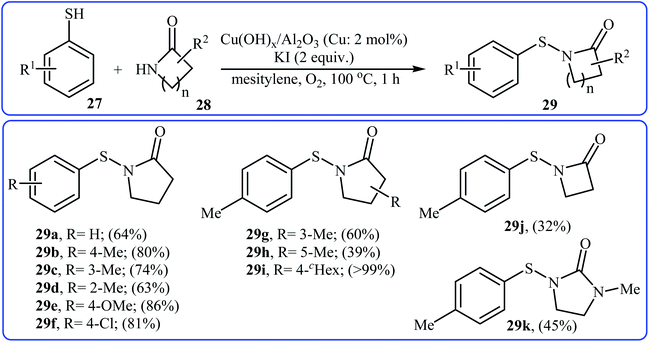 |
| | Scheme 14 Sakagami's synthesis of N-acylsulfenamides 29. | |
4. Synthesis of sulfenylimines
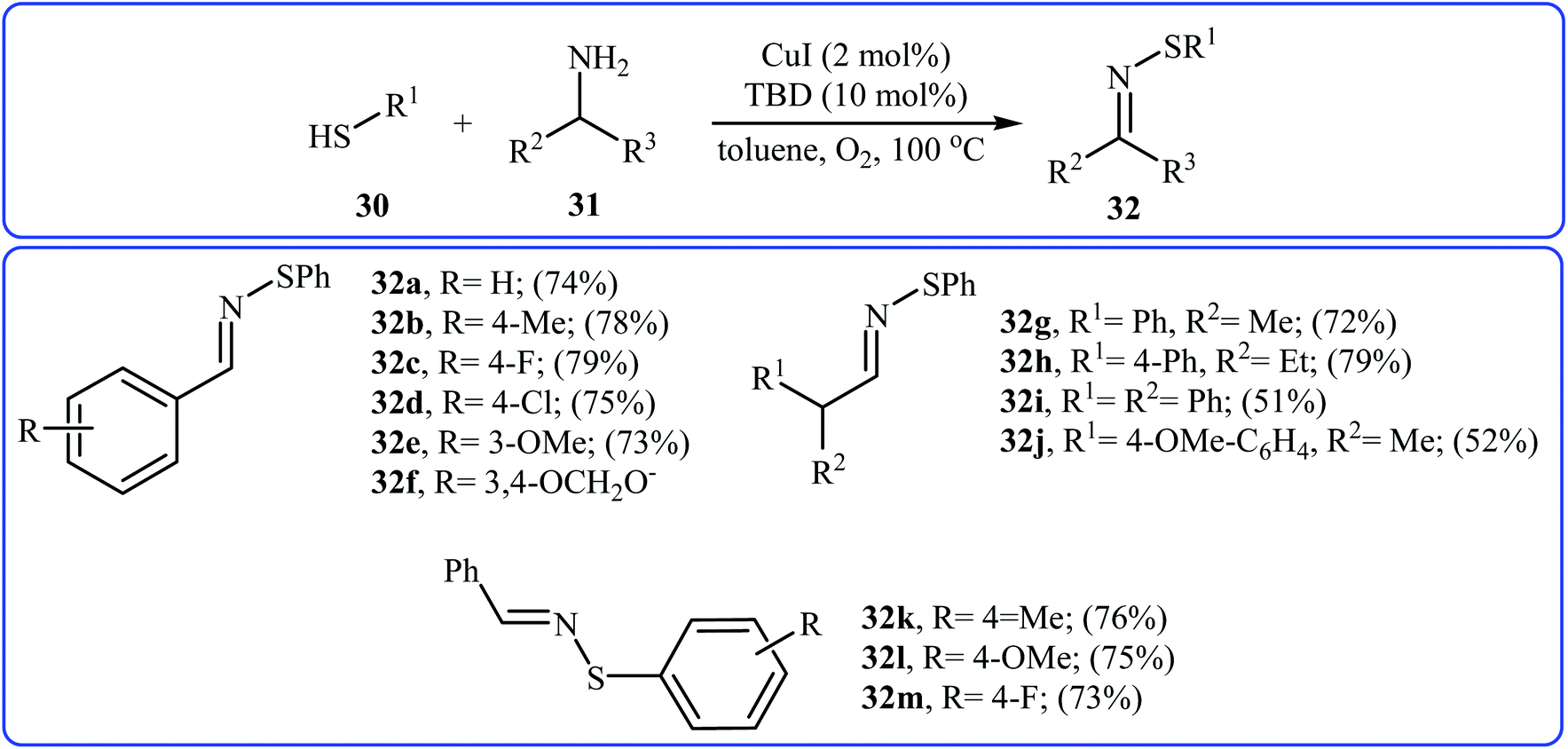
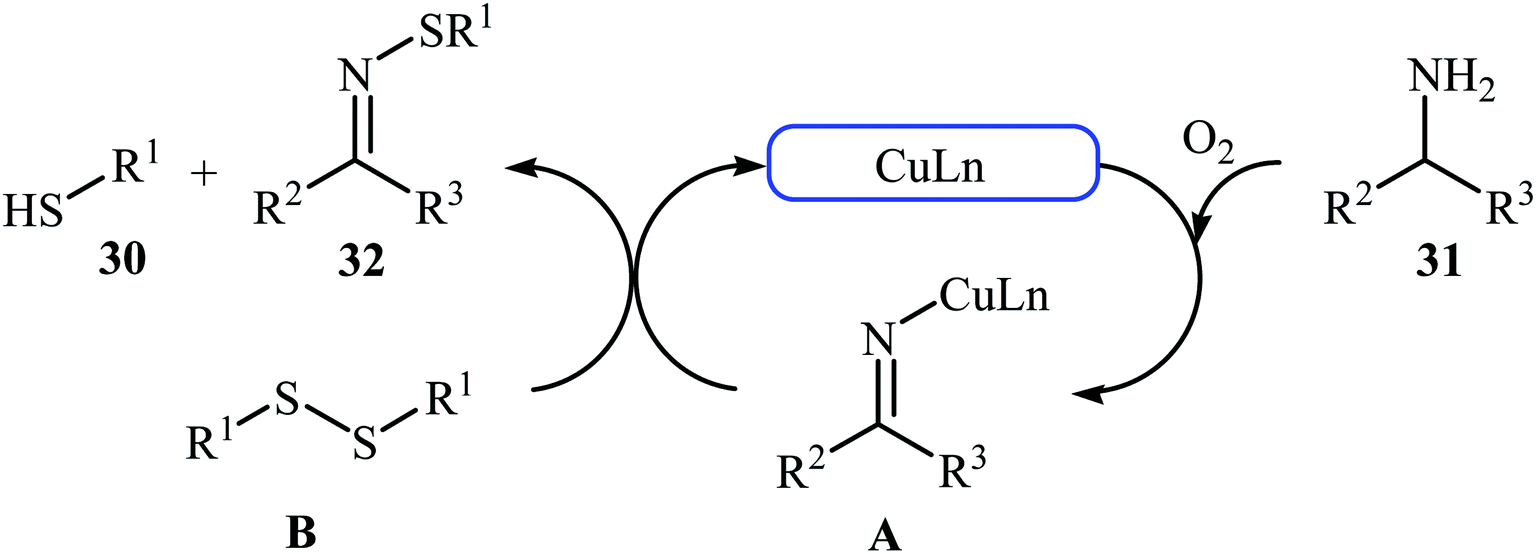
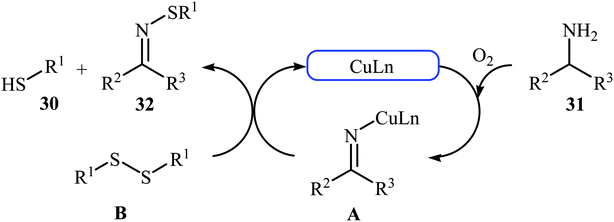
In 2015, Lee, Wang, and Jang developed an elegant Cu(I)-catalyzed oxidative cross-coupling reaction between thiols 30 and benzyl amines 31 for the construction of synthetically important N-sulfenylimines 32 (Scheme 15).27 The best conversion efficiency was obtained for the reactions containing CuI (2 mol%) and TBD (10 mol%) in toluene at 100 °C under O2 atmosphere. This approach provides the first highly practical procedure for synthesizing N-sulfenylimines from commercially available and inexpensive thiols and amines. Functional groups including fluoro, chloro, and ether are tolerated by the reaction conditions employed. It is noteworthy that this Cu(I)-catalyzed reaction is equally efficient for both the primary and secondary amines. According to the authors proposed mechanism, the reaction starts with the generation of an imine–Cu complex A via the aerobic oxidation of benzyl amine 31 by Cu catalyst. Meanwhile, homocoupling of starting thiol 30 affords disulfide intermediate B. Finally, the reaction of complex A with disulfide B yields the expected N-sulfenylimines 32 (Scheme 16).
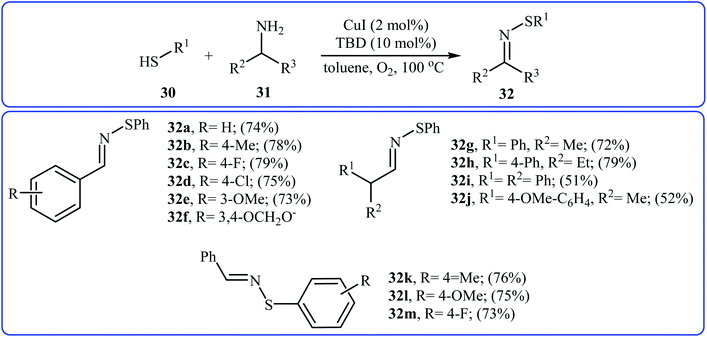 |
| | Scheme 15 Synthesis of N-sulfenylimines 32 from thiols 30 and benzyl amines 31. | |
 |
| | Scheme 16 Plausible mechanism for the formation of 32. | |
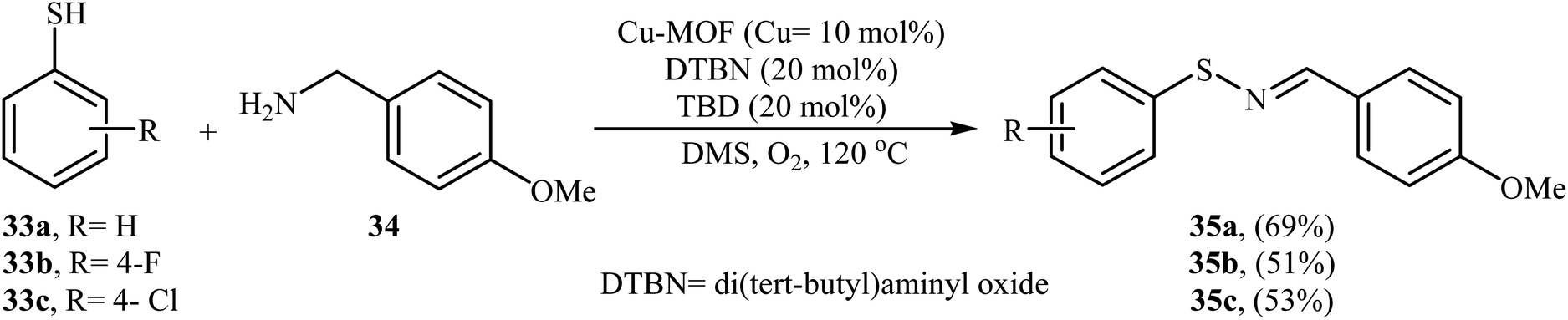
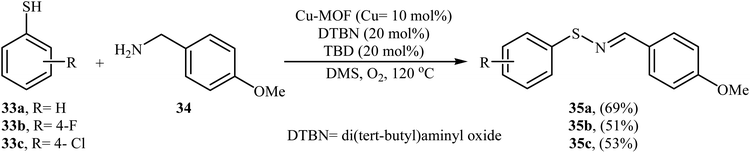
Shortly after this report, Qiu and He along with their co-workers showed that copper-based metal–organic frameworks with pcu-topology (pcu-MOF) are efficient heterogeneous catalyst for the oxidative coupling of thiols and amines. Thus, by using pcu-MOF/DTBN/TBD as a catalytic system in DMSO under an O2 atmosphere, the coupling of thiophenols 33 with 3-methoxybenzylamine 34 afforded corresponding N-sulfenylimines 35 in moderate yields (Scheme 17).28
 |
| | Scheme 17 Qiu–He's synthesis of N-sulfenylimines 35. | |
5. Synthesis of sulfenylsulfoximines
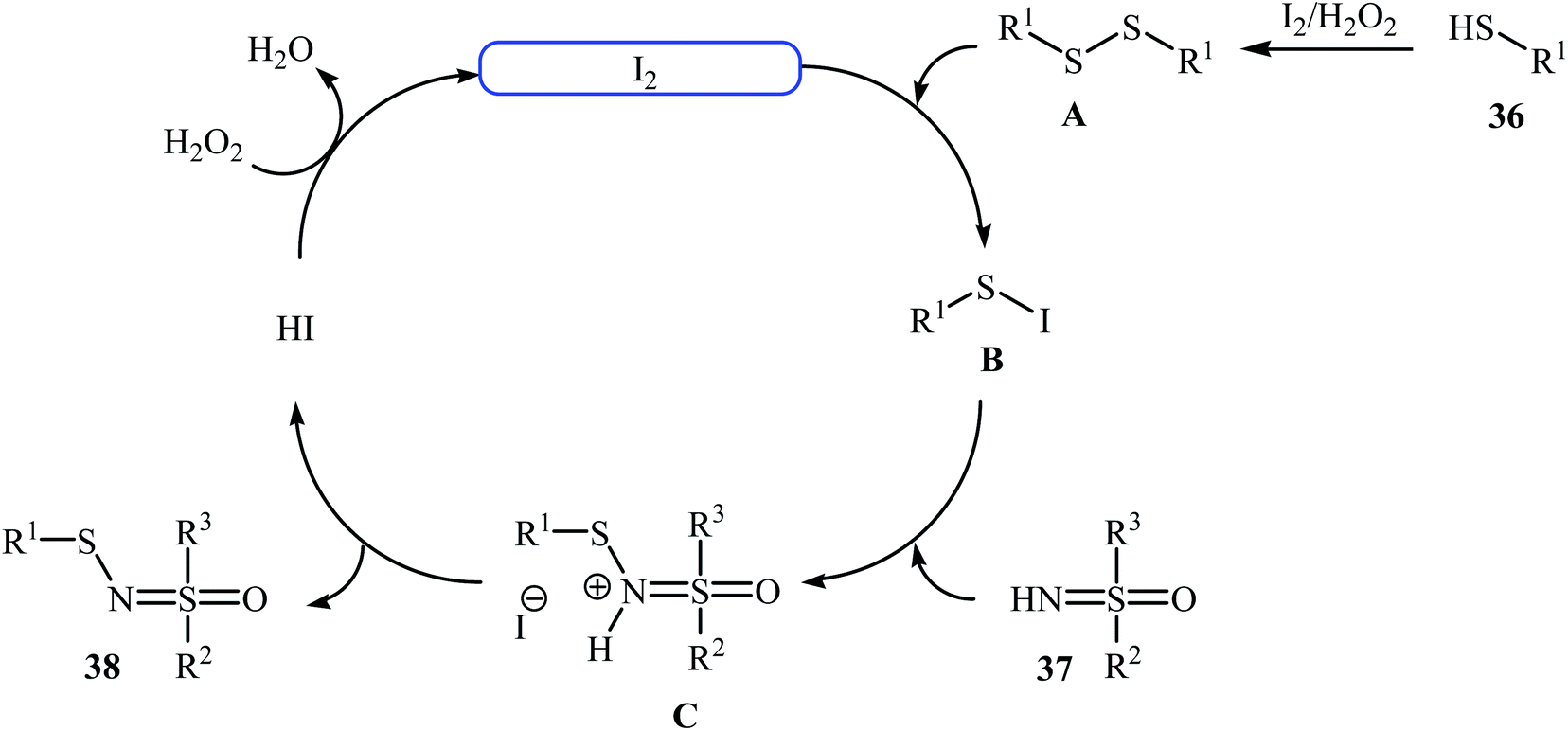
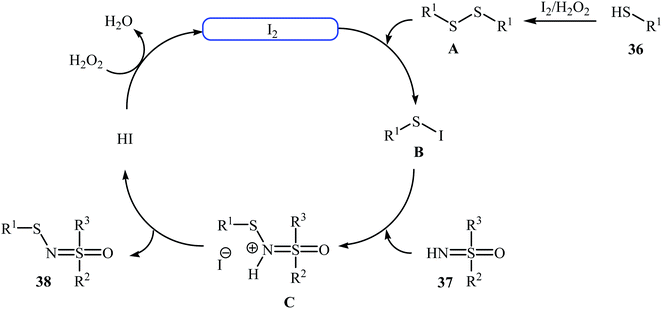
Very recently, Yang and co-workers showed that N-sulfenylsulfoximine derivatives 38 could be synthesized via dehydrocoupling reaction of thiols 36 with sulfoximines 37 in the presence of 1.0 mol% of molecular iodine as the catalyst and 2.5 equiv. of H2O2 as an oxidant in eco-friendly polyethylene glycol-400 (PEG-400) at 50 °C. Generally, target N-sulfenylsulfoximines were obtained in good to excellent yields (Scheme 18).29 The reaction showed excellent functional group tolerance, including fluoro, chloro, bromo, methoxy, and nitro groups that would allow further elaboration of the products. This I2-catalyzed reaction is applicable for both the (hetero)aromatic as well as benzylic thiols and for various substituted sulfoximines. It should be mentioned that the optimized reaction condition was also applied for the cross-dehydrogenative coupling reactions of thiophenol with a diverse set of functionalized anilines, and the reaction gave good yields of the expected sulfonamides. A possible mechanistic pathway was also proposed (Scheme 19), whereby the reaction is initiated with homocoupling of thiol 36 to give disulfide intermediate A, which undergoes reaction with I2 affording sulfenyl iodide intermediate B. Subsequently, nucleophilic attack of sulfoximine 37 onto the activated S–I bond in intermediate B affords the cationic N-sulfenylsulfoximine C, which by deprotonation convert to the desired products 38. The generated hydriodic acid is then oxidized by H2O2 to restore the catalyst I2, and water is the sole by-product. The authors claimed that their report is the first example of the direct cross-dehydrogenative coupling reaction to construct N-sulfenylsulfoximines.
 |
| | Scheme 18 Synthesis of N-sulfenylsulfoximines 37 through metal-free N–H/S–H cross-dehydrogenative coupling reaction. | |
 |
| | Scheme 19 Mechanistic proposal for the reaction in Scheme 18. | |
6. Synthesis of sulfonamides
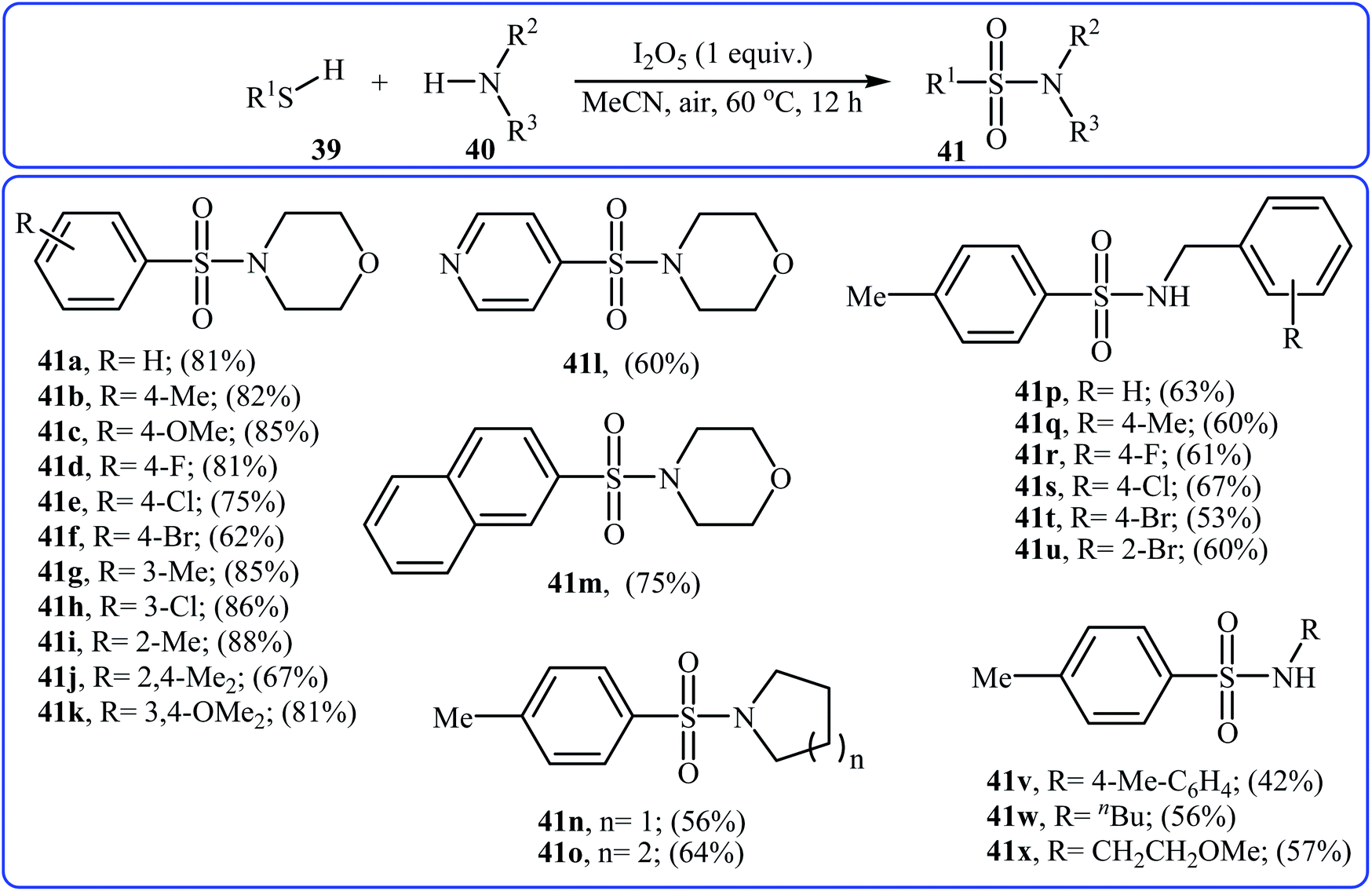
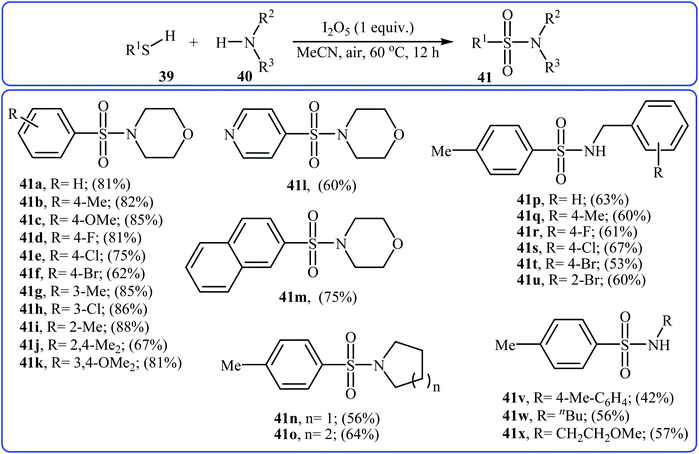
Very recently, the Wei–Wang group have developed an efficient and straightforward approach for the synthesis of biologically important secondary and tertiary sulfonamides 41 via I2O5-mediated oxidative coupling of aryl thiols 39 and amines 40 (Scheme 20).30 The reaction was performed in acetonitrile at 60 °C, tolerated various functional groups, and generally provided corresponding sulfonamides in moderate to high yields. It is noted that the reaction is applicable for various amines like aromatic, benzylic, and aliphatic amines. This protocol was also successfully applied to the preparation of gram quantities of the desired sulfonamide without sacrificing the yield or outcome of the methodology. The proposed reaction mechanism is outlined in Scheme 21 and comprises the following basic steps: (i) initial formation of disulfide A via dimerization of thiol 39; (ii) oxidation of disulfide A by I2O5 to give arylsulfonothioate B with the concomitant formation of molecular iodine; (iii) interaction of intermediate B with iodine to produce sulfonyl iodide C; and (iv) nucleophilic substitution of sulfonyl iodide C by amine 40 and formation of the sulfonamide 41. To the best of our knowledge, this is the only example of direct construction of sulfonamides from readily-available thiols and amines reported so far.
 |
| | Scheme 20 I2O5-mediated direct construction of sulfonamides 41 from thiols 39 and amines 40. | |
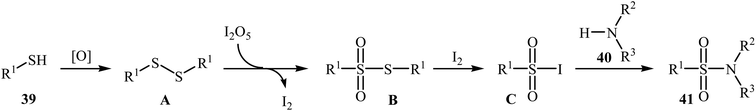 |
| | Scheme 21 Mechanism that accounts for the formation of sulfonamides 41. | |
7. Outlook and conclusion
Sulfur–nitrogen bond-containing compounds have extensive application in organic and medicinal chemistry. Synthetically, such organosulfur compounds are commonly prepared via cross-coupling of S-halo compounds and N–H compounds or cross-coupling reactions between N-halogenated compounds and thiols. However, the limited availability of commercial starting materials, the formation of undesired by-products and/or harsh reaction conditions are the main drawbacks of these methods. Recently, cross-dehydrogenative coupling reaction has become an attractive alternative to traditional S–N cross-coupling reactions due to the waste-minimization and the costs associated with the preparation of starting materials. Shorter synthetic routes and high atom economy are the key features of these eco-friendly green reactions. This new page of sulfur–nitrogen bond formation has been successfully applied in the synthesis of a variety of S–N bond-containing compounds such as sulfenamides, sulfonamides, sulfenylimines, and sulfenyl-sulfoximines. Despite the significant achievements during the past few years in this interesting research arena, there are still many problems remained: (i) almost all of the catalysts and oxidants are limited to the use of copper and hypervalent iodine, respectively. Thus the exploration of different catalytic systems is highly desirable; (ii) the number of reported examples in some reactions such as synthesis of sulfonamides and sulfenylsulfoximines from S–H and N–H compounds are narrow and there is an urgent need to study the scope and limitations of these reactions; and (iii) other reactions such as coupling of thiols with urethanes and ureas should be explored. It is anticipated that the insight provided in this review will be beneficial for eliciting further research in this domain.
Conflicts of interest
There are no conflicts to declare.
References
- R. J. W. Cremlyn, An introduction to organosulfur chemistry, John Wiley & Sons Inc, New York, 1996 Search PubMed.
- N. Goncharov, A. N. Orekhov, N. Voitenko, A. Ukolov, R. Jenkins and P. Avdonin, in Nutraceuticals: Efficacy, Safety and Toxicity, ed. R. Gupta, Academic Press/Elsevier, Amsterdam, 2016, pp. 555–568 Search PubMed.
- C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168–181 CrossRef PubMed.
- S. Arshadi, E. Vessally, L. Edjlali, R. Hosseinzadeh-Khanmiri and E. Ghorbani-Kalhor, Beilstein J. Org. Chem., 2017, 13, 625–638 CrossRef PubMed.
- J. Green, A. Troupin, L. Halpern, P. Friel and P. Kanarek, Epilepsia, 1974, 15, 329–349 CrossRef PubMed.
- F. Engler, M. Maeder-Ingvar, E. Roulet and T. Deonna, Neuropediatrics, 2003, 34, 105–109 CrossRef PubMed.
-
(a) M. J. Brodie, A. Covanis, A. Gil-Nagel, H. Lerche, E. Perucca, G. J. Sills and H. S. White, Epilepsy Behav., 2011, 21, 331–341 CrossRef PubMed;
(b) M. Abdoli, A. Angeli, M. Bozdag, F. Carta, A. Kakanejadifard, H. Saeidian and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2017, 32, 1071–1078 CrossRef PubMed.
-
(a) R. P. Shank, J. F. Gardocki, A. J. Streeter and B. E. Maryanoff, Epilepsia, 2000, 41, 3–9 CrossRef;
(b) H. Saeidian and M. Abdoli, J. Sulfur Chem., 2015, 36, 463–470 CrossRef.
- I. V. Koval', Russ. Chem. Rev., 1996, 65, 421–440 CrossRef.
- Y. Zhu, M. R. Loso, G. B. Watson, T. C. Sparks, R. B. Rogers, J. X. Huang, B. C. Gerwick, J. M. Babcock, D. Kelley and V. B. Hegde, J. Agric. Food Chem., 2010, 59, 2950–2957 CrossRef PubMed.
-
(a) M. Behforouz and J. E. Kerwood, J. Org. Chem., 1969, 34, 51–55 CrossRef;
(b) H. M. Gillis, L. Greene and A. Thompson, Synlett, 2009, 112–116 Search PubMed;
(c) H. Saeidian, M. Abdoli and Z. Mirjafary, Synthesis, 2015, 47, 1057–1075 CrossRef.
-
(a) L. Craine and M. Raban, Chem. Rev., 1989, 89, 689–712 CrossRef;
(b) C. Bohnen and C. Bolm, Org. Lett., 2015, 17, 3011–3013 CrossRef PubMed.
- For selected reviews on carbon-carbon cross-coupling reactions, see:
(a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef;
(b) F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC;
(c) C. Valente, S. Çalimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314–3332 CrossRef PubMed.
- For selected reviews on carbon-heteroatom cross-coupling reactions, see:
(a) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef PubMed;
(b) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596–1636 CrossRef PubMed;
(c) M. Abdoli, Z. Mirjafary, H. Saeidian and A. Kakanejadifard, RSC Adv., 2015, 5, 44371–44389 RSC;
(d) K. Didehban, E. Vessally, A. Hosseinian, L. Edjlali and E. S. Khosroshahi, RSC Adv., 2018, 8, 291–301 RSC;
(e) E. Vessally, K Didehban, R. Mohammadi, A. Hosseinian and M. Babazadeh, J. Sulfur Chem., 2018 DOI:10.1080/17415993.2018.1436711.
- For selected examples on heteroatom-heteroatom cross-coupling reactions, see:
(a) Y. Zhou, S. Yin, Y. Gao, Y. Zhao, M. Goto and L. B. Han, Angew. Chem., Int. Ed., 2010, 49, 6852–6855 CrossRef PubMed;
(b) W. He, X. Hou, X. Li, L. Song, Q. Yu and Z. Wang, Tetrahedron, 2017, 73, 3133–3138 CrossRef;
(c) G. Wang, Q.-Y. Yu, S.-Y. Chen and X.-Q. Yu, Tetrahedron Lett., 2013, 54, 6230–6232 CrossRef;
(d) C. Li, T. Chen and L.-B. Han, Dalton Trans., 2016, 45, 14893–14897 RSC.
- For selected reviews on cross-dehydrogenative coupling reactions, see:
(a) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef PubMed;
(b) J. Yuan, C. Liu and A. Lei, ChemComm, 2015, 51, 1394–1409 RSC;
(c) T. Chen, J.-S. Zhang and L.-B. Han, Dalton Trans., 2016, 45, 1843–1849 RSC.
-
(a) N. Taniguchi, Eur. J. Org. Chem., 2010, 2670–2673 CrossRef;
(b) N. Taniguchi, Synlett, 2007, 1917–1920 CrossRef.
- E. Rattanangkool, W. Krailat, T. Vilaivan, P. Phuwapraisirisan, M. Sukwattanasinitt and S. Wacharasindhu, Eur. J. Org. Chem., 2014, 4795–4804 CrossRef.
- L. Yang, S. Li, Y. Dou, S. Zhen, H. Li, P. Zhang, B. Yuan and G. Yang, Asian J. Org. Chem., 2017, 6, 265–268 CrossRef.
- Y. Dou, X. Huang, H. Wang, L. Yang, H. Li, B. Yuan and G. Yang, Green Chem., 2017, 19, 2491–2495 RSC.
- A. Correa, I. Tellitu, E. Domínguez and R. SanMartin, Org. Lett., 2006, 8, 4811–4813 CrossRef PubMed.
- R. Dahl, Y. Bravo, V. Sharma, M. Ichikawa, R.-P. Dhanya, M. Hedrick, B. Brown, J. Rascon, M. Vicchiarelli and A. Mangravita-Novo, J. Med. Chem., 2011, 54, 3661–3668 CrossRef PubMed.
- Z. Wang, Y. Kuninobu and M. Kanai, J. Org. Chem., 2013, 78, 7337–7342 CrossRef PubMed.
- T.-Q. Yu, Y.-S. Hou, Y. Jiang, W.-X. Xu, T. Shi, X. Wu, J.-C. Zhang, D. He and Z. Wang, Tetrahedron Lett., 2017, 58, 2084–2087 CrossRef.
- C. Lee, Y. N. Lim and H. Y. Jang, Eur. J. Org. Chem., 2015, 5934–5938 CrossRef.
- K. Sakagami, X. Jin, K. Suzuki, K. Yamaguchi and N. Mizuno, Chem. Lett., 2015, 45, 173–175 CrossRef.
- C. Lee, X. Wang and H.-Y. Jang, Org. Lett., 2015, 17, 1130–1133 CrossRef PubMed.
- W. Long, W. Qiu, C. Li, L. Song, G. Bai, G. Zhang and H. He, RSC Adv., 2016, 6, 40945–40952 RSC.
- L. Yang, J. Feng, M. Qiao and Q. Zeng, Org. Chem. Front., 2018, 5, 24–28 RSC.
- M. Zhu, W. Wei, D. Yang, H. Cui, L. Wang, G. Meng and H. Wang, Org. Biomol. Chem., 2017, 15, 4789–4793 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b
b