
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
12-Molybdophosphoric acid anchored on aminopropylsilanized magnetic graphene oxide nanosheets (Fe3O4/GrOSi(CH2)3–NH2/H3PMo12O40): a novel magnetically recoverable solid catalyst for H2O2-mediated oxidation of benzylic alcohols under solvent-free conditions
Saeed Farhadi *a,
Mohammad Hakimib and
Mansoureh Malekib
*a,
Mohammad Hakimib and
Mansoureh Malekib
aChemistry Department, Lorestan University, Khorramabad 68151-44316, Iran. E-mail: sfarhadi1348@yahoo.com; Fax: +98-6633120618; Tel: +98-6633120611
bChemistry Department, Payame Noor University, Tehran 19395-4697, Iran
First published on 12th February 2018
Abstract
In this work, 12-molybdophosphoric acid (H3PMo12O40, HPMo) was chemically anchored onto the surface of aminosilanized magnetic graphene oxide (Fe3O4/GrOSi(CH2)3–NH2) and was characterized using different physicochemical techniques, such as powder X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FT-IR), Raman spectroscopy, energy-dispersive X-ray analysis (EDX), scanning electron microscopy (SEM), BET specific surface area analysis and magnetic measurements. The results demonstrated the successful loading of HPMo (∼31.5 wt%) on the surface of magnetic aminosilanized graphene oxide. XRD patterns, N2 adsorption–desorption isotherms and SEM images confirm the mesostructure of the sample. FT-IR and EDX spectra indicate the presence of the PMo12O403− polyanions in the nanocomposite. The as-prepared Fe3O4/GrOSi(CH2)3–NH2/HPMo nanocomposite has a specific surface area of 76.36 m2 g−1 that is much higher than that of pure HPMo. The selective oxidation of benzyl alcohol to benzaldehyde was initially studied as a benchmark reaction to evaluate the catalytic performance of the Fe3O4/GrOSi(CH2)3–NH2/HPMo catalyst. Then, the oxidation of a variety of substituted primary and secondary activated benzylic alcohols was evaluated with H2O2 under solvent-free conditions. Under the optimized conditions, all alcohols were converted into the corresponding aldehydes and ketones with very high selectivity (≥99%) in moderate to excellent yields (60–96%). The high catalytic performance of the nanocomposite was ascribed to its higher specific surface area and more efficient electron transfer, probably due to the presence of GrO nanosheets. The nanocomposite catalyst is readily recovered from the reaction mixture by a usual magnet and reused at least four times without any observable change in structure and catalytic activity.
1. Introduction
The oxidation of alcohols to the corresponding carbonyl compounds, i.e., benzyl alcohol to benzaldehyde, is one of the most important and vital transformations in synthetic organic chemistry.1 Typically, the chemoselective oxidation of alcohols to aldehydes or ketones without traceable over-oxidation products (i.e., carboxylic acids) is attractive for the preparation of fine chemicals owing to the nature of aldehydes or ketones as an intermediate for producing carbonyl products, which are of great importance and versatility for the chemical industry and pharmaceutical synthesis.2 Numerous efforts on developing efficient catalysts towards these oxidations have been progressed and reported.3 Solvent-free approach is an attractive green process for selective oxidation of benzyl alcohol and oxidants like molecular oxygen, hydrogen peroxide (H2O2) and TBHP are in the order as green options.4 Molecular oxygen as oxidant is not very active, while H2O2 is an active oxygen donor in catalytic oxygen-transfer reactions. However, most of the catalyst systems using H2O2 are based on noble metals such as Pt, Pd, and Ru which are expensive and difficult to synthesize.5–7 From the practical and environmental points of view, there is a strong demand for the screening out efficient catalytic systems using inexpensive and environmentally benign metal catalysts and non-toxic H2O2 or O2 as the sole terminal oxidant. In this context, polyoxometalates (POMs) are definitely an attractive alternative in terms of economic viability and easy-to manufacture alternative with a heterogeneous nature.Polyoxometalates (POMs) are a large family of bulky clusters of transition metal oxide anions with structural diversity.8 Among various POMs, Keggin-type heteropoly acids (e.g. H3PMo12O40 and H3PW12O40) have received much attention and numerous organic transformations can be catalyzed by them, not only due to their controllable and reversible multielectron redox and acidic properties, but also due to their environmentally benign behavior.9,10 Moreover, these compounds have moderately high thermal stability in solid state, relatively simple synthesis procedure, and ability to form pseudo-liquid phases.11 In despite of the above advantages, there exist two major drawbacks in the catalytic systems involving pristine POMs: (i) low surface area in nonpolar solvents (<10 m2 g−1) hindering accessibility of reactants to active sites and (ii) high solubility in polar solvents producing recovery and reuse problems. To overcome these obstacles, many researchers have tried to design heterogeneous catalysts by incorporating HPAs into the structure of solid supports such as SiO2,12,13 Al2O3,14 activated carbon,15 TiO2,16 zeolites,17 organic materials,18,19 clays,20 ZrO2–CeO2 (ref. 21) and metal–organic frameworks (MOFs).22,23 However, most of these supports have some limitations, such as low loading of POM, high leaching of POM especially in polar medium and/or active sites that are unevenly dispersed. Therefore, finding suitable solid supports to overcome the drawbacks is important. Supports modified with functional groups such as carboxylic groups, lactam, amide, imide or amino-groups can allow solving this problem.24,25 Especially, the amino-modified metal oxides supports are commonly used for immobilizing POMs not only because of the exceptionally high stability and surface area but also because the amount and the basicity of anchored functional amino groups can be expected to be important in determining the guest–host interactions of the materials.26–30 This approach allows obtaining high dispersion of the POMs with minimal leaching. Despite facile recovery, such heterogeneous POM catalysts often suffer from the poor accessibility of the H2O2 during the oxidation of alcohols with aqueous H2O2 due to the hydrophobicity of support. Hence, enhancing the accessibility of the oxidant is crucial to the heterogeneous catalyst in the alcohols oxidation with H2O2.
Among various carbon-based nanomaterials, graphene oxide (GrO) has been proven as an effective support for the immobilization of inorganic and organic materials owing to its large theoretical specific surface area (∼2630 m2 g−1) and the presence of numerous oxygen containing functional groups on its surface.31–39 Due to these rich functional groups, GrO can be easily reacted with organic molecules to generate stable chemically functionalized GrO.40 In addition, in comparison with other carbonaceous nanomaterials, GrO may be more environmental friendly and have better biocompatibility.41 However, it is difficult to separate GrO from aqueous solutions using traditional filtration and centrifugation methods during and after the process due to its hydrophilic nature and small particle size which increase the cost of industrial application.42 The magnetic separation method is considered as a rapid and effective technique for separating nanomaterials from aqueous solution.43–45 Hence, magnetic graphene-based composites with large specific surface area and magnetic separation have begun to be used in the field of organic transformations.46–50
On the basis of the above discussions, in this work, amino-organosilane functionalized magnetic graphene oxide (Fe3O4/GrOSi(CH2)3–NH2) was synthesized by a facile method and used as a novel magnetic GrO-based support. Due to relatively high surface area and porosity, insolubility in water and easy magnetically separation, the Fe3O4/GrOSi(CH2)3–NH2 is an appropriate solid support to anchor Keggin-type PMo12O403− polyanion. The ternary magnetic nanocomposite material (abbreviated as Fe3O4/GrOSi(CH2)3–NH2/HPMo) was prepared by a simple acid–base electrostatic interaction between H3PMo12O40 and amino groups of the Fe3O4/GrOSi(CH2)3–NH2 support. The coupling of PMo12O403− anion with Fe3O4/GrOSi(CH2)3–NH2 could improve the surface area and avoid the dissolution of HPMo. This novel magnetically recyclable heterogeneous catalyst was used for selective oxidation of alcohols with H2O2 as a green oxidant under solvent free conditions. Our catalysts showed high catalytic performance in H2O2-mediated alcohol oxidations under solvent-free conditions. The resulting Fe3O4/GrOSi(CH2)3–NH2/HPMo composite could be used as a magnetically separable and efficient catalyst for alcohol oxidation under solvent free conditions.
2. Experimental
2.1 Materials
12-Molybdophosphoric acid (H3PMo12O40, 98%), graphite powder (C, 99.95%) and 3-aminopropyltriethoxysilane (APTES, 99%) were purchased from Merck Chemical Co. All alcohols and other chemicals were commercially purchased and used without further purification.2.2 Preparation of aminosilanized magnetic graphene oxide
Graphene oxide (GrO) was prepared by the modified Hummers method through the oxidation of graphite powder.51,52 Briefly, graphite powder (2.0 g) and NaNO3 (1.0 g) were mixed with 40 mL of concentrated H2SO4 in a 500 mL flask and stirrer for 1 hour in an ice bath. Then KMnO4 (6.0 g) was added into the vigorously stirred suspension slowly below 15 °C. The ice bath was then removed, and the mixture was stirred at room temperature until it slowly became a brownish slurry, and then it was diluted with 100 mL of water. The reaction temperature was rapidly increased to 98 °C with effervescence, and the color changed to brown. After that, 200 mL of water and 20 mL of H2O2 (30 wt%) were added. For purification, the mixture was centrifuged and washed with 10% HCl and then deionized water several times to remove the residual metal ions and acid. After centrifuging and drying at room temperature, GrO was obtained as a powder. To prepare Fe3O4/GrO, 0.25 g of GrO was dispersed in 90 mL water by sonication for 1 hour. Then, 0.84 g of (NH4)2Fe(SO4)2 and 2.08 g of (NH4)Fe(SO4)2 were added to the GrO dispersion and its pH was adjusted at 12 by adding 1 mol L−1 NaOH. The mixture was stirred at 50 °C for 2 h, filtered and washed with water and ethanol three times. The resulting solid was Fe3O/GrO. To prepare amino functionalized magnetic graphene oxide, 0.40 g of the as-synthesized Fe3O4/GrO dispersed in 50 mL of water, 150 mL ethanol and 5 mL of 3-aminopropyltriethoxysilane (APTES) were added to a round bottom flask. The mixture was stirred for 30 min in room temperature and then was refluxed at about 80 °C for 24 h. After the reaction, the solid was separated by a magnet and washed with ethanol to remove the unreacted APTES. The final product was dried at 80 °C in vacuum for 12 h to obtain the Fe3O4/GrOSi(CH2)3–NH2.2.3 Preparation of the Fe3O4/GrOSi(CH2)3–NH2/HPMo nanocomposite catalyst
1 g the as-prepared Fe3O4/GrOSi(CH2)3–NH2 was dispersed in 80 mL water and sonicated for 1 h. To the above suspension, 1 g HPMo in 200 mL ethanol was added and sonicated for another 1 h. The resulting mixture was stirred at room temperature for 24 h, and then filtered, washed with deionized water and ethanol three times to remove the unreacted HPMo. The final product was dried at 60 °C in open air to obtain magnetic Fe3O4/GrOSi(CH2)3–NH2/HPMo hybrid nanomaterial. According to the elemental analysis (ICP-AES) results and molecular weight of H3PMo12O40, the loading amount (wt%) of HPMo in Fe3O4/GrOSi(CH2)3–NH2/HPMo was estimated to be 31.5%.2.4 General procedure for oxidation of benzylic alcohols with H2O2 over the Fe3O4/GrOSi(CH2)3–NH2/HPMo nanocomposite
Benzylic alcohol (10 mmol) and catalyst Fe3O4/GrOSi(CH2)3–NH2/HPMo (0.2 g) were added to a 25 mL flask. Under refluxing conditions, vigorous stirring, and the heating temperature of 100 °C, the aqueous H2O2 (30 wt%, 15 mmol) was added into the above mixture within 5 min, then the reaction mixture was stirred for 4 h. The progress of the reaction was monitored by TLC and/or GC. After reaction, the solid catalyst was removed by an external magnet, and the liquid was analyzed using a gas chromatography (GC SP-6890) equipped with an FID detector and a capillary column (SE-54; internal diameter = 0.32 mm, length = 30 m) using He as the carrier gas. In the GC experiments, n-decane was used as internal standard and the yields were determined by using peak area. The isolated yield was obtained using silica gel plate or column chromatography with a mixture of ethyl acetate/n-hexane as an eluent, and the products was characterized by GC-MS. The recovered catalyst for recycling tests was obtained by magnetic separation, washing with ethanol and dried.2.5 Recyclability test of the catalyst
The activity of the recovered catalyst provides useful information about its stability during the catalytic cycle. In order to recover, the magnetic catalyst was separated from the reaction mixture by a magnet and washed three times with distilled water and ethanol. It was further dried at 100 °C for 2 h. The recovered catalyst was then used in the reaction with a fresh reaction mixture and products were analyzed after the reaction.2.6 Characterization techniques
The infrared spectra were recorded at room temperature on a Schimadzu FT-IR 160 spectrophotometer in the 4000–400 cm−1 region using KBr pellets. The XRD patterns of powder were recorded on a Rigaku D-max C III X-ray diffractometer using Ni-filtered Cu Kα radiation (λ = 1.54184 Å). The morphology of samples was studied by MIRA3 TESCAN scanning electron microscope equipped with energy dispersive X-ray analyzer (EDX) for the elemental analysis. Optical adsorption spectra were obtained from a Cary 100 Varian UV-Vis spectrophotometer in a wavelength range of 200–800 nm. The Brunauer–Emmett–Teller (BET) surface area was measured by N2 adsorption measurements at 77 K using a Nova 2000 instrument. The concentration of Mo in the composite was determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES, model OEC-730). The content of HPMo in the nanocomposite was determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES, model OEC-730). Magnetic measurements were carried out at room temperature using a vibrating sample magnetometer (VSM, Magnetic Daneshpajoh Kashan Co., Iran) with a maximum magnetic field of 10 kOe. Raman spectra were obtained using a Raman microscope (SENTERRA-2009, Germany) with Laser wavenumber of 785 nm at 785 nm. Thin-layer chromatography (TLC) was conducted on glass plates coated with silica gel GF254. GC-MS analysis was carried out on a Shimadzu QP 5050 GC-MS instrument with 5973 MS detector at an ionization voltage of 70 eV and equipped with a HP-INNOWAX capillary column (internal diameter = 0.25 mm, length = 30 m).3. Results and discussion
3.1 Characterization of the hybrid nanocatalyst
In this work, Keggin-type H3PMo12O40 (HPMo) was chemically immobilized onto 3-aminopropyl functionalized graphene oxide nanosheets decorated with magnetic Fe3O4 nanoparticles. The preparation process of Fe3O4/GrOSi(CH2)3–NH2/HPMo hybrid is illustrated in Fig. 1. These suggest that attaching organic bases on GrO might afford an efficient, reusable and environmentally benign base support for heteropoly acid such as HPMo. This novel magnetically recyclable hybrid nanomaterial was constructed by protonating amino groups anchored on the magnetic GrO nanosheets with Keggin-type heteropoly acid HPMo. The surface of GrOSi(CH2)3–NH2 is positively charged by the protonation of –NH2 groups by reacting with HPMo, which is benefit for anchoring polyanion via an electrostatic interaction. To confirm the successful construction of Fe3O4/GrOSi(CH2)3–NH2/HPMo hybrid, element analysis was employed. It reveals that the Fe3O4/GrOSi(CH2)3–NH2/HPMo contains 36.5% Mo, indicating that HPMo was anchored on GrO nanosheets. Along with the anchoring of HPMo on Fe3O4/GrOSi(CH2)3–NH2, the –NH2 groups can be protonated to form the –NH3+ groups, which absorbs the HPMo polyanions via the electrostatic (ionic) attraction interaction.53 And due to the electrostatic attraction, the hydrogen bonds between –NH3+ and HPMo polyanions molecules are reinforced.54,55 The structure and composition of the hybrid nanomaterial was further characterized by XRD, FT-IR, Raman spectra, EDX, SEM, VSM and BET surface area analyses. | ||
| Fig. 1 Preparation process of the magnetic Fe3O4/GrOSi(CH2)3–NH2/HPMo catalyst. | ||
The FT-IR spectra of synthesized materials are shown in Fig. 2. It is well known that Keggin-type polyoxometalate (PMo12O403−) contains a cluster of Mo(VI) ions linked by oxygen atoms with a tetrahedral phosphate group. PMo12O403− has four characteristic vibration bands as shown in Fig. 2(a): 1066 cm−1 for asymmetric stretch vibration of P–Oa (Oa corresponds to oxygen atom of tetrahedral phosphate group), 966 cm−1 for asymmetric stretch vibration of Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ot (Ot corresponds to the terminal oxygen atoms), 870 cm−1 for bending vibration of Mo–Ob–Mo (Ob corresponds to oxygen atom bridging the two tungsten atoms), and 786 cm−1 for bending vibration of Mo–Oc–Mo (Oc represents oxygen atom at the corners of the Keggin structure).56,57 The FT-IR spectrum of the composite sample is shown in Fig. 2(b). The characteristic absorption bands at 1053, 945, 875 and 798 cm−1 corresponding to the P–Oa, MoOt, Mo–Ob–Mo and Mo–Oc–Mo band vibrations confirm the presence of PMo12O403− cluster. The graphitic CC stretching band at about 1505 cm−1 and a strong band at about 588 cm−1 assigned to the grapheme nanosheets and Fe–O stretching vibration of the spinel-type Fe3O4 structure, respectively.58–60 And the presence of the anchored APTES chain to the GrO sheets was confirmed by stretching vibrations of C–H bond (2926 and 2850 cm−1), the stretching and bending modes of –NH2 bonds (3430 and 1630 cm−1) and the C–N stretching vibration (1223 cm−1).61 The Si–O–C stretching vibration (1116 cm−1) shows the successful grafting of APTES onto magnetic GrO nanosheet through covalent bonds. All bands appeared the IR spectrum of the hybrid nanomaterial demonstrate the coexistence of PMo12O403−, Fe3O4 and GrOSi(CH2)3–NH2 in the hybrid nanomaterial. The shift of some peaks of HPMo in the Fe3O4/GrOSi(CH2)3–NH2/HPMo compared to the parent HPMo can be attributed to hydrogen bonding and strong electrostatic attraction between negatively charged PMo12O403− and positively charged Fe3O4/GrOSi(CH2)3–NH3+ surface.62,63
Ot (Ot corresponds to the terminal oxygen atoms), 870 cm−1 for bending vibration of Mo–Ob–Mo (Ob corresponds to oxygen atom bridging the two tungsten atoms), and 786 cm−1 for bending vibration of Mo–Oc–Mo (Oc represents oxygen atom at the corners of the Keggin structure).56,57 The FT-IR spectrum of the composite sample is shown in Fig. 2(b). The characteristic absorption bands at 1053, 945, 875 and 798 cm−1 corresponding to the P–Oa, MoOt, Mo–Ob–Mo and Mo–Oc–Mo band vibrations confirm the presence of PMo12O403− cluster. The graphitic CC stretching band at about 1505 cm−1 and a strong band at about 588 cm−1 assigned to the grapheme nanosheets and Fe–O stretching vibration of the spinel-type Fe3O4 structure, respectively.58–60 And the presence of the anchored APTES chain to the GrO sheets was confirmed by stretching vibrations of C–H bond (2926 and 2850 cm−1), the stretching and bending modes of –NH2 bonds (3430 and 1630 cm−1) and the C–N stretching vibration (1223 cm−1).61 The Si–O–C stretching vibration (1116 cm−1) shows the successful grafting of APTES onto magnetic GrO nanosheet through covalent bonds. All bands appeared the IR spectrum of the hybrid nanomaterial demonstrate the coexistence of PMo12O403−, Fe3O4 and GrOSi(CH2)3–NH2 in the hybrid nanomaterial. The shift of some peaks of HPMo in the Fe3O4/GrOSi(CH2)3–NH2/HPMo compared to the parent HPMo can be attributed to hydrogen bonding and strong electrostatic attraction between negatively charged PMo12O403− and positively charged Fe3O4/GrOSi(CH2)3–NH3+ surface.62,63
 | ||
| Fig. 2 FT-IR spectra of (a) pure HPMo and (d) Fe3O4/GrOSi(CH2)3–NH2/HPMo. | ||
Fig. 3 displays the XRD patterns for Fe3O4 and Fe3O4/GrOSi(CH2)3–NH2/HPMo samples. All of the diffraction patterns in Fig. 3(a) and (b) are similar and can be indexed to the Fe3O4 phase (JCPDS no. 41-1488). No characteristic diffraction peaks of the HPMo appeared which presumably was due to the low content incorporation of HPMo. Also, this result implies that the Keggin unit homogeneously disperses into the GrO nanosheets, which will be benefit to enhance the catalytic activity of the hybrid nanomaterial. Moreover, no characteristic diffraction peaks for GrO are observed in the pattern indicating that the GrO nanosheets do not stack during the synthesis process. The reason can be attributed to that the Fe3O4 nanoparticles, aminopropyl groups and HPMo anchored on the surfaces of GrO prevent the exfoliated GrO nanosheets from restacking.64 However, a broad peak at about 23.5° corresponding to the reduced GrO was appeared, indicating that GrO nanosheets were reduced to graphene during the functionalization process.65
 | ||
| Fig. 3 XRD patterns of (a) pure Fe3O4 and (b) Fe3O4/GrOSi(CH2)3–NH2/HPMo. | ||
Fig. 4(a) and (b) presents the Raman spectra of pure GrO and Fe3O4/GrOSi(CH2)3–NH2/HPMo nanocomposite. Two samples display the characteristic D (sp3 carbon atoms of disorders and defects) and G (sp2 carbon atoms in graphitic sheets) bands of GrO structure.66 As compared with the D and G bands of pure GrO (D, 1318 cm−1; G, 1590 cm−1), the two characteristic bands of GrO in the Fe3O4/GrOSi(CH2)3–NH2/HPMo nanocomposite shift to D, 1311 cm−1 and G, 1592 cm−1, suggesting the successful functionalization of GrO with APTES and HPMo. These shifts in Raman peaks can be attributed to strong interaction between HPMo polyanion and positively charged Fe3O4/GrOSi(CH2)3–NH3+ in the Fe3O4/GrOSi(CH2)3–NH2/HPMo hybrid,67 which is consistent with the FI-IR analysis. Comparing with pristine GrO, the ratio of D and G peaks of Fe3O4/GrOSi(CH2)3–NH2/HPMo becomes higher, suggesting a higher level of disorder of the graphene layers during the functionalization process.68 The peaks of Keggin structure have not been appeared, which indicated that HPMo species on Fe3O4/GrOSi(CH2)3–NH2 sample were in a high-dispersed state. This result was identical to the XRD results.
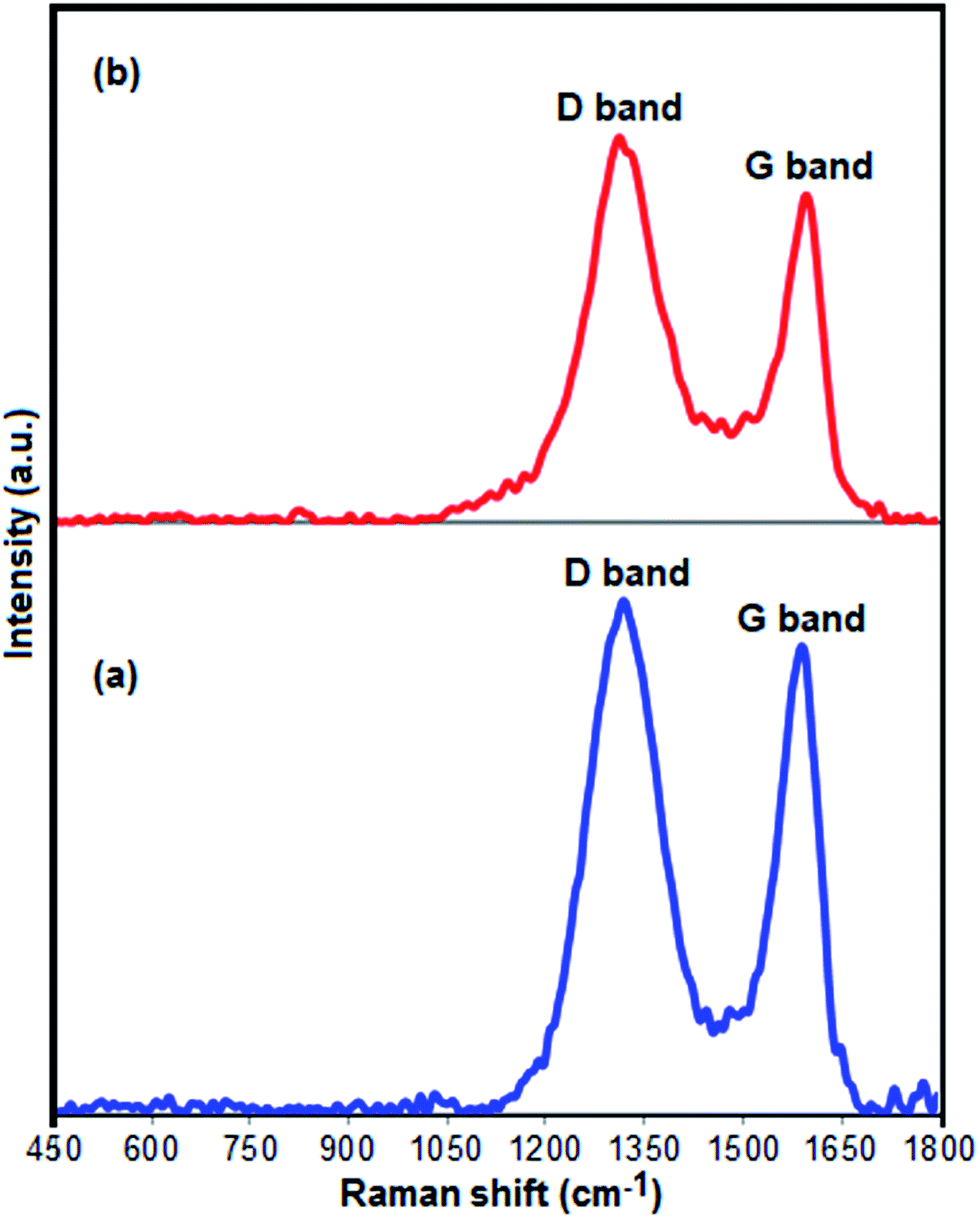 | ||
| Fig. 4 Raman spectra of (a) GO and (b) Fe3O4/GrOSi(CH2)3–NH2/HPMo. | ||
SEM images indicating the microstructural features of GO and Fe3O4/GrOSi(CH2)3–NH2/HPMo nanocomposite are shown in Fig. 5. SEM image of pure GO in Fig. 5(a) shows highly porous and layered structure of GO having large stacks, possibly consisting of hundreds of grapheme oxide nanosheets. It should also be noted that the surfaces of the GO sheets are quite flat and smooth. The SEM image of Fe3O4/GrOSi(CH2)3–NH2/HPMo in Fig. 5(b) and (c) clearly shows graphene oxide nanosheets were successfully decorated with agglomerated Fe3O4 and HPMo particles that completely covered the surfaces of large graphene sheets. From images, it can be clearly seen that the Fe3O4 nanoparticles with a size of about 15–20 nm were well deposited on GO nanosheets which were a flexible interleaved structure. Some wrinkles are found on the surface, which may be important for preventing aggregation of GO nanosheets and maintaining high surface area. All the micrographs of the nanocomposite clearly indicated that the surface properties of modified GO product were strongly affected. In opposite of pure GO, the surfaces of GO nanosheets in the nanocomposite are rough, and the edges are highly crumpled. Thus, the Fe3O4/GrOSi(CH2)3–NH2/HPMo could provide a rough and coarse surface with high porosity for catalytic uses. The results are in good agreement with BET results. The composition of the as-prepared Fe3O4/GrO–NH2/HPMo hybrid nanomaterial was investigated by energy dispersive X-ray spectroscopy (EDX). Fig. 5(d) shows EDX spectrum of the hybrid nanocomposite. The EDX elemental spectrum shows the existence of C, N, Si, O, Fe, P, and Mo elements in the composite. The elements of P and Mo are from HPMo and the results further confirm that the HPMo particles have been successfully supported on the surface of the Fe3O4/GrOSi(CH2)3–NH2.
 | ||
| Fig. 5 SEM images of (a) GO, (b) and (c) Fe3O4/GrOSi(CH2)3–NH2/HPMo nanocomposite at different magnification and (d) EDX spectrum of the Fe3O4/GrO–NH2/HPMo nanocomposite. | ||
The magnetic properties of the Fe3O4 and Fe3O4/GrOSi(CH2)3–NH2/HPMo samples were investigated by vibrating sample magnetometry (VSM) at room temperature. The magnetic hysteresis loops are depicted in Fig. 6. It is clear that both Fe3O4 and the Fe3O4/GrOSi(CH2)3–NH2/HPMo composite are soft-magnetic materials due to their coercivity (Hc) of zero.69 Moreover, they are also superparamagnetic materials, as their magnetic hysteresis loops passed through the origin of the coordinates. The saturation magnetization values of Fe3O4 and Fe3O4/GrOSi(CH2)3–NH2/HPMo are 27.50 and 9.15 emu g−1, respectively. The saturation magnetization of the magnetic nanocomposite decreases by approximately 75% compared with that of pure Fe3O4, which can be attributed to the nano-magnetic components (GrO and HPMo) in the composite sample. However, the saturation magnetization of the composite could satisfy the requirements of easy separation in the suspension solution using an extra magnet after reaction as shown in the inset of Fig. 6. Thus, the Fe3O4/GrOSi(CH2)3–NH2/HPMo composite can be easily separated using a magnetic separation process after being used for the reaction.
 | ||
| Fig. 6 Magnetic hysteresis loop of (a) Fe3O4 and (b) Fe3O4/GrOSi(CH2)3–NH2/HPMo at room temperature. The inset shows the behavior of the nanocomposite under an external magnetic field. | ||
N2 adsorption/desorption measurements were performed to investigate specific surface area and the pore size distribution of the Fe3O4/GrOSi(CH2)3–NH2/HPMo. As can be seen in Fig. 7, the nitrogen adsorption isotherm is a typical IV type curve with an fine H1-type hysteresis loop in the range of ca. 0.8–1.0p/p0, indicating the existence of mesoporous structure.70,71 The BET surface area is measured to be 76.36 m2 g−1 that is much higher than the value of pure HPMo (≤10 m2 g−1).72 In addition, the total pore volume is 0.01 cm3 g−1 and according to the corresponding Barrett–Joyner–Halenda (BJH) pore size distribution curve in the inset of Fig. 7, the pore size distribution of the Fe3O4/GrOSi(CH2)3–NH2/HPMo shows peak centered at around 2.28 nm. Such porosity of Fe3O4/GrOSi(CH2)3–NH2/HPMo composite can improve the catalytic performance. It can be concluded that immobilization of HPMo on the surface of magnetic Fe3O4/GrOSi(CH2)3–NH2 increases its surface area and porosity which they are useful factors for improving the catalytic performance.
 | ||
| Fig. 7 N2 adsorption–desorption isotherm of Fe3O4/GrOSi(CH2)3–NH2/HPMo. The inset shows the pore size distribution plot. | ||
3.2 Catalytic oxidation of alcohols over Fe3O4/GrOSi(CH2)3–NH2/HPMo nanocomposite
The oxidation of benzyl alcohol to obtain benzaldehyde was used as a typical benchmark reaction to verify the catalytic activity of the as-prepared magnetic Fe3O4/GrOSi(CH2)3–NH2/HPMo nanocomposite (see Scheme 1). In an initial experiment, a heterogeneous mixture of benzyl alcohol (10 mmol), aqueous H2O2 (30%, 15 mmol) and a catalytic amount of Fe3O4/GrOSi(CH2)3–NH2/HPMo (0.2 g) was heated for 4 h at 100 °C, the benzaldehyde was formed as the only product in 90% yield (Scheme 1). In control experiments, when pure Fe3O4 and GrO support were used as heterogeneous catalysts for the oxidation of benzyl alcohol, the results showed that about 15% and 18% of benzaldehyde was formed, respectively, after 4 h heating at 100 °C under the same reaction conditions, suggesting that the catalytic activity of the Fe3O4/GrOSi(CH2)3–NH2/HPMo system is mainly due to HPMo heteropoly acid. Using pure and unsupported HPMo, we found that 45% of benzyl alcohol was oxidized to benzaldehyde within 4 h under the same conditions. These findings confirm that the activity of Fe3O4/GrOSi(CH2)3–NH2/HPMo nanocomposite is higher than that of the pure HPMo cluster. This is due to (i) higher specific surface area of composite than that of the starting POM cluster and (ii) the synergistic effect of the HPMo unit and the GO nanosheets support. No noticeable oxidation products were observed when blank experiments were run in the in the absence of Fe3O4/GrOSi(CH2)3–NH2/HPMo or H2O2. | ||
| Scheme 1 | ||
 | ||
| Fig. 8 The conversion of benzyl alcohol and the selectivity to benzaldehyde at different catalyst dosage. Conditions: benzyl alcohol (10 mmol), H2O2 (15 mmol, 30%), catalyst (0.20 g) at reflux temperature 100 °C for 4 h. | ||
 | ||
| Fig. 9 The conversion of benzyl alcohol and the selectivity to benzaldehyde at different reaction time over Fe3O4/GrOSi(CH2)3–NH2/HPMo as the catalyst. Conditions: benzyl alcohol (10 mmol), H2O2 (15 mmol, 30%), catalyst (0.20 g) at 100 °C. | ||
 | ||
| Fig. 10 The conversion of benzyl alcohol and the selectivity to benzaldehyde over the Fe3O4/GrOSi(CH2)3–NH2/HPMo catalyst with different amount of H2O2 (30%). Conditions: benzyl alcohol (10 mmol), catalyst (0.20 g) at 100 °C for 4 h. | ||
 | ||
| Fig. 11 Effect of temperature on the conversion of benzyl alcohol using the Fe3O4/GrOSi(CH2)3–NH2/HPMo catalyst. Conditions: benzyl alcohol (10 mmol), H2O2 (15 mmol, 30%), catalyst (0.20 g) at 100 °C for 4 h. | ||
Thus, optimized conditions for oxidation of benzyl alcohol (90% conversion, 100% selectivity of benzaldehyde) as a model reaction are as follows: amount of the catalyst (0.2 g), temperature (100 °C), H2O2 oxidant to alcohol ratio (1.5) and reaction time (4 h).
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yieldb (%) |
| a Reaction conditions: alcohol (10 mmol), catalyst (0.2 g), H2O2 (15 mmol, 30 wt%), 100 °C under solvent-free conditions for 4 h.b Yields were determined by GC analysis with n-decane as internal standard. | |||
| 1 | 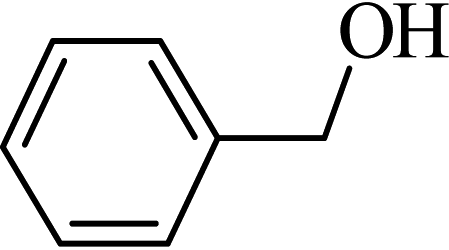 |
 |
90 |
| 2 |  |
 |
96 |
| 3 |  |
 |
94 |
| 4 |  |
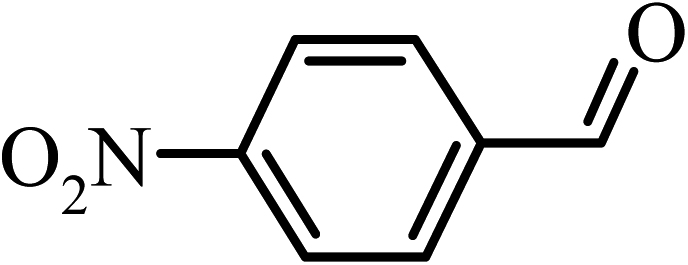 |
86 |
| 5 |  |
 |
60 |
| 6 |  |
 |
82 |
| 7 |  |
 |
92 |
| 8 |  |
 |
94 |
| 9 |  |
 |
82 |
| 10 |  |
 |
90 |
| 11 |  |
 |
90 |
| 12 |  |
 |
90 |
| 13 |  |
 |
90 |
| 14 |  |
 |
88 |
| 15 |  |
 |
65 |
| 16 |  |
 |
65 |
| 17 |  |
 |
60 |
| 18 |  |
 |
68 |

The experiments, usually performed on a 10 mmol scale, can be scaled up to 100 mmol without difficulties. A 50 mmol reaction of 4-methoxybenzyl alcohol provided the corresponding aldehyde in a 92% yield and a 100 mmol reaction of 1-phenylethanol gave acetophenone in an 88% yield.
 | ||
| Fig. 12 Recyclability of the Fe3O4/GrOSi(CH2)3–NH2/HPMo catalyst. Conditions: benzyl alcohol (10 mmol), H2O2 (15 mmol, 30%), catalyst (0.20 g) at 100 °C for 4 h. Yields are for isolated pure benzaldehyde. | ||
The stability of this material is further discussed. As shown in Fig. 13(a)–(c), the XRD pattern, FTIR and Raman spectrum of the recovered catalyst after fourth run are consistent with those of the fresh catalyst (see Fig. 2(b), 3(b) and 4(b)). These observations confirm that the structure of the Fe3O4/GrOSi(CH2)3–NH2/HPMo catalyst is stable under the reaction conditions and is not affected by the reactants. The morphology of the recycled catalyst particles was also analyzed. Fig. 13(d) shows catalyst almost kept initial size and morphology even after four runs (see Fig. 5(b) and (c)). The surface of GO nanosheets is still decorated with spherical Fe3O4 and white HPMo particles revealing the strong binding between the Fe3O4 nanoparticles and HPMo with amino functionalized GO nanosheets. Considering the above mentioned experiment results, we can conclude that the structure of the catalyst remains intact, which further confirms its excellent stability and recyclability.
 | ||
| Fig. 13 (a) XRD pattern, (b) FT-IR spectrum, (c) Raman spectrum and (d) SEM image of the recovered Fe3O4/GrOSi(CH2)3–NH2/HPMo nanocomposite after fourth run. | ||
4. Conclusion
In conclusion, the Fe3O4/GrOSi(CH2)3–NH2/HPMo nanocomposite was prepared and tested as a novel magnetically separable oxidation catalyst under solvent free conditions. Selective oxidation of benzyl alcohol to benzaldehyde was used as a benchmark reaction to evaluate the catalytic performance of the as-prepared catalyst. Under the optimized conditions, H2O2-oxidation of a wide range of alcoholic substrates gave the desired carbonyl compounds in moderate to excellent yields. This reaction is highly selective (≥99%) for benzylic alcohols. In the oxidation of benzylic alcohols, strong electron-withdrawing substituents induced lower yields than others. This indicates that carbocation-type intermediates are involved in the catalytic oxidation reaction due to hydride elimination from the beta proton of substrate. In addition, it is easy to separate by an external magnet and reuse the catalyst for another catalytic recycling. The catalytic performances of the re-used catalyst, even for using four times, were comparable with that of the fresh catalyst, giving this catalyst a good prospect for benzaldehyde production in industry. Also, the present work provides a new type of heterogeneous catalytic materials for selective organic transformations. Work on the detailed mechanism of this catalytic reaction is under investigation in our laboratory.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial supports from the Lorestan University Research Council and Iran Nanotechnology Initiative Council (INIC) are gratefully acknowledged.References
- G. Tojo and M. I. Fernández, Oxidation of alcohols to aldehydes and ketones, Springer, New York, 2006 Search PubMed
.
- N. Lingaiah, K. M. Reddy, N. S. Babu, K. N. Rao, I. Suryanarayana and P. S. Prasad, Catal. Commun., 2006, 7, 245–250 CrossRef CAS
- S. Rana, S. Maddila, R. Pagadala, K. Parida and S. B. Jonnalagadda, Catal. Commun., 2015, 59, 73–77 CrossRef CAS
- R. Borthakur, M. Asthana, M. Saha, A. Kumar and A. K. Pal, RSC Adv., 2014, 4, 21638–21643 RSC
- V. Choudhary, D. Dumbre, B. Uphade and V. Narkhede, J. Mol. Catal. B: Enzym., 2004, 215, 129–135 CrossRef CAS
- V. Mahdavi, M. Mardani and M. Malekhosseini, Catal. Commun., 2008, 9, 2201–2204 CrossRef CAS
- M. Musawir, P. N. Davey, G. Kelly and I. V. Kozhevnikov, Chem. Commun., 2003, 12, 1414–1415 RSC
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS PubMed
- Y. F. Song and R. Tsunashima, Chem. Soc. Rev., 2012, 41, 7384–7402 RSC
- I. Kozhevnikov, Catalysts for fine chemical synthesis, catalysis by polyoxometalates, Wiley, Chichester, 2002 Search PubMed
- N. Mizuno, K. Yamaguchi and K. Kamata, Coord. Chem. Rev., 2005, 249, 1944–1956 CrossRef CAS
- H. Zhao, L. Zeng, Y. Li, C. Liu, B. Hou, D. Wu, N. Feng, A. Zheng, X. Xie and S. Su, Microporous Mesoporous Mater., 2013, 172, 67–76 CrossRef CAS
- Y. Chen, F. Zhang, Y. Fang, X. Zhu, W. Zhen, R. Wang and J. Ma, Catal. Commun., 2013, 38, 54–58 CrossRef CAS
- J. L. García-Gutiérrez, G. A. Fuentes, M. E. Hernández-Terán, F. Murrieta, J. Navarrete and F. Jiménez-Cruz, Appl. Catal., A, 2006, 305, 15–20 CrossRef
- J. Xiao, L. Wu, Y. Wu, B. Liu, L. Dai, Z. Li, Q. Xia and H. Xi, Appl. Energy, 2014, 113, 78–85 CrossRef CAS
- A. Micek-Ilnicka, E. Bielańska, L. Lityńska-Dobrzyńska and A. Bielański, Appl. Catal., A, 2012, 421, 91–98 CrossRef
- K. Zhu, J. Hu, X. She, J. Liu, Z. Nie, Y. Wang, C. H. Peden and J. H. Kwak, J. Am. Chem. Soc., 2009, 131, 9715–9721 CrossRef CAS PubMed
- H. Kim, J. C. Jung, S. H. Yeom, K. Y. Lee and I. K. Song, J. Mol. Catal. A: Chem., 2006, 248, 21–25 CrossRef CAS
- G. Morales, R. Van Grieken, A. Martín and F. Martínez, Chem. Eng. J., 2010, 161, 388–396 CrossRef CAS
- S. K. Bhorodwaj and D. K. Dutta, Appl. Catal., A, 2010, 378, 221–226 CrossRef CAS
- S. Mallick, S. Rana and K. Parida, Ind. Eng. Chem. Res., 2012, 51, 7859–7866 CrossRef CAS
- N. V. Maksimchuk, K. A. Kovalenko, S. S. Arzumanov, Y. A. Chesalov, M. S. Melgunov, A. G. Stepanov, V. P. Fedin and O. A. Kholdeeva, Inorg. Chem., 2010, 49, 2920–2930 CrossRef CAS PubMed
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Chem. Commun., 2017, 53, 10851–10869 RSC
- M. Kooti and E. Nasiri, J. Mol. Catal. A: Chem., 2015, 406, 168–177 CrossRef CAS
- M. Kooti and M. Afshari, Mater. Res. Bull., 2012, 47, 3473–3478 CrossRef CAS
- M. Masteri-Farahani, J. Movassagh, F. Taghavi, P. Eghbali and F. Salimi, Chem. Eng. J., 2012, 184, 342–346 CrossRef CAS
- S. Li, S. R. Zhai, J. M. Zhang, Z. Y. Xiao, Q. D. An, M. H. Li and X. W. Song, Eur. J. Inorg. Chem., 2013, 31, 5428–5435 CrossRef
- J. Zhang, S. Zhai, S. Li, Z. Xiao, Y. Song, Q. An and G. Tian, Chem. Eng. J., 2013, 215, 461–471 CrossRef
- H. Hamadi, M. Kooti, M. Afshari, Z. Ghiasifar and N. Adibpour, J. Mol. Catal. A: Chem., 2013, 373, 25–29 CrossRef CAS
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS PubMed
- A. Tayyebi and M. Outokesh, RSC Adv., 2016, 6, 13898–13913 RSC
- M. Yusuf, F. Elfghi, S. A. Zaidi, E. Abdullah and M. A. Khan, RSC Adv., 2015, 5, 50392–50420 RSC
- Z. Cheng, J. Liao, B. He, F. Zhang, F. Zhang, X. Huang and L. Zhou, ACS Sustainable Chem. Eng., 2015, 3, 1677–1685 CrossRef CAS
- J. Li, C. Chen, R. Zhang and X. Wang, Sci. China: Chem., 2016, 59, 150–158 CrossRef CAS
- G. Zhao, L. Jiang, Y. He, J. Li, H. Dong, X. Wang and W. Hu, Adv. Mater., 2011, 23, 3959–3963 CrossRef CAS PubMed
- J. Wang, Z. Chen and B. Chen, Environ. Sci. Technol., 2014, 48, 4817–4825 CrossRef CAS PubMed
- H. Wang, X. Yuan, G. Zeng, Y. Wu, Y. Liu, Q. Jiang and S. Gu, Adv. Colloid Interface Sci., 2015, 221, 41–59 CrossRef CAS PubMed
- H. Wang, X. Yuan, Y. Wu, H. Huang, X. Peng, G. Zeng, H. Zhong, J. Liang and M. Ren, Adv. Colloid Interface Sci., 2013, 195, 19–40 CrossRef PubMed
- H. Wang, X. Yuan, Y. Wu, H. Huang, G. Zeng, Y. Liu, X. Wang, N. Lin and Y. Qi, Appl. Surf. Sci., 2013, 279, 432–440 CrossRef CAS
- X. Gong, G. Liu, Y. Li, D. Y. W. Yu and W. Y. Teoh, Chem. Mater., 2016, 28, 8082–8118 CrossRef CAS
- H. Chen, B. Gao and H. Li, J. Hazard. Mater., 2015, 282, 201–207 CrossRef CAS PubMed
- X. Deng, L. Lü, H. Li and F. Luo, J. Hazard. Mater., 2010, 183, 923–930 CrossRef CAS PubMed
- S. Zhang, H. Li, Z. Wang, J. Liu, H. Zhang, B. Wang and Z. Yang, Nanoscale, 2015, 7, 8495–8502 RSC
- Z. Zhang, F. Zhang, Q. Zhu, W. Zhao, B. Ma and Y. Ding, J. Colloid Interface Sci., 2011, 360, 189–194 CrossRef CAS PubMed
- X. Xin, Q. Wei, J. Yang, L. Yan, R. Feng, G. Chen, B. Du and H. Li, Chem. Eng. J., 2002, 184, 132–140 CrossRef
- M. Liu, T. Wen, X. Wu, C. Chen, J. Hu, J. Li and X. Wang, Dalton Trans., 2013, 42, 14710–14717 RSC
- J. Zhu, S. Wei, H. Gu, S. B. Rapole, Q. Wang, Z. Luo, N. Haldolaarachchige, D. P. Young and Z. Guo, Environ. Sci. Technol., 2012, 46, 977–998 CrossRef CAS PubMed
- Y. Liu, C. Luo, G. Cui and S. Yan, RSC Adv., 2015, 5, 54156–54164 RSC
- X. Bai, R. Feng, Z. Hua, L. Zhou and H. Shi, Environ. Eng. Sci., 2015, 32, 370–378 CrossRef CAS
- F. Gu, M. Liang, D. Han and Z. Wang, RSC Adv., 2015, 5, 39964–39972 RSC
- Y. Yao, S. Miao, S. Liu, L. P. Ma, H. Sun and S. Wang, Chem. Eng. J., 2012, 184, 326–332 CrossRef CAS
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771–778 CrossRef CAS
- L. Zhao, Y. Chi, Q. Yuan, N. Li, W. Yan and X. Li, J. Colloid Interface Sci., 2013, 390, 70–77 CrossRef CAS PubMed
- L. Qin, Y. Zheng, D. Li, Y. Zhou, L. Zhang and Z. Zuhra, Fuel, 2016, 181, 827–835 CrossRef CAS
- Y. Fu and X. Wang, Ind. Eng. Chem. Res., 2011, 50, 7210–7218 CrossRef CAS
- J. C. Juan, J. Zhang and M. A. Yarmo, J. Mol. Catal. A: Chem., 2007, 267, 265–271 CrossRef CAS
- C. Rocchiccioli-Deltcheff, M. Fournier, R. Franck and R. Thouvenot, Inorg. Chem., 1983, 22, 207–216 CrossRef CAS
- L. Guo, P. Ye, J. Wang, F. Fu and Z. Wu, J. Hazard. Mater., 2015, 298, 28–35 CrossRef CAS PubMed
- Q. Zhao, D. Chen, Y. Li, G. Zhang, F. Zhang and X. Fan, Nanoscale, 2013, 5, 882–885 RSC
- J. Huang, S. Ding, W. Xiao, Y. Peng, S. Deng and N. Zhang, Catal. Lett., 2015, 145, 1000–1007 CrossRef CAS
- S. K. Singh, M. K. Singh, P. P. Kulkarni, V. K. Sonkar, J. J. Grácio and D. Dash, ACS Nano, 2012, 6, 2731–2740 CrossRef CAS PubMed
- H. Zhang, A. Xie, Y. Shen, L. Qiu and X. Tian, Phys. Chem. Chem. Phys., 2012, 14, 12757–12763 RSC
- D. Zhou and B. H. Han, Adv. Funct. Mater., 2010, 20, 2717–2722 CrossRef CAS
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I. C. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979–3986 CrossRef CAS PubMed
- G. H. Moon, Y. Park, W. Kim and W. Choi, Carbon, 2011, 49, 3454–3462 CrossRef CAS
- Y. Sun, D. Shao, C. Chen, S. Yang and X. Wang, Environ. Sci. Technol., 2013, 47, 9904–9910 CrossRef CAS PubMed
- Y. X. Ma, Y. F. Li, G. H. Zhao, L. Q. Yang, J. Z. Wang, X. Shan and X. Yan, Carbon, 2012, 50, 2976–2986 CrossRef CAS
- S. Alwarappan, A. Erdem, C. Liu and C. Z. Li, J. Phys. Chem. C, 2009, 113, 8853–8857 CAS
- W. Zhang, M. Wang, W. Zhao and B. Wang, Dalton Trans., 2013, 42, 15464–15474 RSC
- X. Tian, C. Cheng, L. Qian, B. Zheng, H. Yuan, S. Xie, D. Xiao and M. M. Choi, J. Mater. Chem., 2012, 22, 8029–8035 RSC
- H. Pang, Y. Ma, G. Li, J. Chen, J. Zhang, H. Zheng and W. Du, Dalton Trans., 2012, 41, 13284–13291 RSC
- S. Zhang, G. Zhao, S. Gao, Z. Xi and J. Xu, J. Mol. Catal. A: Chem., 2008, 289, 22–27 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2018 |