
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An efficient route to N-alkylated 3,4-dihydroisoquinolinones with substituents at the 3-position†
Aoi Tazawaab,
Junki Andoa,
Kohei Ishizawaa,
Isao Azumaya *b,
Hidemasa Hikawab and
Minoru Tanaka*a
*b,
Hidemasa Hikawab and
Minoru Tanaka*a
aSohyaku Innovative Research Division, Mitsubishi Tanabe Pharma Corporation, 1000 Kamoshida-cho, Aoba-ku, Yokohama-shi, Kanagawa 227-0033, Japan. E-mail: tanaka.minoru@mw.mt-pharma.co.jp
bFaculty of Pharmaceutical Sciences, Toho University, 2-2-1 Miyama, Funabashi, Chiba 274-8510, Japan. E-mail: isao.azumaya@phar.toho-u.ac.jp
First published on 7th February 2018
Abstract
A facile synthetic procedure for the production of N-alkylated 3,4-dihydroisoquinolinone derivatives is described. The desired products were obtained by N-alkylation of 3,3′-dimethyl-3,4-dihydroisoquinoline derivatives followed by oxidation of the resulting iminium salts. Reaction conditions for both steps were very mild and the desired cyclization products could be obtained in good yield. This strategy allows the generation of N-substituted 3,4-dihydroisoquinolinone derivatives with substituents at the 3-position.
Introduction
3,4-Dihydroisoquinolinone 1 is an important member of the class containing a core structure found in compounds that exhibit biological and pharmacological properties, such as anti-nausea/vomiting,1 antidiabetic,2,3 and antiallergy/antiasthmatic4 activities. Some examples of pharmacologically active compounds that include the dihydroisoquinolinone moiety are shown in Fig. 1. Its suitable size and moderate polarity as a pharmacophore make 3,4-dihydroisoquinolinone a suitable scaffold that has been widely used in various drug candidates. For 3,4-dihydroisoquinolinone derivatives, the introduction of substituents at the 3-position generally improve their biostability5 because the substituent prevents oxidation of the unsubstituted 3-position. Nevertheless, only a few methods for effective preparation of N-alkylated 3,4-dihydroisoquinolinone with substituents at the 3-position are available, most of which are related to the synthesis of N-methyl analogues.6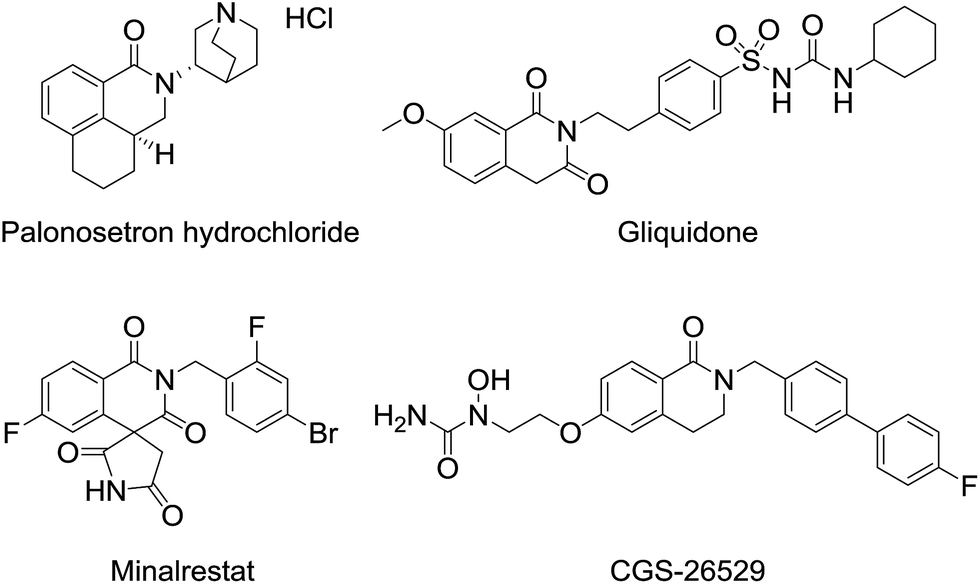 | ||
| Fig. 1 Representative pharmacologically active dihydroisoquinolinones. | ||
One of the simplest synthetic methods of the N-alkylated dihydroisoquinolinone skeleton is direct N-alkylation of N-unsubstituted dihydroisoquinolinone A (Scheme 1).7 The N-alkylation reaction, however, often does not proceed in the case of substrates with some substituents at the 3-position. Another method for preparing the skeleton involves benzylic oxidation of N-alkylated tetrahydroisoquinoline B-1.8 However, this route cannot be applied to substrates in which another position can be easily oxidized. Using this approach, the precursor is easy to synthesize because the nitrogen atom of tetrahydroisoquinolines B-2 is more nucleophilic than that of 3,4-dihydroisoquinolinones A. However, in the case of 3-substituted analogues, precursors B-2 are not obtained via cyclization reactions of the corresponding phenethylamine derivatives B-3.9
 | ||
| Scheme 1 Retrosynthetic analysis of 3-substituted 3,4-dihydroisoquinolinones. | ||
Thus, we have focused on the oxidation reaction of N-alkylated iminium salts C-1. As previous reports are limited to 3-unsubstituted analogues,10 we examined whether this strategy was compatible with 3-substituted dihydroisoquinolinone derivatives. In the course of the reaction, N-alkylation proceeds via a less sterically hindered transition state when compared with that of direct N-alkylation of dihydroisoquinolinone A. Therefore, this method has an apparent advantage for N-alkylation of sterically hindered analogues. In this work, we developed an effective synthetic method of 3-substituted N-alkyl-dihydroisoquinolinone derivatives via iminium intermediates.
Results and discussion
Preparation of 3,3-dimethyl-dihydroisoquinolinium salt
3,3-Dimethyl-3,4-dihydroisoquinoline 8, was prepared by referring to a previously described procedure (Scheme 2).11 Our synthesis began with commercially available 2-bromo-phenylacetic acid 2, which was converted into an ester and reacted with methyl Grignard reagent to afford the tertiary alcohol 4. Nucleophilic substitution of 4 with chloroacetonitrile followed by reaction with thiourea afforded the tertiary amine 612 in high yield. Amidation of the tertiary amine 6 with ethyl formate under neat conditions produced the formamide 7 in 85% yield. Tandem cyclization of 7 using oxalyl chloride followed by iron(III) chloride afforded a tricyclic intermediate, which was pyrolyzed by MeOH–H2SO4 to give 3,3-dimethyl-3,4-dihydroisoquinoline in 89% yield. | ||
Scheme 2 Reagents and conditions: (i) conc. H2SO4 (5 mol%), MeOH, reflux: (ii) MeMgBr (2.5 equiv.), THF, 0 °C to rt, 98% (2 steps); (iii) ClCH2CN (2 equiv.), conc. H2SO4 (1.3 equiv.), AcOH (3 equiv.), 0 °C to rt; (iv) thiourea (1.2 equiv.), EtOH–AcOH (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 100 °C, quant (2 steps); (v) EtOCHO (2.0 M), 60 °C, 85%; (vi) (COCl)2 (1.1 equiv.), CH2Cl2; (vii) FeCl3 (1.5 equiv.); (viii) MeOH–H2SO4 (19:1), reflux, 89%. :1), 100 °C, quant (2 steps); (v) EtOCHO (2.0 M), 60 °C, 85%; (vi) (COCl)2 (1.1 equiv.), CH2Cl2; (vii) FeCl3 (1.5 equiv.); (viii) MeOH–H2SO4 (19:1), reflux, 89%. | ||
After obtaining 3,3-dimethyl-3,4-dihydroisoquinoline 8, we examined the formation of isoquinolinium key precursors 9. For the preparation of the 3,3-dimethyl-dihydroisoquinolinium salt 9 (Scheme 3), dihydroisoquinoline 8 was reacted with methyl bromoacetate (2 equiv.) in acetonitrile at 60 °C. After 6 h, the desired product 9 was produced and precipitated as a colorless powder. The obtained powder was sufficiently pure for use in the next reaction without further purification.
 | ||
| Scheme 3 Alkylation of 3,3-dimethyl-3,4-dihydroisoquinoline. | ||
Synthesis of 3-substituted dihydroisoquinolinones
We next examined the oxidation of the 3,4-dihydro-isoquinolinium salt 9. The reaction was first examined in the presence of hydrochloric acid and dimethyl sulfoxide with reference to a previously described procedure (Table 1, entry 1),10a but the desired product was not obtained. Next, we focused on optimizing the oxidation conditions. When 9 was reacted with m-chloroperoxybenzoic acid, dealkylation of 9 to dihydroisoquinoline 8 occurred predominantly (entry 2). Employing Dess–Martin periodinane as the oxidant, the reaction became messy and a complex product mixture that included 8 was obtained (entry 3). When oxone was employed as the oxidant, no reaction was observed (entry 4). However, when 9 was reacted with oxone in the presence of sodium bicarbonate, desired product 10 was obtained as the major product (entry 5). This suggested that basic conditions were preferable for 3-substituted dihydroisoquinolinones. As the yield was not satisfactory, further experiments were conducted. Treatment of 9 with potassium ferricyanide in the presence of potassium hydroxide gave the desired product as the hydrolyzed carboxylic acid 11 in 69% yield (entry 6).13 Dealkylated compound 8 was also obtained as a minor product.
|
||
|---|---|---|
| Entry | Conditionsa | Yields (%) |
| a 3,3-dimethyl-3,4-dihydroisoquinolinium salt 9 (0.1 mmol), oxidants, and solvent (0.1 M) were reacted for 12 h at ambient temperature. | ||
| 1 | Conc. HCl, DMSO (1:7) |
N.R. |
| 2 | mCPBA (1.5 equiv.), CH2Cl2 | 8: 88% |
| 3 | Dess–Martin periodinane (1.2 equiv.), CH2Cl2 | Decomposed |
| 4 | Oxone (2 equiv.), MeCN/H2O (3:2) |
N.R. |
| 5 | Oxone (2 equiv.), NaHCO3 (2 equiv.), MeCN/H2O (3:2) |
10: 39% |
| 6 | K3Fe(CN)6 (6 equiv.), KOH (24 equiv.), dioxane/H2O (1:2) |
11: 69%, 8: 25% |

We also examined the oxidation reaction of isoquinolinium salts, including those with other alkyl substituents on the nitrogen atom.14 As expected, under optimal conditions, the desired dihydroisoquinolinones were obtained in high yields (Scheme 4). However, when dihydroisoquinolinium salt 16 was employed in the oxidation reaction, the desired product was obtained only in trace amounts, while dealkylated compound 8 was generated as the major product (Scheme 5). This was probably due to steric hindrance at the oxidation site.
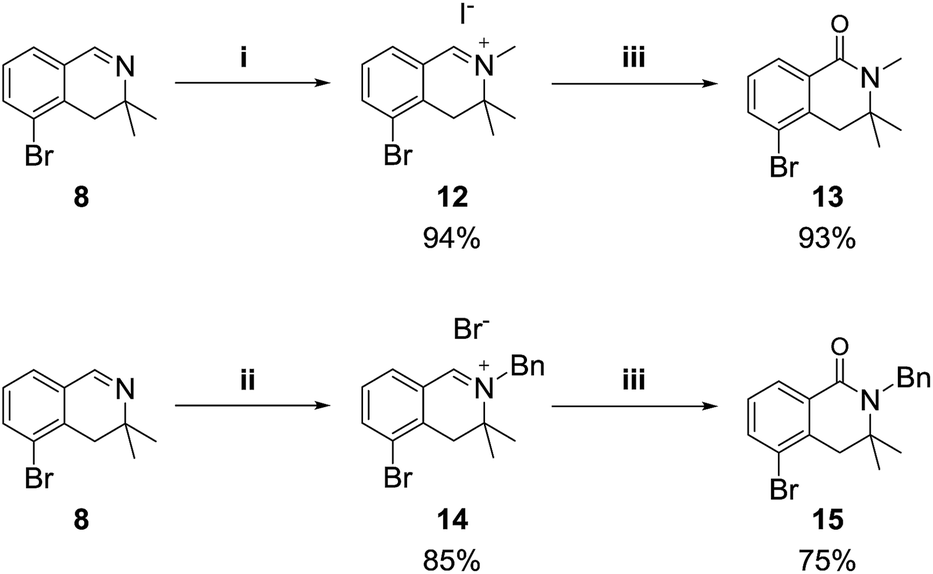 | ||
| Scheme 4 Reagents and conditions: (i) MeI (10 equiv.), acetone, rt; (ii) BnBr (10 equiv.), toluene, 70 °C; (iii) K3Fe(CN)6 (6 equiv.), KOH (24 equiv.), dioxane/H2O (1:2), rt. | ||
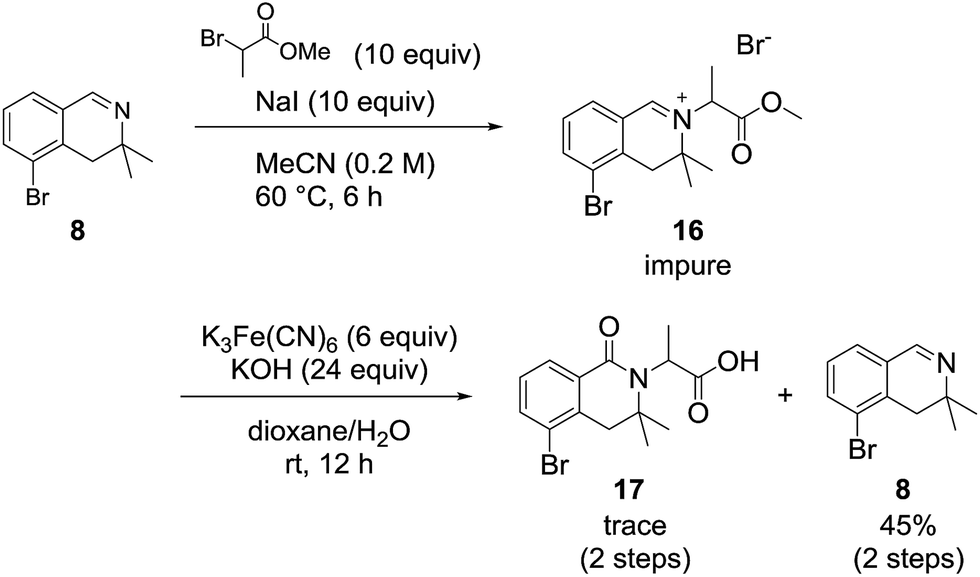 | ||
| Scheme 5 Oxidation of a 3,3-dimethyl-3,4-dihydroisoquino-linium derivative. | ||
Investigation of the reaction mechanism
We hypothesize that the mechanism of the reaction involves a hydroxide ion as an oxygen source and proceeds through a radical pathway, because hexacyanoferrate(III) is a typical one-electron oxidizing agent.15To verify this hypothesis, we conducted the oxidation reaction of isoquinolinium salt 12 under a nitrogen atmosphere. As a result, the reaction proceeded smoothly and the desired product 13 was obtained in excellent yield (Scheme 6, eqn (1)), suggesting that atmospheric oxygen (O2) was not the oxygen source. Next, we investigated the reaction of 12 with only potassium hydroxide solution to confirm that hydroxide ions were the oxygen source. The tetrahydroisoquinolinol intermediate 18 was determined by LC/MS and 1H NMR analysis16 of the crude mixture. Furthermore, when potassium ferricyanide was added to the solution of intermediate 18, generation of the desired product was detected (eqn (2)). When the reaction was conducted in the presence of a radical scavenger, the reaction mixture became messy and the desired product was not detected (eqn (3)). Based on these experimental results, we concluded that the oxidation reaction proceeded through a radical process via the tetrahydroisoquinolinol intermediate 18.
 | ||
| Scheme 6 Control experiments for investigating the mechanism. | ||
Conclusions
We have developed a novel and efficient approach for the preparation of 3-substituted N-alkyl-3,4-dihydroisoquinolinones via the oxidation of iminium intermediates. Using this method, N-alkyl-3,4-dihydroisoquinolinones bearing substituents at the 3-position, which had not been obtained by N-alkylation of the amide nitrogen, could be obtained in moderate yield. We believe this method will contribute to the development of pharmacologically active compounds based on this skeleton. However, there is currently a limitation of the substituent patterns on the nitrogen atom such as a reactive methylene moiety, which we are currently trying to overcome by searching for various reaction conditions including oxidizing reagents.Experimental
General information
1H and 13C NMR spectra were recorded on a Bruker 400 ULTRASHIELD PLUS. 1H and 13C chemical shifts are reported in ppm downfield from tetramethylsilane (TMS, δ scale) with the solvent resonances as internal standards. The following abbreviations were used to explain the multiplicities: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; band, several overlapping signals; br, broad. IR spectra were recorded on a PerkinElmer Spectrum One FT-IR Spectrometer using attenuated total reflection (ATR). Melting points (mp) were recorded on a BÜCHI Melting Point B-545. Mass spectra were provided at the DMPK Research Laboratory, Mitsubishi Tanabe Pharma Corporation.:EtOAc = 3:1) to give methyl 2-(5-bromo-3,3-dimethyl-1-oxo-3,4-dihydroisoquinolin-2(1H)-yl)acetate 10 (48.2 mg, 0.148 mmol) in 39% yield. 1H NMR (400 MHz, CDCl3) δH: 8.04 (1H, dd, J = 7.7, 1.5 Hz), 7.69 (1H, dd, J = 8.2, 1.0 Hz), 7.22 (1H, dd, J = 8.2, 7.7 Hz), 4.30 (2H, s), 3.76 (3H, s), 3.12 (2H, s), 1.35 (6H, s); 13C NMR (100 MHz, CDCl3) δC: 170.4, 163.9, 136.1, 135.9, 130.1, 128.2, 127.7, 123.0, 56.2, 52.3, 43.4, 42.2, 26.9, 26.9; IR (ATR cm−1) νmax: 2950, 1750, 1643, 1562, 1455, 1413, 1386, 1199, 1167, 1012, 970, 754, 653; HRMS (ESI) [M + H]+ calculated for C14H17BrNO3: 326.03863, found: 326.03908.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the members of this team for stimulating discussions, in particular Dr Masayuki Watanabe, Mr Kazunari Shimooka and Ms Kaoru Marukawa. We also thank Ms Kaori Murakoshi and Ms Saori Tahara for technical support of LC-MS/MS and FT-IR analysis. We thank Mr Iwao Takamuro for helpful advice. This research was supported by the Mitsubishi Tanabe Pharma Corporation.Notes and references
- R. D. Clark, A. B. Miller, J. Berger, D. B. Repke, K. K. Weinhardt, B. A. Kowalczyk, R. M. Eglen, D. W. Bonhaus, C. H. Lee, A. D. Michael, W. L. Smith and E. H. Wong, J. Med. Chem., 1993, 36, 2645–2657 CrossRef CAS PubMed.
- E. Kutter, G. Griss, W. Grell and M. Kleemann, US Pat., US1973/3708486, 1973.
- M. S. Malamas, US Pat., US1992/5106978, 1992.
- J. H. Van Duzer and D. M. Roland, EP Pat., EP0526402, 1993.
- (a) Y. Lou, X. Han, A. Kuglstatter, R. K. Kondru, A. K. Sweeney, M. Soth, N. Dewdney, H. Zecic, J. P. Taygerly, K. Sarma, J. Hong, R. J. Hill, T. Gabriel, D. M. Goldstein and T. D. Owens, J. Med. Chem., 2015, 58, 512–516 CrossRef CAS PubMed; (b) G. Szántó, A. Makó, I. Vágó, T. Hergert, I. Bata, B. Farkas, S. Kolok and M. Vastag, Bioorg. Med. Chem. Lett., 2016, 26, 3905–3912 CrossRef PubMed; (c) P. Qian, Y.-B. Zhang, Y.-F. Yang, W. Xu and X.-W. Yang, Molecules, 2017, 22, 214–232 CrossRef PubMed.
- (a) D. M. Bailey and C. G. DeGrazia, J. Org. Chem., 1970, 35, 4088–4093 CrossRef CAS; (b) D. M. Bailey and C. G. DeGrazia, Tetrahedron Lett., 1970, 9, 633–636 CrossRef; (c) W. Liu, Q. Yu, L. Hu, Z. Chen and J. Huang, Chem. Sci., 2015, 6, 5768–5772 RSC; (d) T. Miura, M. Morimoto, M. Yamauchi and M. Murakami, J. Org. Chem., 2010, 75, 5359–5362 CrossRef CAS PubMed; (e) A. G. Schultz, T. J. Guzi, E. Larsson, R. Rahm, K. Thakkar and J. M. Bidlack, J. Org. Chem., 1998, 63, 7795–7804 CrossRef CAS.
- (a) M. J. Fisher, B. P. Gunn, S. Um and J. A. Jakubowski, Tetrahedron Lett., 1997, 38, 5747–5750 CrossRef CAS; (b) D. Zhou, J. L. Gross, A. B. Adedoyin, S. B. Aschmies, J. Brennan, M. Bowlby, L. Di, K. Kubek, B. J. Platt, Z. Wang, G. Zhang, N. Brandon, T. A. Comery and A. J. Robichaud, J. Med. Chem., 2012, 55, 2452–2468 CrossRef CAS PubMed; (c) F. A. Davis and Y. W. Andemichael, Tetrahedron Lett., 1998, 39, 3099–3102 CrossRef CAS; (d) J. M. Fevig, J. Feng, K. A. Rossi, K. J. Miller, G. Wu, C.-P. Hung, T. Ung, S. E. Malmstrom, G. Zhand, W. J. Keim, M. J. Cullen, K. W. Rohrbach, Q. Qu, J. Gan, M. A. Pelleymounter and J. A. Robl, Bioorg. Med. Chem. Lett., 2013, 23, 330–335 CrossRef CAS PubMed.
- (a) A. G. Schultz, T. J. Guzi, E. Larsson, R. Rahm, K. Thakkar and J. M. Bidlack, J. Org. Chem., 1998, 63, 7795–7804 CrossRef CAS; (b) W.-J. Huang, O. V. Singh, C.-H. Chen, S.-Y. Chiou and S.-S. Lee, Helv. Chim. Acta, 2002, 85, 1069–1078 CrossRef CAS; (c) A.-R. Song, J. Yu and C. Zhang, Synthesis, 2012, 22, 2903–2909 Search PubMed; (d) M. Nakanishi and C. Bolm, Adv. Synth. Catal., 2007, 349, 861–864 CrossRef CAS; (e) M. Ochiai, D. Kajishima and T. Sueda, Tetrahedron Lett., 1999, 40, 5541–5544 CrossRef CAS; (f) D. Crich and V. Krishnamurthy, Tetrahedron, 2006, 62, 6830–6840 CrossRef CAS.
- When 6 was reacted with paraformaldehyde (100 wt%) and AcOH–H2SO4 (0.8 M, 3:1) from ambient temperature to 80 °C to generate the tetrahydroisoquinoline, no reaction was observed.
- (a) S. Ruchirawat, M. Chuankamnerdkarn and S. Thianpatanagul, Tetrahedron Lett., 1984, 25, 3479–3480 CrossRef CAS; (b) A. P. Venkov and S. M. Statkova-Abeghe, Tetrahedron, 1996, 52, 1451–1460 CrossRef CAS; (c) B. Bechi, S. Herter, S. Mckenna, C. Riley, S. Leimkühler, N. J. Turner and A. J. Carnell, Green Chem., 2014, 16, 4524–4529 RSC.
- R. D. Larsen, R. A. Reamer, E. G. Corley, P. Davis, E. J. J. Grabowski, P. J. Reider and I. Shinkai, J. Org. Chem., 1991, 56, 6034–6038 CrossRef CAS.
- S. D. Brown and D. J. Matthews, US Pat., US2012/0252802, 2012.
- (a) N. Itou and S. Sugasawa, Tetrahedron, 1957, 1, 45–48 CrossRef; (b) E. Dominguez, E. M. Marigorta, L. Carrillo and R. Fañanás, Tetrahedron, 1991, 47, 9253–9258 CrossRef CAS.
- A. G. Mikhailovskii, V. S. Shklyaev and E. V. Feshina, Chem. Heterocycl. Compd., 1998, 34, 211–215 CrossRef CAS.
- B. S. Thyagarajan, Chem. Rev., 1958, 58, 439–460 CrossRef CAS.
- In 1H NMR analysis, we confirmed the disappearance of the iminium proton peak and detected the doublet peaks of the OH and O–CH–N moieties. See the ESI† for more details.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13627g |
| This journal is © The Royal Society of Chemistry 2018 |