
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUp-conversion white emission and other luminescence properties of a YAG:Yb2O3·Tm2O3·Ho2O3@SiO2 glass-nanocomposite†
Andrzej M. Kłonkowski *a,
Benedykt Kuklińskib,
Mariusz Kubusc,
Jacek Ryld,
Karol Szczodrowskib,
Dorota Wileńskaa and
H.-Jürgen Meyerc
*a,
Benedykt Kuklińskib,
Mariusz Kubusc,
Jacek Ryld,
Karol Szczodrowskib,
Dorota Wileńskaa and
H.-Jürgen Meyerc
aFaculty of Chemistry, University of Gdańsk, Wita Stwosza, 80-308 Gdańsk, Poland. E-mail: andrzej.klonkowski@ug.edu.pl
bInstitute of Experimental Physics, University of Gdańsk, Wita Stwosza, 80-308 Gdańsk, Poland
cInstitute of Inorganic Chemistry, University of Tuebingen, Auf der Morgenstelle 18, 72076 Tuebingen, Germany
dChemical Faculty, Gdańsk University of Technology, G. Narutowicza 11/12, 80-233 Gdańsk, Poland
First published on 20th March 2018
Abstract
We report on a glass-nanocomposite material consisting of yttrium aluminum garnet (Y3Al5O12, YAG) nanocrystals co-doped with Yb3+, Tm3+ and Ho3+ ions as well as entrapped into a SiO2 xerogel. This 94YAG·5Yb2O3·0.8Tm2O3·0.2Ho2O3@SiO2 (abbr. YAG:YbTmHo@SiO2) nanocomposite material has been prepared by sol–gel procedure. Its structure and morphology has been characterized by means of X-ray diffraction (XRD) and scanning electron microscope (SEM) techniques as well as energy dispersive X-ray (EDX), X-ray photoelectron (XPS) and luminescence spectroscopies. The luminescent glass-nanocomposite exhibited an up-conversion effect under λexc = 980 nm and emission when excited under 355 nm in steady-state conditions. Then time-resolved luminescence emission was observed, when the sample was excited at 290 and 355 nm by a pulse laser. Average decay times for the SiO2 matrix and for some transitions of the Tm3+ and Ho3+ dopants present in the YAG:YbTmHo@SiO2 material have been evaluated. The luminescent nanocomposite when excited under 290 or 355 nm wavelengths in both conditions emits blue light. However, the nanocomposite is promising as a single-source white-light phosphor owing to its up-conversion luminescence under 980 nm excitation. Such optical features make the studied material an alternative phosphor.
Introduction
Up-conversion is an anti-Stokes process in which a near infrared photon is converted into visible or even ultraviolet light owing to multi-photon action. Among various near infrared excitation lasers, the 980 nm one is the most commonly used excitation source generating up-conversion emission.1 Actually there are many up-conversion processes with widely different conversion efficiencies. The processes require energy levels which are resonant with the incoming or outgoing radiation. Lanthanide(III) ions (Ln3+) are very suitable as the emitting centers owing to their numerous electronic levels and narrow emission bands. Among the Ln(III) ions Ho3+ can be applied to obtain red and green up-conversion luminescence and Tm3+ acts as a blue emitting activator under infrared (980 nm) excitation. While, Yb3+ can act as the sensitizer enhancing the emission efficiency due to appropriate structure of energy levels and longer decay lifetime of the excited states.2The efficiency of the system depends strongly on the choice of the host matrix. It is known that oxides are less suitable than fluorides, since lifetimes in the former are shorter than in the latter due to stronger interaction between the luminescent ion and its coordination environment. Since in the up-conversion oxide material co-doped, e.g. with Yb3+ and Er3+ the lifetime of the intermediary 4I11/2 (Er3+) is shorter, in consequence the total efficiency of the up-conversion process will be lower, in contrast to fluoride material.3 Irrespective of this fact there are many papers devoted to up-conversion in oxide materials, among them also works considering yttrium aluminum garnet (e.g. ref. 2 and 4–11).
Yttrium aluminum garnet (YAG) is a prominent crystalline material of the garnet group. The choice of YAG relies on this material exhibiting exceptional optical transparency, having a cubic crystallographic structure that can well accommodate such dopants as lanthanide ions.12,13 YAG nanocrystals doped with Ln3+ ions were prepared using many different methods.7,14,15
A nanocomposite is defined as a material consisting of at least two different solid phases. In our case glassy silica as the host phase of larger extent plays a matrix role. Then the nano-sized YAG guest phase doped with Ln(III) ions is dispersed into the matrix. Obviously, the constituent phases of the composite are separated by distinct interface. In the nanocomposites, the guest nanoparticles have dimension in the range 1–100 nm. In this type of nanocomposite materials glass is an excellent host owing to its several inherent superior properties compared to those of other encapsulating hosts. There are many types of glass-nanocomposites depending on the compositions, sizes and shapes of the nano-guests as well as processing parameters.16
One of the encapsulating methods used in nanocomposite preparation is sol–gel processing. This method involves the generation of colloidal suspensions (sols) which are subsequently converted to viscous gels and then to rigid xerogels.17 Thus, by sol–gel procedure crystalline particles can be immobilized in a solid xerogel. Wide range of inorganic and hybrid organic–inorganic composite materials share this common preparation procedure. In contrast to bulk materials prepared by the melting method, sol–gel materials can be obtained as thin films and coatings. If these materials are transparent, then they gain a competitive advantage with respect to bulk glasses and crystals. It is important especially in glass-nanocomposite luminescent materials doped with such luminescent species as Ln3+ ions, where the nanosize of the Ln3+ doped crystals is crucial to avoid light scattering. The nanocrystals play important role, when activated by lanthanide ions. The presence of the crystalline environment around the lanthanide ion allows high absorption and emission cross sections as well as tailoring of the ion–ion interaction by the control of the lanthanide ion partition. This last point is crucial and still an object of intense experimental and theoretical studies.
The integration of luminescent nanocrystals into vitreous matrix can lead to new optical nanocomposites. There is growing interest to integrate up-conversion nanocrystals with an optical matrix. Not only glasses but also rigid vitreous xerogels can serve as indispensable optical matrix materials owing to their high optical transparency, thermomechanical strength and the ability (especially in the case of xerogels) to be shaped into an almost unlimited range of geometrical structures.18 However, it remains a preparation challenge to integrate up-conversion nanoparticles with tailored nanophotonic properties into a vitreous matrix to create a new hybrid optical material. Wet chemistry is the satisfied synthetic technique in the case of up-conversion nanocrystals because it enables control over crystallite phase, size, shape, composition and nanostructure.19–21 This control gives a clear advantage over in situ glass ceramic procedure by synthesizing nanocrystals and glass separately, and then integrate them into a hybrid material. This preparation method can overcome the limitations of the glass-ceramic processing. Among the limitations is an extremely high risk of completely dissolving the nanosized crystals at higher temperatures.
Up-conversion nanocrystals containing thousands of Ln3+ ions emit high brightness up-conversion by utilizing high-irradiance excitation to enrich the effective emitters19,20,22 or clustering Yb3+ sensitizers in arrays at sublattice level to promote localized excited states.23
In our study glass-nanocomposites consisting of the Ln3+ co-doped YAG nanocrystals incorporated into glassy silica matrix by sol–gel procedure were synthesized. We demonstrate that this approach allows incorporation of a controlled amount of YAG:Ln up-conversion nanocrystals with well-defined concentrations of Ln3+ ions into amorphous xerogel. The vitreous materials doped with up-conversion nanocrystals exhibit high transparency and identical glassy xerogel structure as a blank silica xerogel. Aim of the study is to compare luminescence of the glass nanocomposite excited in different conditions, viz. in steady-state (λexc = 355 and 980 nm) or by pulse laser (λexc = 290 and 355 nm) spectroscopy.
Experimental
Synthetic procedures
Pure Y3Al5O12 (YAG) and co-doped 94Y3Al5O12·6Ln2O3 (in mol%) materials (abbr. YAG:Ln, where Ln = Yb, Tm and Ho), were synthesized by glycol modification of the sol–gel procedure after Veith et al.24 The pure YAG as well as doubly co-doped 94YAG·5Yb2O3·1Ho2O3 (abbr. YAG:YbHo) and triply co-doped 94YAG·5Yb2O3·0.8Tm2O3·0.2Ho2O3 (abbr. YAG:YbTmHo) agglomerated crystals were powdered in an agate mortar and then pulverized in methanol slurry to nanosize particles by an ultrasonic technique for 3 h.25 The chemicals for the doped YAG synthesis such as yttrium trioxide (Y2O3), aluminum nitrate [Al(NO3)3·5H2O] and appropriate lanthanide salts Ln(NO3)3·5H2O (where Ln = Yb, Tm and Ho) of p.a. grade were purchased from Aldrich Co. While acetic acid (CH3COOH), methanol (CH3OH) and 1,2-ethanediol (glycol, HOCH2CH2OH) of p.a. grade were from Polish Chemical Reagents.The co-doped YAG:YbHo and YAG:YbTmHo nanopowders were immobilized into silica xerogel by sol–gel processing in a one-pot method.17 2.5 cm3 of tetramethoxysilane (TMOS was) dissolved in 5 cm3 of methanol and 1 cm3 of water was added. The mixture was stirred and 1.2 cm3 of NH3(aq) catalyst dropwise was added. After that the MeOH sol consisting of the co-doped YAG nanoparticles together with the sol–gel mixture was stirred for 15 min. The molar ratio of TMOS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O:MeOH was established as 1:4:8. After two days, the co-doped YAG nanocrystals YAG:YbHo@SiO2 and YAG:YbTmHo@SiO2 incorporated into rigid wet gels with the materials were obtained. The nanocomposites were dried at 200 °C, then crushed and sieved (0.25–0.50 mm).25 After that they were calcined at 200, 600, 800 and 1000 °C for 3 hours. The chemicals for the silica matrix preparation such as tetramethoxysilane (TMOS) was purchased from Aldrich Co. but methanol (CH3OH) and ammonia NH3(aq) were from Polish Chemical Reagents.
:H2O:MeOH was established as 1:4:8. After two days, the co-doped YAG nanocrystals YAG:YbHo@SiO2 and YAG:YbTmHo@SiO2 incorporated into rigid wet gels with the materials were obtained. The nanocomposites were dried at 200 °C, then crushed and sieved (0.25–0.50 mm).25 After that they were calcined at 200, 600, 800 and 1000 °C for 3 hours. The chemicals for the silica matrix preparation such as tetramethoxysilane (TMOS) was purchased from Aldrich Co. but methanol (CH3OH) and ammonia NH3(aq) were from Polish Chemical Reagents.
Apparatus
The ultrasonic equipment for the crystals crushing to nanoparticles was Sonorex Super RK 103H. After straining and drying at 120 °C the nanocrystals were thermally treated at 200 °C in a programmable oven equipped with an SM-946 temperature controller.Powder X-ray diffraction (XRD) analysis of the YAG, doped and co-doped YAG:Ln nanocrystals as well as YAG:Ln@SiO2 nanocomposites was made using a Bruker diffractometer of the D2Phaser model employing CuKα radiation operated at 30 kV and 0.2 mA. The XRD patterns were collected using scanning steps of 0.02 deg. and counting time 0.4 s per step. The average crystalline size as well as percentage of crystallinity were calculated using Diff. EVA software provided by Bruker.
The topography of the samples was characterized using scanning electron microscope SEM S-3400N (Hitachi, Japan), equipped with a tungsten source. A secondary electron (SE) detector was used and analysis was performed under 20 kV accelerating voltage.
Chemical composition of the investigated materials was examined by means of energy dispersive X-ray spectroscopy (EDX) equipped with ThermoFisher Scientific detector attached to (SEM) and X-ray photoelectron spectroscopy (XPS) fitted with ThermoFisher Scientific: Escalab 250Xi utilizing monochromatic Al-Kα source with charge neutralization implemented by means of flood gun. High-resolution XPS spectra were recorded at energy step size of 0.1 eV at pass energy 10 eV. In order to normalize the spectroscopic measurements, the X axis (binding energy) from XPS spectrum was calibrated for the peak characteristics for neutral carbon C1s (284.6 eV).26 Data analysis was performed using Avantage software provided by the manufacturer.
Photoluminescence spectra were recorded using a monochromators SPM2 produced by Carl Zeiss Jena for excitation and emission. An excitation source in this system was Osram 250 W ozone-free Xenon lamp and laser produced by Optoelectronics Tech. Co., Ltd MDL-III-980L-1000 mW. Fluorescence intensity was measured using a Hamamatsu R928 Side-on photomultiplier. All spectra were recorded at 300 K.
To follow the luminescence kinetics, we used a system consisting of a YAG:Nd (PL 2143 A/SS) laser and system OPG (optical paramagnetic generators). This system can generate 30 ps laser pulses, with the frequency of 10 Hz with wavelengths ranging 290 and 355 nm. The emission signal was analyzed by a Bruker Optics 2501S spectrometer and the Hamamatsu Streak Camera model C4334-01 with a final spectral resolution 0.47 nm. Luminescence spectra were collected by integration of the streak camera pictures over time intervals, whereas luminescence decays were obtained by the integration of streak camera images over the wavelength intervals.27
Results and discussion
XRD patterns
XRD patterns of the pure YAG (curve a), YAG doped with 6 mol% Yb3+ (YAG:Yb, curve b) and YAG triply co-doped with Yb3+, Tm3+ and Ho3+ (YAG:YbTmHo, curve c) dried at 200 °C are presented in Fig. S1.† All the patterns are consistent with the standard data corresponding to cubic Y3Al5O12 nanocrystals (PDF 01-082-3500), i.e. no secondary crystalline phase is observed and presence of the dopants did not change the crystalline structure. The calculated by the Scherrer formula average size of the nanocrystals was equal to 27 ± 4 nm.XRD patterns of the thermally treated YAG:YbTmHo@SiO2 nanocomposite is shown in Fig. S2.† The sample was dried at 200 °C and then calcined at 600, 800 and 1000 °C. As previously, the XRD patterns are in accordance with the YAG standard data. The average crystallite sizes of the triply co-doped YAG nanoparticles in the nanocomposite collected in Table S1† are within experimental error close to the crystallite size of the un-doped, doubly and triply co-doped YAG nanoparticles. The crystallinity percentage of the samples were estimated. Its values change a little with temperature of the thermal treatment (see Table S1†).
SEM images
SEM micrographs of the pure YAG and doped YAG:Yb powder samples presented in Fig. 3S(a and b,† respectively) exhibit agglomerated crystals. Then in images of greater enlargement in insets are shown nanoparticles which average size could be estimated to ca. 30 nm. It is within experimental error close to the size calculated by the Scherrer formula (vide supra). On the other hand, SEM image of the YAG:YbTmHo@SiO2 nanocomposite powder thermally treated at 600 °C shows agglomerates (Fig. S4†). Image of greater enlargement in inset shows the sphere-like nanoparticles consisted of the doped YAG nanocrystals capped with amorphous SiO2. Size of the nanoparticles is near two-fold greater than size of the YAG and YAG:Yb nanocrystals.EDS and XPS measurements
EDS measurement confirm the composition of the YAG and Yb-doped YAG nanocrystals (Fig. S5,† inset). Whereas high-resolution XPS spectra of the YAG and YAG:Yb demonstrated in Fig. S6† allow to calculate the Y/Al/O ratio for both the samples. The results give a close match to Y3Al5O12 (3.0/6.4/11.6 and 3.0/6.1/11.9 for YAG and YAG:Yb samples, respectively) proving their high purity (see Table S2†).Luminescence spectroscopy
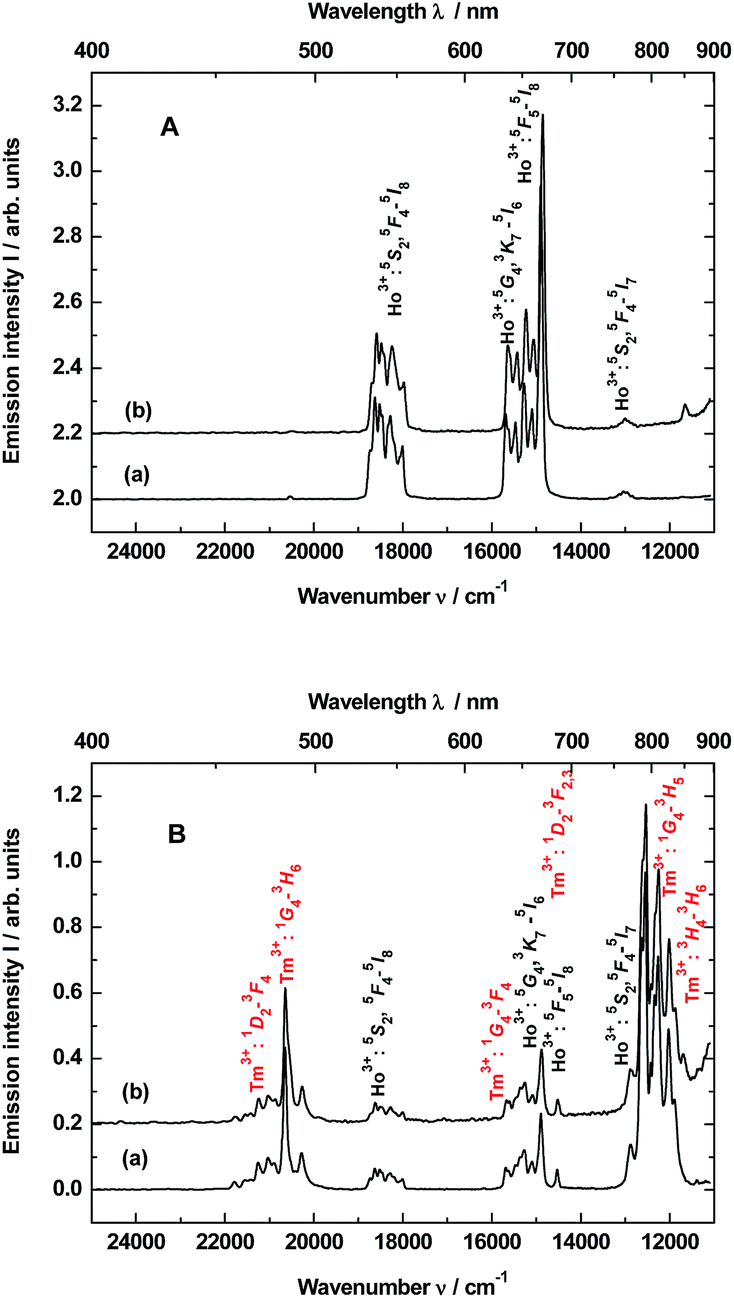 | ||
| Fig. 1 Up-conversion emission spectra of the samples: (A) YAG:YbHo nanocrystals (curve a) and YAG:YbHo@SiO2 glass nanocomposite (curve b) as well as (B) YAG:YbTmHo nanocrystals (curve a) and YAG:YbTmHo@SiO2 glass nanocomposite (curve b). The samples dried at 200 °C were excited under λexc = 980 nm by the laser in steady-state condition. | ||
The up-conversion emissions shown in Fig. 1A are generated by two- and three-photon processes such as energy transfer (ET) and excited state absorption (ESA) illustrated in the known schematic energy level diagrams presented in Fig. 2 for Ho3+ ions. In the first step Yb3+ sensitizer is excited under 980 nm from the ground state 2F7/2 to the 2F5/2 one. Then, occurs the Yb3+:2F5/2 → Ho3+:5I6 transfer. In this situation either the non-radiative transition Ho3+:5I6 → 5I7 takes place and subsequently by step (2) the Ho3+ is excited to the 5F5 state or directly occurs the Ho3+:5I6 → 5F4, 5S2 transition by two-photon process (step 2) from Yb3+. In former case the red emission about 670 nm (14870 cm−1) corresponds to the 5F5 → 5I8 transition. Whereas in the latter case can be present two transitions, viz. 5F4, 5S2 → 5I8 and 5F4, 5S2 → 5I7. Owing these transitions are observed such emission bands as green one at about 550 nm (18180 cm−1) and near-infra red (NIR) band centered at 764 nm (13004 cm−1), respectively. However, after the 5F5 → 5I5 non-radiative decay three-photon process (step 3) takes place the 5F5 → 5G4, 3K7 transition. The 5G4, 3K7 → 5I6 relaxation generates an emission band peaked at ca. 650 nm (15380 cm−1).4,22
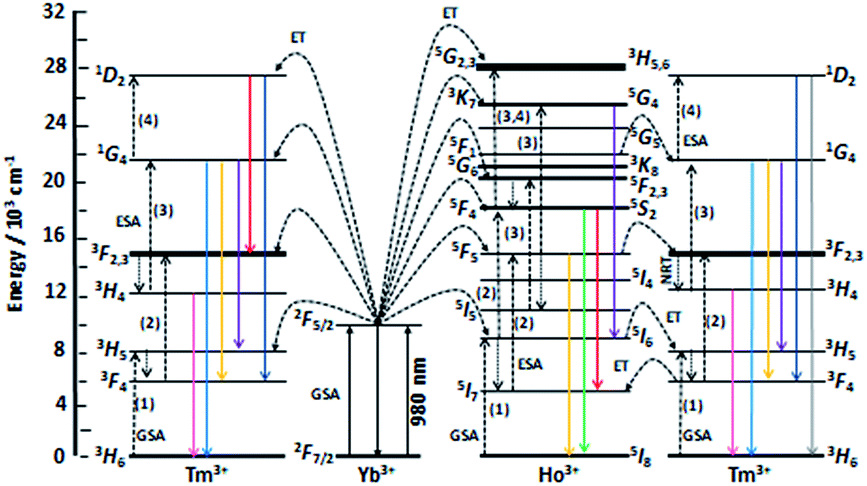 | ||
| Fig. 2 Scheme of energy levels of Yb3+ (sensitizer) as well as Tm3+ and Ho3+ (activators) in YAG:YbTmHo@SiO2 nanocomposite. The scheme demonstrates up-conversion mechanisms of the emissions under 980 nm laser excitation. GSA means ground state absorption, ESA – excited state absorption, ET – energy transfer and NRT – non-radiative transition. After ref. 22 and 28. | ||
Emission spectra shown in Fig. 1B are superpositions of spectra consisting of lines assigned to transitions of Ho3+ and Tm3+ dopants in YAG:YbTmHo and its glass nanocomposite counterpart YAG:YbTmHo@SiO2. The observed emission bands of Tm3+ are due to two-, three- and four-photon up-conversion processes. As previously, the up-conversion mechanism starts when the excited Yb3+ ion transfers its energy to the Tm3+ and the latter ion reaches the excited 3H5 level. As in the case of Ho3+ it is a phonon assisted energy transfer because there is energy difference between the excited state of Yb3+ and the excited 3H5 level of Tm3+ (see left part of the energy diagram in Fig. 7). Then after the non-radiative relaxation Tm3+:3H5 → 3F4 takes place ET from the Yb3+ (step 2) that causes the 3F4 → 3F2,3 transition of Tm3+. From this energy level the Tm3+ ions can firstly non-radiatively relax to 3H4 and then radiatively relax to the ground state (3H6). The latter transition generates the NIR emission band centered at 826 nm (11990 cm−1) in the spectra shown in Fig. 1B. Step (3) allows to excite Tm3+ to the 1G4 state. From this level start three emission transitions of Tm3+, viz. 1G4 → 3H5, 1G4 → 3F4 and 1G4 → 3H6 which correspond to bands at 810 nm (12250 cm−1), 637 nm (15635 cm−1) and 484 nm (20640 cm−1), respectively. After the next ESA (step 4) the Tm3+ ions are transferred to the 1D2 state from which they can relax by photon emissions of wavelengths of 792 nm (12540 cm−1) and 470 nm (21220 cm−1). In this case the emission lines are related to 1D2 → 3F2,3 and 1D2 → 3F4 transitions, respectively.22
The up-conversion mechanism in the triply co-doped nanocrystals and nanocomposite could be more complicated because such pairs of energy levels as (Ho3+:5I7 and Tm3+:3F4), Ho3+:5I6 and Tm3+:3H5, Ho3+:5F5 and Tm3+:3F2,3 as well as Ho3+:5F1 and Tm3+:1G4 possessing small energy differences between them (compare in Fig. 2). Thus, a quasi-resonant ET Ho3+ → Tm3+, e.g. Ho3+:5F5 → Tm3+:3F2,3 and next multiphonon non-radiative transitions (NRTs) to the Ho3+:5I7 and Tm3+:3H4: levels could be taken into account (see right part of the diagram in Fig. 2). In this case population of the Tm3+:1G4 state can be reached in the three-photon processes as follows: Yb3+:2F7/2 → GSA Yb3+:2F5/2 → ESA Ho3+:5I6 → NRT Ho3+:5I7 → ESA Ho3+:5F5 → ET Tm3+:3F2,3 → NRT Tm3+:3H4 → ESA Tm3+:1G4. The other observed bands are assigned to the same Ho3+ electronic transitions as in the doubly co-doped samples YAG:YbHo and YAG:YbHo@SiO2 (Fig. 1A).28
The material consisting of the triply co-doped YAG nanocrystals (YAG:YbTmHo) embedded into silica (YAG:YbTmHo@SiO2) was excited under 355 nm, i.e. the excitation wavelength typical for Tm3+, Ho3+ ions and additionally for silica matrix (see Fig. 3). The appropriate emission spectra of the YAG:YbTmHo@SiO2 phosphor dried at 200 °C and then calcined at 600, 800 and 1000 °C are demonstrated in Fig. 3. Characteristic feature of the spectra is a band wing penetrated deeply from UV into the visible range. This wing is the long wavelength part of the spectrum (Fig. 3, inset) attributed to electronic transitions between structural defect levels of amorphous SiO2.29,30 Emission lines of Tm3+ and Ho3+ are observed against the wing background. The distinct Ho3+ bands attributed to 5S2, 5F4 → 5I8 (peaked at 541 nm, 18484 cm−1) and 5G4 → 5I6 (at 585 nm, 17094 cm −1) transitions are present only in the spectra of the material thermally treated in the 200–800 °C range (curves a–c). While the Ho3+ bands centered at 466 nm (21459 cm−1) and 771 nm (12970 cm−1) ascribed to 3K8 → 5I8 and 5S2, 5F4 → 5I7 transitions, respectively, are present also after calcination at 1000 °C (curve d). The Tm3+ bands corresponding to 1D2 → 3F4 (466 nm, 21459 cm−1), 1G4 → 3H6 (491, 20367), 1G4 → 3F4 (608, 16447) and 3H4 → 3H6 (832, 12019) transitions are also present in all the spectra (a–d). One can assume that the faint red emission related to the 5S2, 5F4 → 5I7 transition of Ho3+ is present by additional population for the (5S2, 5F4) levels by energy transfer from Tm3+ to Ho3+ ions.31 However, the Ho3+ band quenching in range between 530 and 600 nm signifies that the Tm3+ → Ho3+ energy transfer in the material thermally treated at 1000 °C did not exist in this case.32,33 The set of the emission spectra in Fig. 3 suggests that 600 °C as the lowest calcination temperature applied for 3 h allows to obtain optimal luminescence properties of the nanocomposite material.
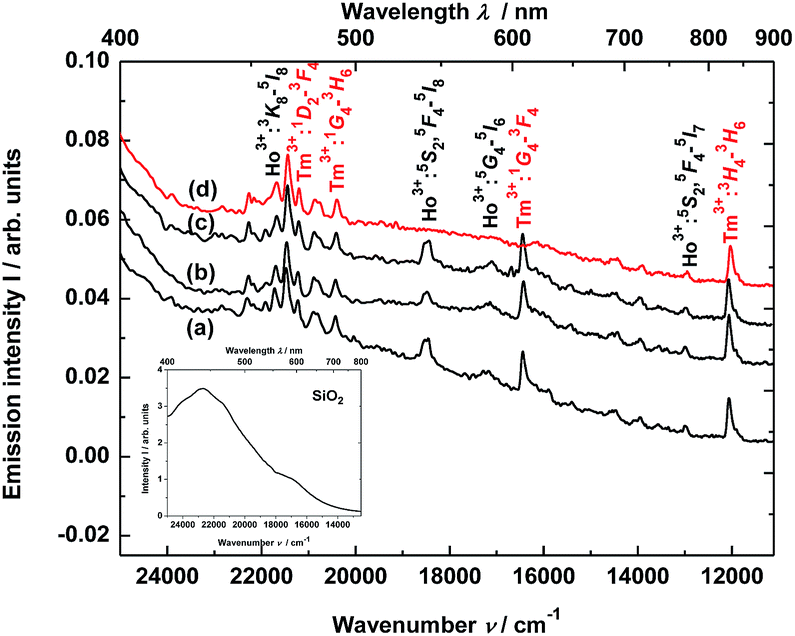 | ||
| Fig. 3 Steady-state emission spectra of the YAG:YbTmHo@SiO2 nanocomposite excited under 355 nm after drying at (a) 200 °C and calcined at: (b) 600, (c) 800 and (d) 1000 °C for 3 h. Inset presents emission spectrum of SiO2 xerogel dried at 200 °C and excited under 355 nm. | ||
Thus, the luminescence excitation spectrum of the YAG:YbTmHo@SiO2 nanocomposite calcined at 600 °C for 3 h is demonstrated in Fig. 4. The spectrum monitored at 466 nm (corresponding to Tm3+:1D2 → 3F4 and Ho3+:3K8 → 5I8 transitions as well as simultaneously transition in the amorphous silica) consists of lines centered at: 284 nm (35211 cm−1, corresponding to transitions Ho3+:5I8 → 3G3, 3L8 and Tm3+:3H6 → 3P0), 290 (34483, Ho3+:5I8 → 3D3), 319 (31348, Ho3+:5I8 → 5G2), 343 (29155, Ho3+:5I8 → 5G3), 355 (28169, Ho3+:5I8 → 3H6 and Tm3+:3H6 → 1D2), 384 (26042, Ho3+:5I8 → 5G4), 399 (25063, Ho3+:5I8 → 3K7), 415 (24096, Ho3+:5I8 → 5G5), 425 (23529, Ho3+:5I8 → 5G6) and 435 (22989, Ho3+:5I8 → 5F1). In the inset is presented excitation spectrum of glassy SiO2. This spectrum is attributed to many transitions between defects states in the amorphous silica matrix.
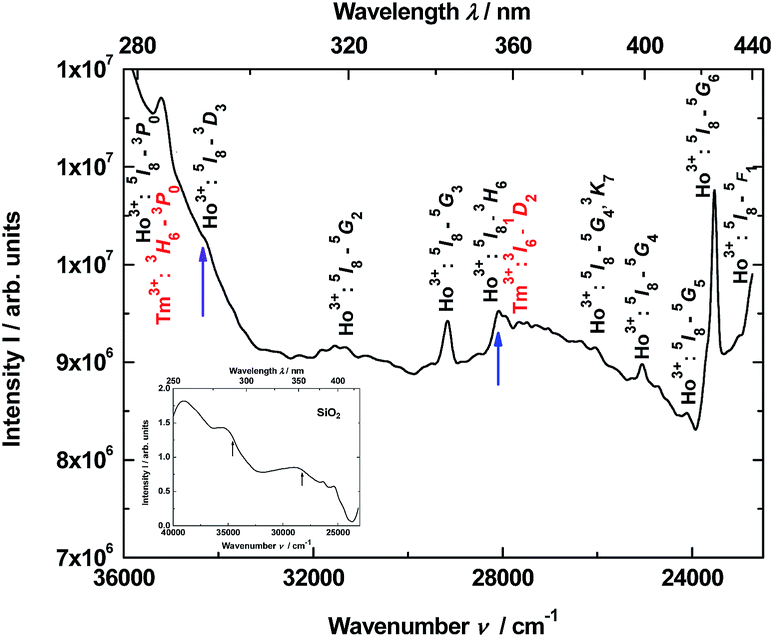 | ||
| Fig. 4 Excitation spectrum of the YAG:YbTmHo@SiO2 nanocomposite monitored at 466 nm. The nanocomposite was calcined at 600 °C for 3 h. The inset demonstrates excitation spectrum of pure SiO2 glass monitored at 440 nm. The arrows indicate the excitation wavelengths (290 and 355 nm) used in steady-state condition. | ||
Time-resolved luminescence
To obtain time-resolved spectra, the YAG:YbTmHo@SiO2 nanocomposite was excited using of a YAG:Nd laser and system OPG (see Apparatus). The apparatus allows to integrate luminescence in the 0–200 μs time interval. The spectra are collected in Fig. 5. Obviously, they are superpositions consisting of Tm3+ and Ho3+ bands. When the sample is excited under 290 nm (see curve a), among the lines there are four most intensive bands, viz. the resultant emission band peaked at 371 nm (26963 cm−1) attributed to Tm3+:1D2 → 3H6 and Ho3+:3H6 → 5I8 transitions. The next band placed at 466 nm (21459 cm−1) is a result of overlapping three bands related to both transitions of Tm3+:1D2 → 3F4 and 1G4 → 3H6 as well as the Ho3+:3K8 → 5I8 transition. Whereas lines centered at 652 (15337 cm−1) and 736 nm (13592 cm−1) are owing to the 1G4 → 3F4 transition of Tm3+ and the 5S2, 5F4 → 5I7 transitions of Ho3+, respectively. Similar bands or their traces are present in the emission spectrum excited under 355 nm (see Fig. 5, curve b). However, mutual intensity ratios of the bands are quite other and two lines centered at 393 (25434) and 407 (24593) as well as a shoulder at ca. 615 nm (16345 cm−1) are absent in comparison to the spectrum excited under 290 nm (curve a). Moreover, the spectrum (curve b) differs from the emission spectrum excited under the same wavelength but in steady-state condition (see Fig. 3, curve b).
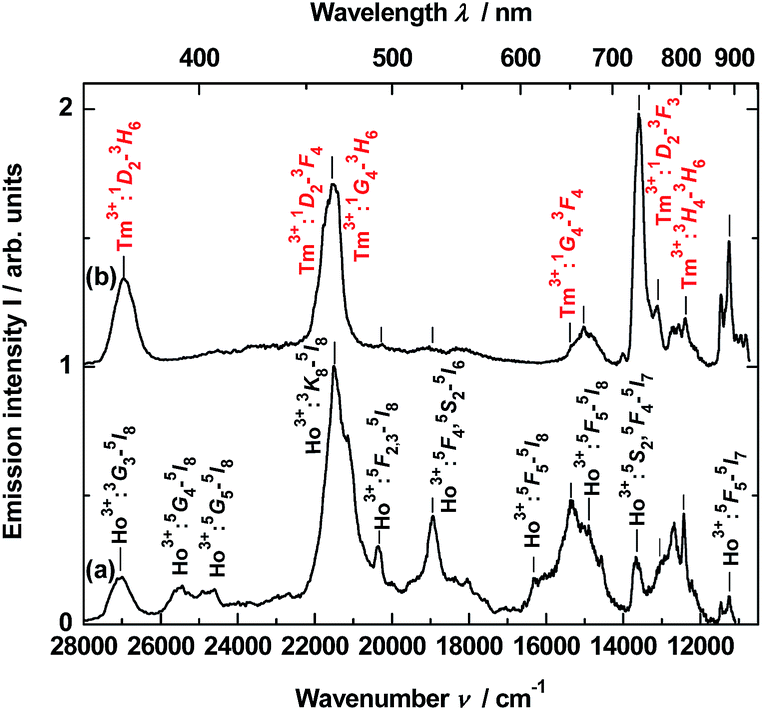 | ||
| Fig. 5 Time-resolved emission spectra of the YAG:YbTmHo@SiO2 calcined at 600 °C for 3 h. The material was excited by the pulse laser under: (a) 290 and (b) 355 nm and the luminescence was integrated for the time interval 0–200 μs. | ||
Photoluminescence lifetime measurements for YAG:YbTmHo@SiO2 were performed by the pulsed laser under excitation at 290 nm (see decay curves in Fig. 6). The average lifetime values for electronic transitions related to the four most intense lines mentioned above (see Fig. 5) are presented in Table 1. The average decay times τav were evaluated using the equation:34
 | (1) |
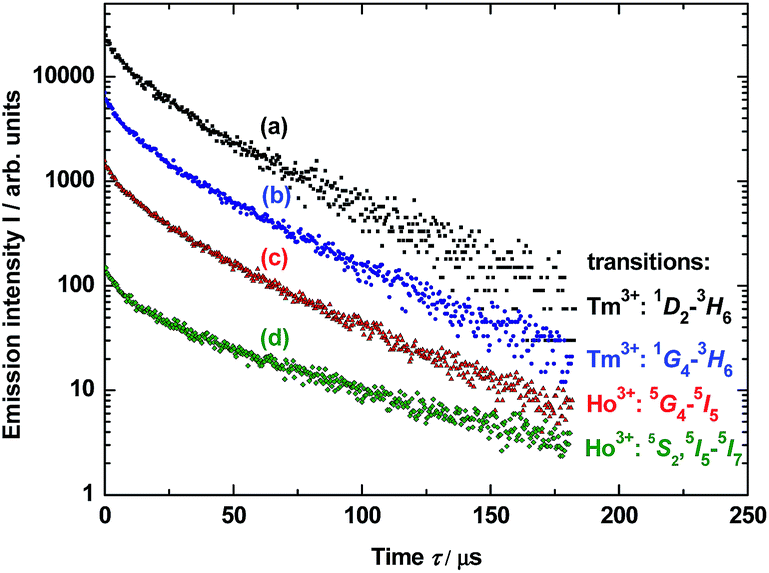 | ||
| Fig. 6 Decay luminescence curves for electronic transitions related to four emission lines of Tm3+ and Ho3+ dopants in YAG nanocrystals incorporated into SiO2 matrix (YAG:YbTmHo@SiO2) calcined at 600 °C for 3 h and excited under 290 nm. The lines are presented in the spectra in Fig. 3 (curve b). For details vide supra. | ||
| Ln3+ | Transition | Average decay time τav/μs | |
|---|---|---|---|
| Excitation wavelength λexc/nm | |||
| 290 | 355 | ||
| Tm3+ | 1D2 → 3H6 | 41 ± 3 | 30 ± 2 |
| Tm3+ | 1G4 → 3H6 | 36 ± 3 | 28 ± 2 |
| Ho3+ | 5G4 → 5I5 | 33 ± 2 | 36 ± 3 |
| Ho3+ | 5S2, 5F4→ 5I7 | 30 ± 2 | 41 ± 3 |
Calculated average decay times τav of the above mentioned transitions are listed in Table 1 for λexc = 290 and 355 nm. One notes that independently on the excitation wavelength the decay times demonstrate approximate values for the observed transitions in both lanthanide dopants of YAG nanoparticles incorporated into silica matrix.
In much shorter time interval (0–50 ns) and under 290 nm excitation was observed for the YAG:Yb,TmHo@SiO2 only a time-resolved spectrum of the vitreous silica matrix (Fig. 7). The spectrum consists of a strong and broad emission band centered at ca. 440 nm (22738 cm−1). Its origin is attributed to photon recombinations from numerous defects (as dangling bonds) in the amorphous silica.29,30
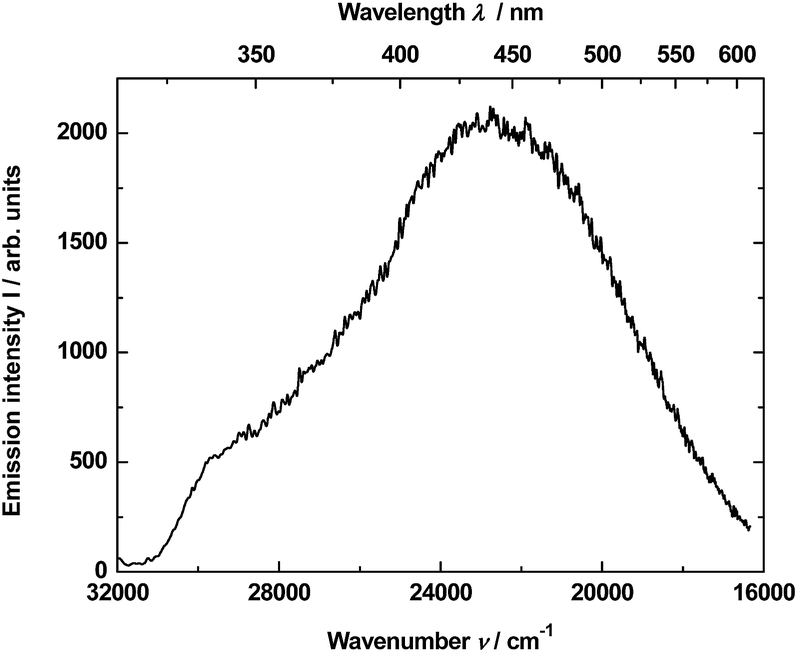 | ||
| Fig. 7 Time-resolved emission spectrum of the YAG:YbTmHo@SiO2 nano-composite calcined at 600 °C for 3 h and excited at 290 nm by the pulse laser. The luminescence was integrated in the time interval 0–50 ns. | ||
The measured decay profile of the silica luminescence under excitation by the pulse laser at 290 nm is shown in Fig. 8. The curve allows to estimate average emission decay time value for silica equal to 5.6 ± 0.6 ns.
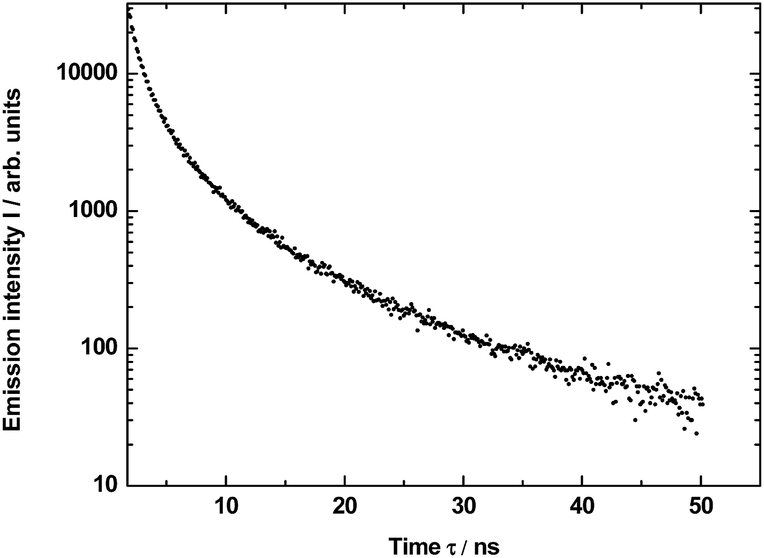 | ||
| Fig. 8 Decay of the luminescence excited under 290 nm and integrated in the band 450–470 nm for SiO2 matrix in YAG:YbTmHo@SiO2 material calcined at 600 °C for 3 h. Average lifetime τav = 5.6 ± 0.6 ns. | ||
Chromaticity
The emission spectra of the YAG:YbTmHo@SiO2 glass-nanocomposite under different excitation wavelengths and luminescence techniques (such as the steady state and pulse laser measurements) are demonstrated in Fig. 9. Under the 355 nm excitation (curve a) in the steady-state condition, the nanocomposite material emits simultaneously reddish, green and blue (RGB) luminescent light (see Fig. 3 curve b).35 The emissions are ascribed to the transitions of Tm3+ and Ho3+ ions, while blue emission is especially intense due to electronic transition in silica (see Fig. 3, inset). The up-conversion spectrum (curve b) obtained under 980 nm excitation by the laser in steady-state condition supply with red, green and bluish green light owing to the transitions of Tm3+ and Ho3+ ions (compare Fig. 1B). On the other hand, under excitations 355 and 290 nm by the pulse laser (curves c and d, respectively) also the both lanthanide ions are responsible for the emission in visible range. However, the 355 nm excitations delivers only red and blue emission (Fig. 5, curve b), while under 290 nm excitation is observed light consisting of emissions of primary colours (RGB) (Fig. 5, curve a). | ||
| Fig. 9 Comparison of the steady-state emission spectra excited under: (a) 355 nm and (b) 980 nm (up-conversion) as well as excited by the pulse laser under: 355 (c) and 290 nm (d) for the YAG:YbTmHo@SiO2 nanocomposite calcined at 600 °C for 3 h. The luminescence in the cases (c) and (d) was integrated in the time interval 0–200 μs. | ||
The CIE chromaticity coordinates and diagram of YAG:YbTmHo@SiO2 under the different excitation wavelengths and luminescent techniques are presented in Table 2 and Fig. 10, respectively. The luminescent nanocomposite emits cold white light with bluish tone (point A), when excited by 355 nm in steady state-condition. In this case the correlated colour temperature (Tcc) is rather high (ca. 8100 K).36 Whereas the up-conversion emission (point B) excited at 980 nm in the same condition is white and its Tcc ≈ 4800 K is in between temperatures of mean noon sunlight and household tungsten-filament light bulb (100 W).35,36 Both the emissions excited under 290 and 355 nm by pulse laser emitted blue light which Tcc values in the former case (point D) is equal to 14000 K but in the latter case (point C) Tcc is too high to evaluate them.
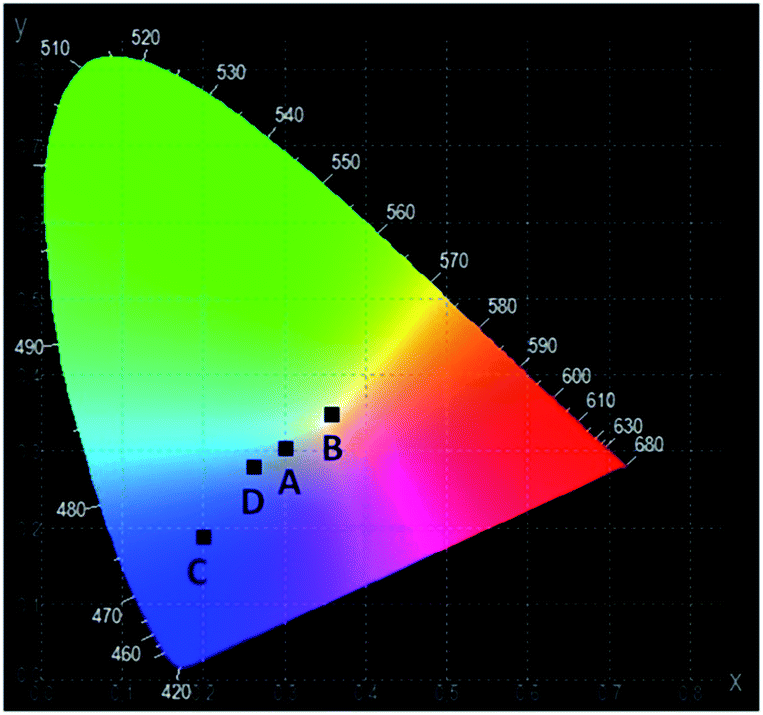 | ||
| Fig. 10 The CIE chromaticity diagram of the YAG:YbTmHo@SiO2 nanocomposite calcined at 600 °C for 3 h. For details see Table 2. | ||
Conclusions
The glass nanocomposite, i.e. nanocrystalline YAG co-doped with Yb3+, Tm3+ and Ho3+ entrapped in glassy silica (YAG:YbTmHo@SiO2), was successively prepared by sol–gel method. Presence of the specific lanthanide ions such as Yb3+, Tm3+ and Ho3+ in the nanocomposite allows to observe up-conversion effect, when the composite sample was excited under 980 nm in steady-state condition. Other steady-state emission was also recorded, when λexc = 355 nm. The nanocomposite material excited by pulse laser shows dissimilar time-resolved luminescence spectra for different excitation wavelengths (λexc = 290 and 355 nm). Luminescence property based on up-conversion effect of the glass-nanocomposite excited under 980 nm in steady-state condition allows to recognize the material as a white phosphor candidate.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to express our gratitude to Prof. Dr Marek Grinberg (Institute of Experimental Physics, University of Gdańsk, Poland) and Prof. Dr Wiesław Wiczk (Faculty of Chemistry, University of Gdańsk, Poland) for making the apparatus accessible for the measurements. K. S. has been supported by the National Centre for Research and Development by grant NOPLID PL-TWII/8/2015.References
- J. Xu, M. Sun, Y. Kuang, H. Bi, B. Liu, D. Yang, R. Lv, S. Gai, F. He and P. Yang, Dalton Trans., 2017, 46, 1495–1501 RSC.
- J. Jin, K. Yang, J. Su and Z. Si, J. Lumin., 2015, 159, 178–182 CrossRef CAS.
- G. Blasse and B. C. Grabmaier, Luminescent Materials, Springer-Verlag, Berlin, 1994, ch. 10 Search PubMed.
- C. S. Lim, V. V. Atuchin, A. S. Aleksandrovsky, M. S. Molokeev and A. S. Oreshonkov, J. Alloys Compd., 2017, 695, 737–746 CrossRef CAS.
- H. W. Zheng, S. J. Liu, G. S. Yin, W. C. Wang, C. L. Diao, Y. Z. Gu and W. F. Zhang, J. Sol-Gel Sci. Technol., 2011, 59, 290–296 CrossRef CAS.
- X. Mateos, M. C. Pujol, F. Güell, R. Sole, J. Gavalda, J. Massons, M. Aguilo and F. Diaz, Opt. Mater., 2004, 27, 475–479 CrossRef CAS.
- M. Nakielska, J. Sarnecki, M. Malinowski and R. Piramidowicz, J. Alloys Compd., 2008, 451, 190–193 CrossRef CAS.
- J. Zhang and J. Jia, J. Lumin., 2016, 174, 1–5 CrossRef CAS.
- F. G. Yang, F. P. Yan, Z. Y. You, C. Y. Tu, C. L. Sun, Y. Wang, Z. J. Zhu and J. F. Li, Laser Phys. Lett., 2010, 7, 867–869 CrossRef CAS.
- L. Marciniak, A. Bednarkiewicz and W. Stręk, J. Lumin., 2017, 184, 179–184 CrossRef CAS.
- S. Hu, J. Yang, C. Li and J. Lin, Mater. Chem. Phys., 2012, 133, 751–756 CrossRef CAS.
- A. Ikesue, T. Kinoshita, K. Kamata and K. Yoshida, J. Am. Chem. Soc., 1995, 78, 1033–1040 CAS.
- S. Pfeifer, M. Bischoff, R. Niewa, B. Clauss and M. R. Buchmeister, J. Eur. Ceram. Soc., 2014, 34, 1321–1328 CrossRef CAS.
- X.-X. Ge, Y.-H. Sun, C. Liu and W.-K. Qi, J. Sol-Gel Sci. Technol., 2009, 52, 179–187 CrossRef CAS.
- R. Muenchausen, L. G. Jacobsohn, B. L. Bennett, E. A. McKigney, J. F. Smith, J. A. Valdez and D. W. Cooke, J. Lumin., 2007, 126, 838–842 CrossRef CAS.
- B. Karmakar, Fundamentals of Glass and Glass Nanocomposites in Glass Nanocomposites, Synthesis, Properties and Applications, ed. B. Karmakar, K. Rademann, A. L. Stepanov, Elsevier, Amsterdam, 2016 Search PubMed.
- C. J. Brinker and G. W. Scherer, Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing, Academic Press, Boston, 1990 Search PubMed.
- J. Zhao, X. Zheng, E. P. Schartner, P. Ionescu, R. Zhang, T.-L. Nguyen, D. Jin and H. Ebendorff-Heidepriem, Adv. Opt. Mater., 2016, 4, 1507–1517 CrossRef CAS.
- J. Zhao, D. Jin, E. P. Schartner, Y. Lu, Y. Liu, A. V. Zvyagin, L. Zhang, J. M. Dawes, P. Xi, J. A. Piper, E. M. Goldys and T. M. Monro, Nat. Nanotechnol., 2013, 8, 729–734 CrossRef CAS PubMed.
- D. J. Gargas, E. M. Chan, A. D. Ostrowski, S. Aloni, M. V. P. Altoe, E. S. Barnard, B. Sanii, J. J. Urban, D. J. Milliron, B. E. Cohen and P. J. Schuck, Nat. Nanotechnol., 2014, 9, 300–305 CrossRef CAS PubMed.
- D. Liu, X. Xu, Y. Du, X. Qin, Y. Zhang, C. Ma, S. Wen, W. Ren, E. M. Goldys, J. Pieper, S. Dou, X. Liu and D. Jin, Nat. Commun., 2016, 7, 10432–10438 CrossRef PubMed.
- J. Zhou, G. Chen, Y. Zhu, L. Huo, W. Mao, D. Zou, X. Sun, E. Wu, H. Zeng and J. Zhang, J. Mater. Chem. C, 2015, 3, 364–369 RSC.
- J. Wang, R. Deng, M. A. MacDonald, B. Chen, J. Yuan, F. Wang, D. Chi, T. S. Andy Hor, P. Zhang, G. Liu, Y. Han and X. Liu, Nat. Mater., 2014, 13, 157–162 CrossRef CAS PubMed.
- M. Veith, S. Mathur, A. Kareiva, M. Jilavi, M. Zimmer and V. Huch, J. Mater. Chem., 1999, 9, 3069–3079 RSC.
- M. Kubus, H.-J. Meyer, L. Kienle and A. M. Kłonkowski, J. Non-Cryst. Solids, 2009, 355, 1333–1337 CrossRef CAS.
- D. Pawlak, K. Wozniak, Z. Frukacz, T. L. Barr, D. Fiorentino and S. Seal, J. Phys. Chem. B, 1999, 103, 1454–1461 CrossRef CAS.
- A. Kubicki, P. Bojarski, M. Grinberg, M. Sadownik and B. Kukliński, Opt. Commun., 2006, 263, 275–280 CrossRef.
- J. Żmojda, D. Dorosz, M. Kochanowicz, P. Miluski and J. Dorosz, Acta Phys. Pol., A, 2013, 124, 598–601 CrossRef.
- L. N. Skuja, J. Non-Cryst. Solids, 1992, 149, 77–95 CrossRef CAS.
- A. M. Kłonkowski, W. Wiczk, J. Ryl, K. Szczodrowski and D. Wileńska, J. Alloys Compd., 2017, 724, 649–658 CrossRef.
- A. C. Yanes, J. J. Velazquez, J. del-Castillo, J. Mendez-Ramos and V. D. Rodriguez, J. Sol-Gel Sci. Technol., 2009, 51, 4–9 CrossRef CAS.
- D. Wang, Y. Guo, G. Sun, J. Li, L. Zhao and G. Xu, J. Alloys Compd., 2008, 451, 122–124 CrossRef CAS.
- D. Chen, Y. Wang, Y. Yu, P. Huang and F. Weng, J. Solid State Chem., 2008, 181, 2763–2769 CrossRef CAS.
- S. Shinoya and W. M. Yen, Phosphor Handbook, CRC Press, Boca Raton, 1999 Search PubMed.
- R. J. D. Tilley, Colour and the optical properties of materials, J. Wiley, Chichester, 2 edn, 2011, ch 3 Search PubMed.
- http://www.vendian.org/mncharity/dir3/blackbody/UnstableURLs/bbr_color.html.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00118a |
| This journal is © The Royal Society of Chemistry 2018 |