
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Discovery of N-cyclobutylaminoethoxyisoxazole derivatives as novel sigma-1 receptor ligands with neurite outgrowth efficacy in cells†
Hao
Sun‡
a,
Yun-Jie
Wang‡
b,
Wen-Wen
Shi‡
a,
Fan
Yang
a,
Jie
Tang
a,
Tao
Pang
*b and
Li-Fang
Yu
 *a
*a
aShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, School of Chemistry and Molecular Engineering, East China Normal University, 3663 North Zhongshan Road, Shanghai 200062, China. E-mail: lfyu@sat.ecnu.edu.cn; Tel: +86-021-622-31385
bJiangsu Key Laboratory of Drug Screening, State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing 210009, PR China. E-mail: tpang@cpu.edu.cn; Tel: +86-25-832-71043
First published on 14th February 2018
Abstract
Herein we reported a series of 14 novel derivatives based on the N-cyclobutylaminoethoxyisoxazole scaffold. In vitro binding studies of these compounds demonstrated their low nanomolar to subnanomolar potencies as σ1 receptor ligands, with moderate to excellent selectivity over the σ2 receptor as represented by compounds 17–30. The majority of the derivatives scored high (>4.7) in the CNS MPO appraisal system, indicating their high likelihood in penetrating the blood–brain barrier. A number of these compounds exhibited significant neurite outgrowth efficacy in N1E-115 neuronal cells and displayed excellent selectivity for σ1 receptors over the selected endogenous neurotransmitter transporters, such as DAT, NET and SERT. Among the mini-series, compound 28 (Ki σ1 = 0.2 nM, Ki σ2 = 198 nM, CNS MPO score = 5.4) emerged as a promising selective σ1 receptor ligand that warrants its further evaluation as a potential therapeutic for neurodegenerative diseases.
Introduction
Neurodegenerative diseases, such as Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS) and Huntington's disease (HD) are caused by the progressive loss of neuronal integrity or acute neuron injury such as stroke or trauma in the brain and spinal cord.1–3 Current therapeutic strategies for neurodegenerative diseases are mainly aimed to decrease CNS neuron damage or brain dysfunction by conferring neuroprotection and neurogenesis.4,5 Consequently, the development of effective therapeutic medicine or discovery of new biological targets with neurogenesis activities remains an urgent need in the treatment of neurodegenerative diseases.During the past decades, various kinds of receptors, ion channels and signaling pathways associated with neurogenesis activities have been identified as potential therapeutic targets for neurodegenerative diseases. Among these, the σ1 receptor has attracted wide attention.6 σ1 receptor is one of the subtype belonging to the σ receptors family which was first discovered by Martin in 1976.7 The σ1 receptor has been cloned and encoded as a protein of 223 amino acids, and specifically localized at the mitochondrial-associated endoplasmic reticulum membrane.8,9 In 2016, the crystal structure of the human σ1 receptor was reported in complex with two divergent ligands. The structure indicated that the human σ1 receptor has a trimeric architecture with a single transmembrane domain.10 The literature reports to date indicate that σ1 receptor is considered as chaperonin to modulate signaling pathways of ER–mitochondrion such as Ca2+, K+, and NMDA and IP3 receptors, playing an important role in healthy CNS functioning.11–13 In particular, neurogenesis efficacy of brain penetrant σ1 receptor ligands is of a considerable interest, which has been attributed to the σ1 receptor's role in the modulation of cellular trafficking.14,15 The activation of σ1 receptor leads to the transfer of cholesterol, ceramides and essential amino acids that are essential for the growth and proliferation of the neurons, along with an increased gene expression of specific protein and synthesis of various growth factors involved in neuronal outgrowth.16–18 Furthermore, neurogenesis function was reported to be subdued in the hippocampus of σ1 receptor-knockout animals.19
An increasing number of σ1 receptor ligands that stimulate neurite outgrowth in cellular screening models have been reported to date. Pentazocine (1) is a classical σ1 receptor ligand, capable of enhancing the neurite outgrowth in PC-12 cellular model caused by the activation of σ1 receptor and this activity was antagonized by NE-100 (2), a σ1 receptor antagonist.20 Ruscher et al. reported that the cortical neurons outgrowth upon treatment with SA4503 (3) could be prevented with σ1 receptor silencing.16 In addition, some reports in the literature suggested that the neuronal outgrowth activity by σ1 receptor activation is synergistic with some growth factors. PRE-084 (4) is an example of high affinity σ1 receptor ligand that can enhance neurite outgrowth and elongation induced by NGF or EGF in PC12 cells.21 Subsequently, PRE-084 could enhance the survival of the motoneurons and improve locomotor function in murine models of ALS.22 RC-33 (5), as a σ1 receptor ligand, also exhibited the neurite outgrowth effects and good in vitro metabolic stability.23–25 The (R)-enantiomer of RC-33 (6) has been selected as a lead compound for studies in multiple sclerosis.26 Therefore, the development of novel, brain-penetrant σ1 receptor ligands with neurogenesis efficacy could find some therapeutic use for neurodegenerative diseases (Fig. 1).
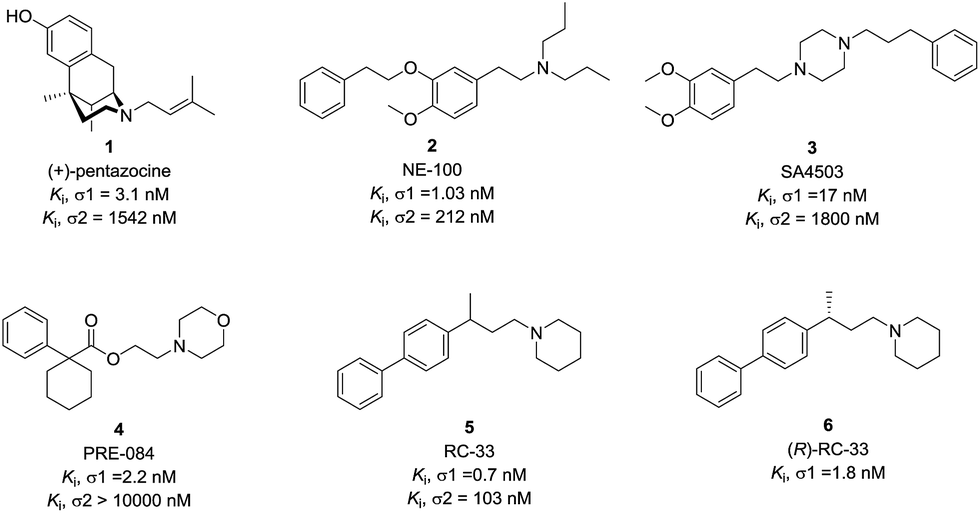 | ||
| Fig. 1 Representative σ1 receptor ligands. | ||
In our preliminary work in the σ receptors drug discovery, we developed a series of alkoxyisoxazole derivatives that were found to be σ1 receptor ligands with high affinity and selectivity over the σ2 receptor.27 Based on this work,27 we further screened some of the most potent derivatives in a cellular model of neurite outgrowth. During the course of this investigation, alkoxyisoxazole 7 exhibited synergistic neurite outgrowth effects at a level similar to that of the positive control RC-33 (Fig. 2). Encouraged by this finding, we performed further structure–activity relationship (SAR) studies on this scaffold, incorporating N-cyclobutylaminoethoxy linker that was hypothesized to be a crucial structural element for neurite outgrowth. With 7 as the starting compound, herein we reported our continued efforts in the SAR studies of the N-cyclobutylaminoethoxyisoxazole as σ1 receptor ligands and their cellular evaluation in neurite outgrowth assay.
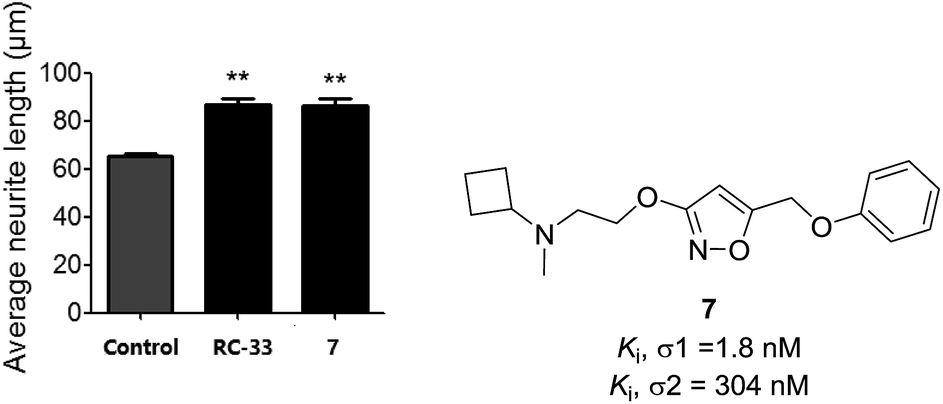 | ||
| Fig. 2 The potential effect of compound 7 (10 μM) and RC-33 (10 μM) on NGF (2.5 ng mL−1)-induced neurite outgrowth model in PC12 cells. aSee Experimental section. Each histograms represent the mean ± SEM of at least five different experiments. Statistically significant differences: **p < 0.01 vs. control using NGF alone. | ||
Results and discussion
Chemistry
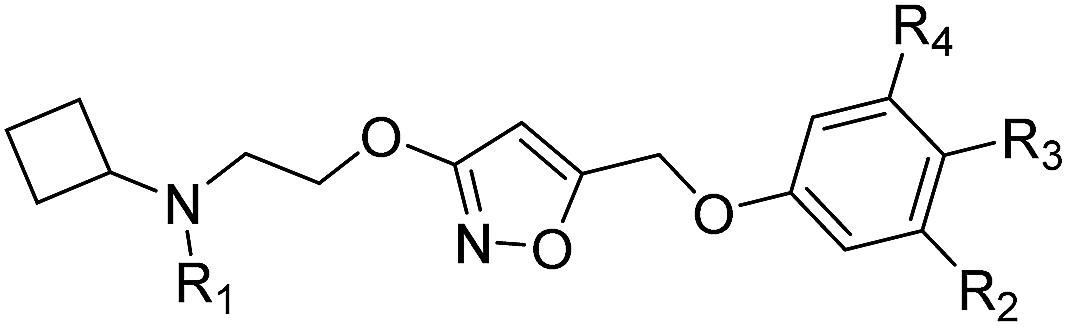
The synthesis of the new compounds was accomplished as shown in Scheme 1. The starting material 8 or 9 underwent a Mitsunobu reaction with previously reported methyl 3-hydroxyisoxazole-5-carboxylate (10) to obtain the intermediate esters 11 and 12. Lithium borohydride reduction afforded the alcohols 13 and 14, respectively, which were converted to the corresponding iodides 15 and 16 using iodine, triphenylphosphine and imidazole as a base. Subsequent reaction of the iodides with phenol or substituted phenols under alkaline conditions gave the Boc-protected isoxazole intermediates. Finally, treatment with hydrogen chloride and reductive amination with cyclobutanone and sodium cyanoborohydride resulted in final compounds 17–30. All final compounds were recrystallized by HCl/ethyl acetate as hydrochloride salts.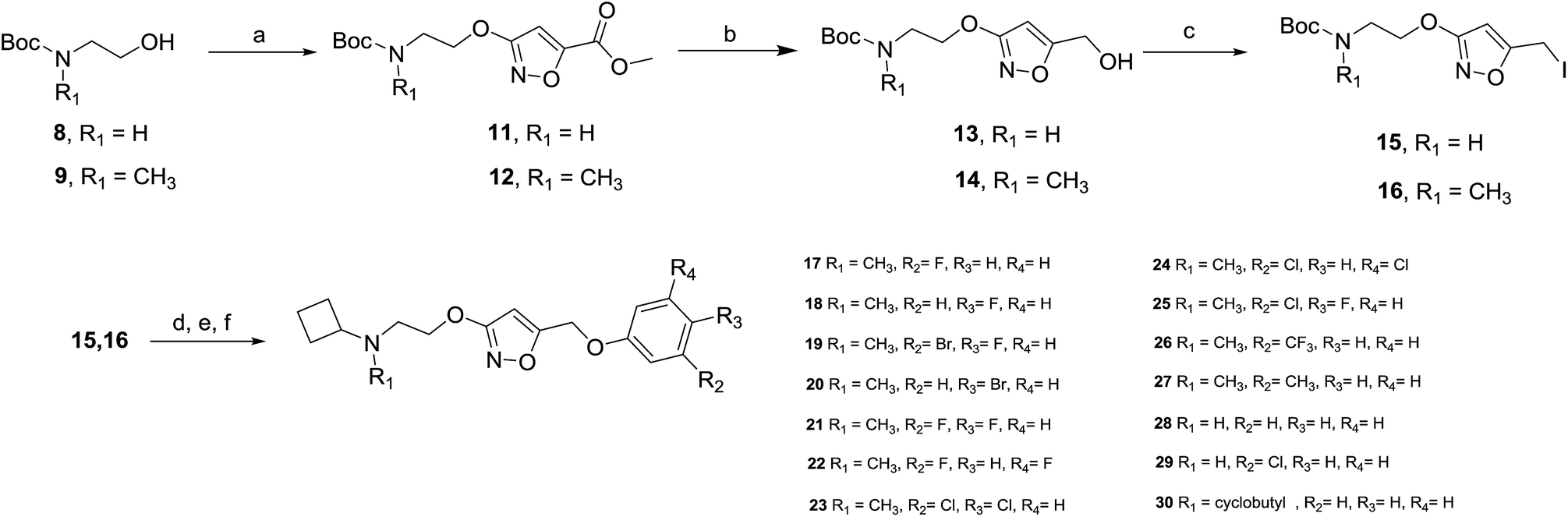 | ||
| Scheme 1 aReagents and conditions: (a) methyl 3-hydroxyisoxazole-5-carboxylate (10), diisopropyl azodicarboxylate, PPh3, THF, 0 °C to rt; (b) LiBH4, THF, 0 °C to rt; (c) I2, PPh3, imidazole, CH2Cl2, 0 °C to rt; (d) phenol, or substituted phenol, K2CO3, DMF, rt; (e) HCl/EtOAc, rt; (f) cyclobutanone, Na(CN)BH3, CH3CO2H, CH3OH, rt. | ||
Radioligand binding studies at σ1 and σ2 receptors.28
In this study, [3H] pentazocine and [3H]DTG (6) were used as the radioligands at σ1 and σ2 receptors. The binding affinity assays of compounds 17–30 are shown in Table 1. Firstly, we examined the effects of changing the substituents on the benzene ring. 3-Methyl substituted compound 27 retained high affinity to parent compound 7 at σ1 receptor but a 3-fold increase in the binding affinity for σ2 receptor. Compound 26 with a 3-trifluoromethyl substituent showed similar affinity for σ1 receptor and significantly decreased selectivity over the σ2 receptor when compared with compound 7. Mono-halogen substitution on the benzene ring exemplified by compounds 17–20 generally retained affinity for σ1 receptor compared with compound 7. However, all of these mono-halogen substituted derivatives also exhibited subdued selectivity over the σ2 receptor. Notably, the 3-bromo substituted compound 19 displayed a better selectivity (Ki σ2/Ki σ1 = 131) than the 4-bromo substituted compound 20. The derivatives 21–25 also maintained similar high affinities for the σ1 receptor upon incorporation of two halogen atoms on the benzene ring. However, this is also accompanied with higher binding affinity to the σ2 receptor. Among the disubstituted compounds, 3,5-dichloro substituted compound 24 showed a 4-fold decreased affinity for σ1 receptor and more than 10-fold increased binding affinity for σ2 receptor. Next, we examined the effects of removing the methyl group on the basic nitrogen atom. Gratifyingly, compound 28 showed a high affinity for σ1 receptor (Ki σ1 = 0.2 nM) and excellent selectivity on σ2 receptor (Ki σ2 = 198 nM). Compound 29 with a 3-chloro substituted benzene ring also exhibited high binding affinity for σ1 receptor (Ki σ1 = 0.4 nM) but slightly lower selectivity over σ2 receptor (Ki σ2 = 64.3 nM). Compound 30 was a by-product obtained during the synthesis of compound 28, which showed a decreased binding affinity and selectivity indicating an unfavourable steric repulsion by the cyclobutyl group. In summary, except for compound 30, most of the derivatives exhibited high binding affinities for σ1 receptor (Ki σ1 < 10 nM). However, all the benzene ring substituted derivatives showed various degree of decreased selectivity over the σ2 receptor, implying that the non-substituted benzene ring was a crucial structural element for selectivity. The results for the secondary amines 28 and 29 indicate that the hydrogen bond donor on basic nitrogen atom was beneficial for increased binding affinity to the σ1 receptor.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | K i σ1 (nM) | K i σ2 (nM) | Selectivity (Ki σ2/Ki σ1) |
| a See Experimental section. Radioligands: σ1: [3H]-(+)-pentazocine; σ2: [3H] DTG (ditolylguanidine). Evaluation platform: σ1: Guinea pig homogenate; σ2: PC12 cells. b The Ki values for compound 7 are cited from the literature.27 Data are expressed as the mean ± SEM and each performed from three independent experiments. | |||||||
| 7 b | –CH3 | –H | –H | –H | 1.8 ± 0.2 | 307 ± 56 | 171 |
| 17 | –CH3 | –F | –H | –H | 1.9 ± 0.0 | 91.5 ± 7.5 | 48 |
| 18 | –CH3 | –H | –F | –H | 0.9 ± 0.1 | 78.5 ± 19.5 | 87 |
| 19 | –CH3 | –Br | –H | –H | 0.8 ± 0.1 | 105 ± 21 | 131 |
| 20 | –CH3 | –H | –Br | –H | 5.5 ± 1.3 | 86.6 ± 16.8 | 16 |
| 21 | –CH3 | –F | –F | –H | 1.7 ± 0.3 | 153 ± 37 | 90 |
| 22 | –CH3 | –F | –H | –F | 2.4 ± 1.2 | 57.0 ± 20.1 | 24 |
| 23 | –CH3 | –Cl | –Cl | –H | 3.2 ± 1.6 | 36.0 ± 9.7 | 11 |
| 24 | –CH3 | –Cl | –H | –Cl | 7.8 ± 3.2 | 22.8 ± 11.1 | 3.0 |
| 25 | –CH3 | –Cl | –F | –H | 1.3 ± 0.4 | 32.0 ± 6.0 | 25 |
| 26 | –CH3 | –CF3 | –H | –H | 4.0 ± 1.2 | 38.0 ± 2.0 | 9.5 |
| 27 | –CH3 | –CH3 | –H | –H | 4.2 ± 1.9 | 124 ± 68 | 29.5 |
| 28 | –H | –H | –H | –H | 0.2 ± 0.0 | 198 ± 64 | 990 |
| 29 | –H | –Cl | –H | –H | 0.4 ± 0.1 | 64.5 ± 13.2 | 161 |
| 30 |
|
–H | –H | –H | 16.0 ± 5.0 | 53.5 ± 0.5 | 3.3 |
Blood–brain barrier permeability studies by CNS MPO
In this study, all of the novel σ1 receptor ligands were tested their blood–brain barrier permeability by using the central nervous system multiparameter optimization (CNS MPO) desirability tool which was reported by Travis T. Wager et al.29,30 In CNS MPO appraisal system, six structural properties of candidates would be used such as Clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, ClogD, MW, TPSA, HBDs and pKa that were determined as important factors in the BBB permeability. Each property was valued between 0 to 1 and weighted equally. Final collective score ranged from 0 to 6, with the higher scores of CNS MPO correlating with desirable brain permeability. Through the analysis of CNS MPO scores on more than hundreds of the drugs and original candidates acting on the CNS, a majority of them had CNS MPO desirability scores greater than 4.30 As showed in Table 2, majority of the newly synthesized alkoxyisoxazoles obtained CNS MPO scores greater than 4. In particular, compound 28 scored the highest CNS MPO scores (5.4), in addition to the observed high binding affinities at σ1 receptor (Table 1).
P, ClogD, MW, TPSA, HBDs and pKa that were determined as important factors in the BBB permeability. Each property was valued between 0 to 1 and weighted equally. Final collective score ranged from 0 to 6, with the higher scores of CNS MPO correlating with desirable brain permeability. Through the analysis of CNS MPO scores on more than hundreds of the drugs and original candidates acting on the CNS, a majority of them had CNS MPO desirability scores greater than 4.30 As showed in Table 2, majority of the newly synthesized alkoxyisoxazoles obtained CNS MPO scores greater than 4. In particular, compound 28 scored the highest CNS MPO scores (5.4), in addition to the observed high binding affinities at σ1 receptor (Table 1).
| Compound | ClogP |
ClogD |
MW | TPSA | HBDs | pKa | CNS MPO scoresb |
|---|---|---|---|---|---|---|---|
|
a The data sources and explanations. ClogP: calculated partition coefficient; ClogD: calculated distribution coefficient at pH 7.4; MW: molecular weight; TPSA: topological polar surface area; HBDs: number of hydrogen-bond donors: pKa: most basic center. The calculated physicochemical properties of the derivatives were obtained using standard commercial software: ACD/Laboratories, version 6.0 for ClogP, pKa, HBDs and ClogD at PH 7.4, ChemBioDraw, version 14.0 for MW and TPSA.
b Last CNS MPO scores were calculated by the tools reported by Travis T. Wager et al.30
|
|||||||
| 7 | 3.27 | 2.08 | 302.37 | 43.29 | 0 | 8.89 | 5.4 |
| 17 | 3.55 | 2.18 | 320.36 | 43.29 | 0 | 8.89 | 5.2 |
| 18 | 3.44 | 2.17 | 320.36 | 54.15 | 0 | 8.94 | 5.3 |
| 19 | 4.27 | 2.17 | 381.07 | 43.29 | 0 | 8.89 | 4.7 |
| 20 | 4.27 | 3.11 | 381.27 | 43.29 | 0 | 8.89 | 4.2 |
| 21 | 3.68 | 2.22 | 338.35 | 43.29 | 0 | 8.89 | 5.1 |
| 22 | 3.75 | 2.33 | 338.35 | 43.29 | 0 | 8.89 | 5.0 |
| 23 | 4.77 | 3.47 | 371.26 | 43.29 | 0 | 8.89 | 3.9 |
| 24 | 4.89 | 3.75 | 371.26 | 43.29 | 0 | 8.89 | 3.7 |
| 25 | 4.32 | 3.15 | 354.80 | 43.29 | 0 | 8.89 | 4.3 |
| 26 | 4.40 | 3.14 | 370.37 | 43.29 | 0 | 8.89 | 4.2 |
| 27 | 3.77 | 2.53 | 316.18 | 43.29 | 0 | 8.89 | 4.9 |
| 28 | 2.68 | 0.86 | 288.15 | 52.08 | 1 | 8.94 | 5.4 |
| 29 | 3.52 | 1.68 | 322.11 | 52.08 | 1 | 8.94 | 5.2 |
| 30 | 3.64 | 1.97 | 342.19 | 43.29 | 0 | 8.75 | 5.3 |
Neuritogenesis studies in N1E-115 cells
Following the radioligand binding studies and CNS MPO calculations, all of the synthesized derivatives were further tested in cellular neuritogenesis studies. In this study, N1E-115 neuronal cells were used as a screening platform for assessing the neuritogenesis effects of candidate compounds.31,32 All the derivatives and positive control (compound 7 and RC-33) were assessed at 10 μM concentration in DMSO, and vehicle group using DMSO alone. The test results are shown in Fig. 3. Gratifyingly, a large proportion of the drug administration significantly increased the average neurite length of N1E-115 cells at 10 μM concentration vs. vehicle group, suggestive of a neuritogenesis effect. Through the analysis of test results, most of the disubstituted derivatives such as compound 22–25 exhibited greater neuritogenesis efficacy when compared with vehicle control. The 4-fluoro substituted compound 18 and 3-trifluoromethyl substituted compound 26 displayed better efficacy than other mono-substituted compounds. The higher efficacies observed for compounds 28 and 29 indicated that the secondary amine moiety was beneficial for promoting neuritogenesis efficacy within this series. Particularly, we obtained six compounds (18, 22, 24, 25, 28 and 29) that showed preferable neurite outgrowth effects compared to the parent compound 7 and positive control RC-33 in N1E-115 neuronal cells.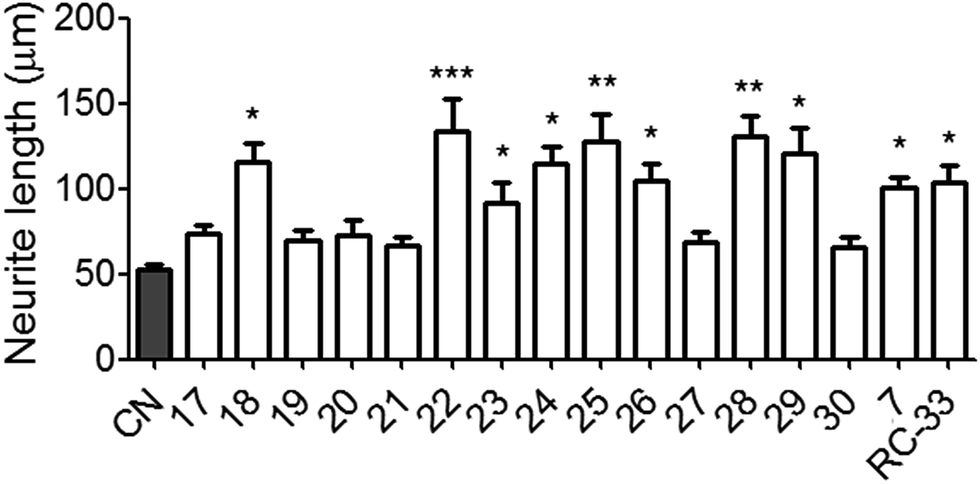 | ||
| Fig. 3 Neurite outgrowth effects of all derivatives in N1E-115 neuronal cells. aSee Experimental section. Each histograms represent the mean ± SEM of at least five different experiments. Statistically significant differences: *p < 0.05, **p < 0.01, ***p < 0.001 vs. control (CN) using DMSO alone. | ||
Selectivity studies at selected neurotransmitter transporters
In our previous work, we found that some derivatives based on the alkoxyisoxazole scaffold not only bind to σ1 receptor but also to common neurotransmitter transporters, such as DAT, NET and SERT.27,33 In order to exclude the potential interference of other receptor pathways to the observed neuritogenesis efficacy, we assessed the binding affinities of compounds 7, 18, 22, 24, 25, 28 and 29 at the biogenic amine transporters (NET, DAT, and SERT). Gratifyingly, no appreciable binding affinity was observed for these transporters using 10 μM concentration of tested compounds. This set of results indicated that the selected compounds had excellent selectivity for σ1 receptor over DAT, NAT and SERT (Table 3).| Compound | K i, DAT (nM) | K i, NET (nM) | K i, SERT (nM) | K i, σ1 (nM) |
|---|---|---|---|---|
| a See Experimental section. DAT: dopamine transporter; NET: norepinephrine transporter; SERT: serotonin transporter. Radioligands: DAT: [3H] WIN35428; NET: [3H] nisoxetine; SERT: [3H] citalopram. b The Ki values for GBR12909, desipramine, amitriptyline and compound 7 are cited from the literature.27 c NA: not active, defined as <50% binding in the primary assay at 10 μM. Ki values were determined for those targets where the binding efficacy at 10 μM was greater than 50%. d Data are expressed as average of Ki values and each performed from three independent experiments. | ||||
| GBR12909 | 8.8 ± 3.8b | — | — | — |
| Desipramine | — | 2.5 ± 0.4 | — | — |
| Amitriptyline | — | — | 5.4 ± 0.9 | — |
| 7 | NAc | NA | NA | 1.8 ± 0.2 |
| 18 | 5631d | NA | NA | 0.9 ± 0.1 |
| 22 | >10000 |
2878 | NA | 2.4 ± 1.2 |
| 24 | 1811 | 1606 | NA | 7.8 ± 3.2 |
| 25 | >10000 |
1780 | NA | 1.3 ± 0.4 |
| 28 | NA | 7102.5 | NA | 0.2 ± 0.0 |
| 29 | 1518 | 600.5 | NA | 0.4 ± 0.1 |
Conclusions
In summary, 14 novel derivatives based on the N-cyclobutylaminoethoxyisoxazole scaffold were designed and synthesized. Most of the derivatives showed potent binding affinities to the σ1 receptor (Ki < 10 nM) and good selectivity over the σ2 receptor in radioligand binding tests. CNS MPO scores were calculated for predicting their blood–brain barrier permeability and the results showed most of derivatives obtained favourable CNS MPO scores greater than 4. All of the compounds were assessed for their neurite outgrowth efficacy in N1E-115 cells. Compounds 18, 22, 24, 25, 28 and 29 exhibited significant neuritogenesis effects at 10 μM concentration vs. vehicle group and superior efficacy of increasing the average neurite length compared with compound 7 and RC-33. Moreover, compounds 18, 22, 24, 25, 28 and 29 showed great selectivity for σ1 receptor over DAT, NET, and SERT transporters. Among these derivatives, compound 28 was found as the most promising compound with excellent in vitro binding profile (Ki σ1 = 0.2 nM, Ki σ2 = 198 nM), high CNS MPO score (5.4) and significant neuritogenesis efficacy, which will be further developed as potential therapeutics for neurodegenerative diseases.Experimental section
Chemistry
General procedures for vitro binding studies
Radioligand competition studies were carried out by the National Institute of Mental Health's Psychoactive Drug Screening Program, Contract #HHSN-271-2008-00025-C (NIMH PDSP). The general procedures are listed as follows:All the test compounds solution (1 mg mL−1) were prepared by dissolving in DMSO or buffer solvent (50 mM Tris–HCl, pH = 8.0). The filter-mats were presoaked in 0.5% aqueous polyethyleneimine solution for 2 h at rt before use. All binding experiments were carried out in poly-L-lysine-coated 96-well plate and the final test concentration (50 μM, 5 μM, 1.5 μM, 500 nM, 150 nM, 50 nM, 15 nM, 5 nM, 1.5 nM, 0.5 nM, 0.05 nM) were prepared by addition of Tris–HCl solution (50 mM, pH = 8.0). The following specific radioligands and tissue source were used: (a) σ1 receptor, [3H]-(+)-pentazocine, Guinea pig brain homogenate; (b) σ2 receptor, [3H]-DTG, PC-12 cells. 100 μL of corresponding radioligand solution, 100 μL test compound solution and 50 μL of the respective receptor preparation into each well of the plate (total volume 250 μL). During the incubation, the plates were shaken at a speed of 500–600 rpm at the specified temperature (σ1 receptor, 37 °C, 150 min; σ2 receptor, rt, 120 min). The incubation was followed by a rapid vacuum filtration through Whatman GF/B glass filters, and the filtrates were washed twice with 10 mL of cold buffer and transferred to the 6 mL scintillation vials. EcoScint scintillation fluid (4.0 mL) was added, and the radioactivity bound was measured using a Beckman LS 6500 liquid scintillation counter. All experiments were performed at least three times.
For experimental details please refer to the PDSP website http://pdsp.med.unc.edu/.28
General procedures for cellular evaluation
RC-33(5). White solid; 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 7.64 (m, 4H), 7.46 (t, J = 7.6 Hz, 2H), 7.36 (d, J = 8.1 Hz, 3H), 3.03 (m, 2H), 2.88–2.68 (m, 4H), 2.06 (m, 2H), 1.72 (m, 6H), 1.43–1.28 (m, 1H), 1.25 (d, J = 6.9 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 144.9, 139.9, 138.2, 128.9, 127.4, 127.2, 126.8, 126.5, 54.7, 51.8, 51.8, 36.7, 30.7, 22.3, 21.4.
Compound 7. White solid; 1H NMR (400 MHz, DMSO-d6) δ 10.99 (s, 1H), 7.36–7.28 (m, 2H), 7.03 (d, J = 8.0 Hz, 2H), 6.99 (t, J = 8.0 Hz, 1H), 6.45 (s, 1H), 5.19 (s, 2H), 4.56 (m, 2H), 3.81–3.68 (m, 1H), 2.67 (s, 3H), 2.38–2.24 (m, 2H), 2.17 (m, 2H), 1.68 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 170.9, 169.6, 157.0, 129.9, 122.3, 115.0, 95.4, 63.6, 61.1, 59.9, 52.0, 36.8, 25.9, 12.6.
Conflicts of interest
There are no conflicts of interest to declare.Abbreviations
| AD | Alzheimer's disease |
| PD | Parkinson's disease |
| ALS | Amyotrophic lateral sclerosis |
| HD | Huntington's disease |
| CNS | Central nervous system |
| ER | Endoplasmic reticulum |
| NMDA | N-Methyl-D-aspartate |
| IP3 | Inositol 1,4,5-trisphosphate |
| NGF | Nerve growth factor |
| SAR | Structure–activity relationship |
| NIMH-PDSP | National Institute of Mental Health-Psychoactive Drug Screening Program |
| CNS MPO | Central nervous system multiparameter optimization |
| DAT | Dopamine transporter |
| NET | Norepinephrine transporter |
| SERT | Serotonin transporter |
| CC | Column chromatography |
| rt | Room temperature |
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81402780), the Natural Science Foundation of Shanghai (14ZR1411000), the Natural Science Foundation of Jiangsu Province (BK20160032), and Innovation Program of East China Normal University (78210263). We thank the PDSP for performing binding affinity assays. We thank Dr Hendra Gunosewoyo for proofreading the manuscript.References
- I. Solanki, P. Parihar and M. S. Parihar, Neurochem. Int., 2016, 95, 100–108 CrossRef CAS.
- J. Hardy, Ann. N. Y. Acad. Sci., 1999, 74, 835–837 CAS.
- U. Dirnagl, Ann. N. Y. Acad. Sci., 2012, 1268, 21–25 CrossRef PubMed.
- V. Francardo, Y. Schmitz, D. Sulzer and M. A. Cenci, Exp. Neurol., 2017, 298, 137–147 CrossRef CAS.
- T. Wieloch and K. Nikolich, Curr. Opin. Neurobiol., 2006, 16, 258–264 CrossRef CAS.
- S. Kourrich, T. P. Su, M. Fujimoto and A. Bonci, Trends Neurosci., 2012, 35, 762–771 CrossRef CAS.
- W. R. Martin, C. G. Eades, J. A. Thompson, R. E. Huppler and P. E. Gilbert, J. Pharmacol. Exp. Ther., 1976, 197, 517–532 CAS.
- M. Hanner, F. F. Moebius, A. Flandorfer, H. G. Knaus, J. Striessnig, E. Kempner and H. Glossmann, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8072–8077 CrossRef CAS.
- T. Hayashi, R. Rizzuto, G. Hajnoczky and T. P. Su, Trends Cell Biol., 2009, 19, 81–88 CrossRef CAS.
- H. R. Schmidt, S. D. Zheng and E. Gurpinar, et al. , Nature, 2016, 532, 527–530 CrossRef CAS.
- T. Hayashi and T. P. Su, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 491–496 CrossRef CAS.
- T. Hayashi and T. P. Su, Cell, 2007, 131, 596–610 CrossRef CAS.
- T. Hayashi, J. Pharmacol. Sci., 2015, 127, 2–5 CrossRef CAS.
- M. V. Sofroniew, Trends Neurosci., 2009, 32, 638–647 CrossRef CAS.
- T. P. Su, T. Hayashi, T. Maurice, S. Buch and A. E. Ruoho, Trends Pharmacol. Sci., 2010, 31, 557–566 CrossRef CAS.
- K. Ruscher, M. Shamloo and M. Rickhag, et al. , Brain, 2011, 134, 732–746 CrossRef.
- M. Pabba, A. Y. Wong and N. Ahlskog, et al. , J. Neurosci., 2014, 34, 11325–11338 CrossRef.
- M. Fujimoto, T. Hayashi, R. Urfer, S. Mita and T. P. Su, Synapse, 2012, 66, 630–639 CrossRef CAS.
- S. Sha, W. J. Qu, L. Li, Z. H. Lu, L. Chen, W. F. Yu and L. Chen, CNS Neurosci. Ther., 2013, 19, 705–713 CrossRef CAS.
- M. Takebayashi, T. Hayashi and T. P. Su, Synapse, 2004, 53, 90–103 CrossRef CAS.
- T. Ishima, T. Nishimura, M. Iyo and K. Hashimoto, Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 2008, 32, 1656–1659 CrossRef CAS.
- R. Mancuso, S. Oliván and A. Rando, et al. , Neurotherapeutics, 2012, 9, 814–826 CrossRef CAS PubMed.
- D. Rossi, A. Pedrali, M. Urbano, R. Gaggeri and S. Collina, et al. , Bioorg. Med. Chem., 2011, 19, 6210–6224 CrossRef CAS.
- D. Rossi, A. Marra, P. Picconi, S. Pricl and S. Collina, et al. , Bioorg. Med. Chem., 2013, 21, 2577–2586 CrossRef CAS.
- A. Marra, D. Rossi, L. Pignataro and S. Collina, et al. , Future Med. Chem., 2016, 8, 287–295 CrossRef CAS PubMed.
- S. Collina, M. Rui, G. Rossino and S. D. Volpe, et al. , Future Med. Chem., 2017, 9, 2029–2051 CrossRef CAS PubMed.
- H. Sun, M. Shi, W. Zhang and L. F. Yu, et al. , J. Med. Chem., 2016, 59, 6329–6343 CrossRef CAS.
- K i determinations were generously provided by the National Institute of Mental Health's Psychoactive Drug Screening Program, contract no. HHSN-271-2008-00025-C (NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth MD, PhD, at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda, MD, USA.
- T. T. Wager, R. Y. Chandrasekaran and X. Hou, et al. , ACS Chem. Neurosci., 2010, 1, 420–434 CrossRef CAS PubMed.
- T. T. Wager, X. Hou and P. R. Verhoest, et al. , ACS Chem. Neurosci., 2016, 7, 767–775 CrossRef CAS.
- P. N. M. Konings, Neurosci. Lett., 1994, 178, 115–118 CrossRef CAS.
- S. Chang, W. C. Ruan, Y. Z. Xu, H. Liao and T. Pang, et al. , Acta Pharmacol. Sin., 2017, 38, 29–40 CrossRef CAS.
- L. F. Yu, H. K. Zhang, H. Gunosewoyo and A. P. Kozikowski, ACS Med. Chem. Lett., 2012, 3, 1054–1058 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectrum of the corresponding compounds (1H and 13C NMR of all final compounds). See DOI: 10.1039/c8ra00072g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |