
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A short review of recent advances in CO2 hydrogenation to hydrocarbons over heterogeneous catalysts
Wenhui Lia,
Haozhi Wanga,
Xiao Jiangc,
Jie Zhua,
Zhongmin Liub,
Xinwen Guo *a and
Chunshan Song*ac
*a and
Chunshan Song*ac
aState Key Laboratory of Fine Chemicals, PSU-DUT Joint Center for Energy Research, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, P. R. China
bNational Engineering Laboratory for Methanol to Olefins, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, P. R. China
cClean Fuels & Catalysis Program, EMS Energy Institute, PSU-DUT Joint Center for Energy Research, Departments of Energy and Mineral Engineering and Chemical Engineering, Pennsylvania State University, University Park, PA 16802, USA. E-mail: guoxw@dlut.edu.cn; csong@psu.edu
First published on 16th February 2018
Abstract
CO2 hydrogenation to hydrocarbons is a promising way of making waste to wealth and energy storage, which also solves the environmental and energy issues caused by CO2 emissions. Much efforts and research are aimed at the conversion of CO2 via hydrogenation to various value-added hydrocarbons, such as CH4, lower olefins, gasoline, or long-chain hydrocarbons catalyzed by different catalysts with various mechanisms. This review provides an overview of advances in CO2 hydrogenation to hydrocarbons that have been achieved recently in terms of catalyst design, catalytic performance and reaction mechanism from both experiments and density functional theory calculations. In addition, the factors influencing the performance of catalysts and the first C–C coupling mechanism through different routes are also revealed. The fundamental factor for product selectivity is the surface H/C ratio adjusted by active metals, supports and promoters. Furthermore, the technical and application challenges of CO2 conversion into useful fuels/chemicals are also summarized. To meet these challenges, future research directions are proposed in this review.
1. Introduction
Continuing consumption of fossil fuels worldwide has led to an increasing CO2 concentration in the atmosphere, and global climate change caused by greenhouse gases has become a major challenge. Mitigation of CO2 concentration in the atmosphere is in urgent need due to the continuing rise in atmospheric CO2 concentration (e.g., exceeding 400 ppm in 2016 (ref. 1)) and its negative and even possibly irreversible impact on the climate system. A recent report by UNEP (United Nations Environment Programme) estimated that if no firm global action is taken against climate change, temperatures might increase by more than 2 °C by 2050, and more than 4 °C by 2100.2 In order to avoid this outcome, scientists indicate that global greenhouse gas emissions need to be reduced by at least 50% by 2050 compared to 1990, while the European Commission objective aims to achieve a reduction of 80–95% greenhouse gas emissions by 2050 compared to 1990.2 A number of Europe's key partners from all over the world, such as China, Brazil, and Korea, are addressing these issues through concrete actions to promote the “low carbon economy”.2 The oil company TOTAL has generated its climate strategy based on the International Energy Agency's 2 °C scenario which aims to limit emissions to approximately 15 Gt CO2-eq. per year in 2050 with the objective to achieve carbon neutrality in the second half of the century.At present, CO2 can be reduced in three ways: control of CO2 emissions, CO2 capture and storage, and chemical conversion and utilization of CO2.3,4 Carbon storage is important for cutting CO2 emissions quickly, but has an issue of potential leakage of CO2.4,5 CO2 can be regarded as a carbon source to offer an alternative to produce carbon-containing value-added products and feedstocks. CO2 obtained by capture not only can provide a pure carbon source for hydrogenation, but also can avoid the leakage problem caused by CO2 storage. In addition, the National Aeronautics and Space Administration (NASA) also regarded the Sabatier reaction (CO2 methanation) as a step in reclaiming oxygen within closed cycle life support systems.6,7 Even the CO2 in industrial flue gas can be used directly as a feed for hydrogenation.8 Therefore, an efficient utilization of renewable carbon resources is crucial and beneficial to maintain a long-term and sustainable development of our society. CO2 conversion requires energy input, and its conjunction with renewable energy would make this strategy more promising in terms of sustainability and environmental friendliness.
CO2 reduction can be catalyzed through electrocatalysis,9 photocatalysis,10 and thermal catalysis. Among them, thermal catalysis receives significant attention due to its fast kinetics and flexible combination of active components. Carbon dioxide is a highly stable molecule, the activation and subsequent conversion of which alone are energy demanding. The addition of another substance with relatively higher Gibbs energy will make the CO2 conversion more favorable thermodynamically. However, electrocatalysis and photocatalysis have the fatal flaw of low energy efficiency. Therefore, CO2 hydrogenation11–14 using H2 produced with renewable energy sources15,16 is a promising research direction to produce chemicals and fuels,17–24 which not only reduces the CO2 emissions, but also covers the shortage of fossil fuels.
Catalytic hydrogenation of CO2 using H2 produced with renewable energy is considered as a potential path forward for the sustainable production of lower olefins,25 higher hydrocarbons,26 formic acid,27 methanol,28,29 and higher alcohols30 (Fig. 1). Considering the depletion of fossil fuels, CO2 hydrogenation to hydrocarbons is a promising way to covert CO2 into fuels among the other CO2 hydrogenation paths. Yet, we need to confront two challenges along with it: (1) sustainable hydrogen source and (2) dispersed product distribution. Much effort has been devoted to solving the former challenge, and scientists have already made great progress in water electrolysis to produce H2 using electricity generated with solar or wind or other renewable energy, and water splitting using photocatalytic, photoelectrochemical or other photochemical processes. There are established industrial technologies for water electrolysis with energy efficiencies of around 70%.31 However, the C2+ hydrocarbons have a wide distribution. For example, CH4, C2–C4, and C5+ are targeted regions for production, while the selectivity was spread in a wide range, which becomes an obstruction to meet the requirement for real applications in industry. However, to date few reviews have dealt with the CO2 conversion mechanism and hydrocarbon chain growth both experimentally and with density functional theory (DFT) calculations. This review provides an overview of advances in CO2 hydrogenation to hydrocarbons that have been achieved recently in terms of catalyst design, catalytic performance and reaction mechanism from both experiments and DFT calculations. The review is organized based on some apparent factors which affect catalyst performance and unified to the essential reason for CO2 hydrogenation that is the chemical state of the catalysts. The fundamental factor for product selectivity is the surface H/C ratio adjusted by the use catalysts. In addition, DFT research advances are summarized from the point of view of C–O bond cleavage and C–C bond formation which gave a deep insight into CO2 activation and conversion. Guidance as to how to adjust the catalysts to promote hydrocarbon chain growth in CO2 conversion is also given in this review.
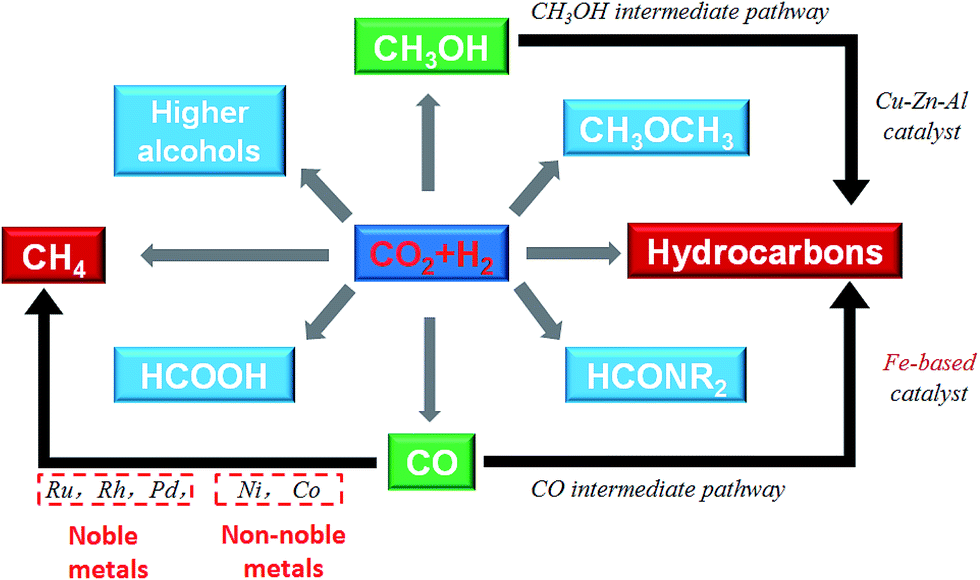 | ||
| Fig. 1 Conversion of CO2 to chemicals and fuels through hydrogenation. | ||
2. CO2 hydrogenation to CH4
CO2 also can be identified as an energy carrier for the transformation of renewable energy. As aforementioned, CO2 hydrogenation to value-added products is one of the promising approaches to combat the CO2-induced climate change, wherein the electrolysis of water to generate H2 with renewable energy is a potential energy storage approach, and would definitely add more credits to the establishment of such a sustainable carbon-based cycle. However, currently uses of renewable energy sources are limited by their inherent intermittency, and require scalable means of storage.32 Therefore, the production of synthetic natural gas or liquid fuels is the most feasible and convenient way to store large amounts of intermittent energy produced from renewable sources for long periods. Among them, the so-called ‘‘power to gas’’ (PtG) concept has garnered significant attention (Fig. 2),33 in which CO2 reacts with H2, generated by water electrolysis with renewable wind or solar energy, to produce CH4 as an alternative source of natural gas. In Copenhagen, a commercial scale operation PtG project with 1.0 MW capacity was running successfully using transformation of the energy system toward a sustainable system in 2016.34 From 2009 to 2013, there were five projects in Germany involving CO2 methanation at pilot plant or commercial scale with capacity ranging from 25 kW to 6300 kW.35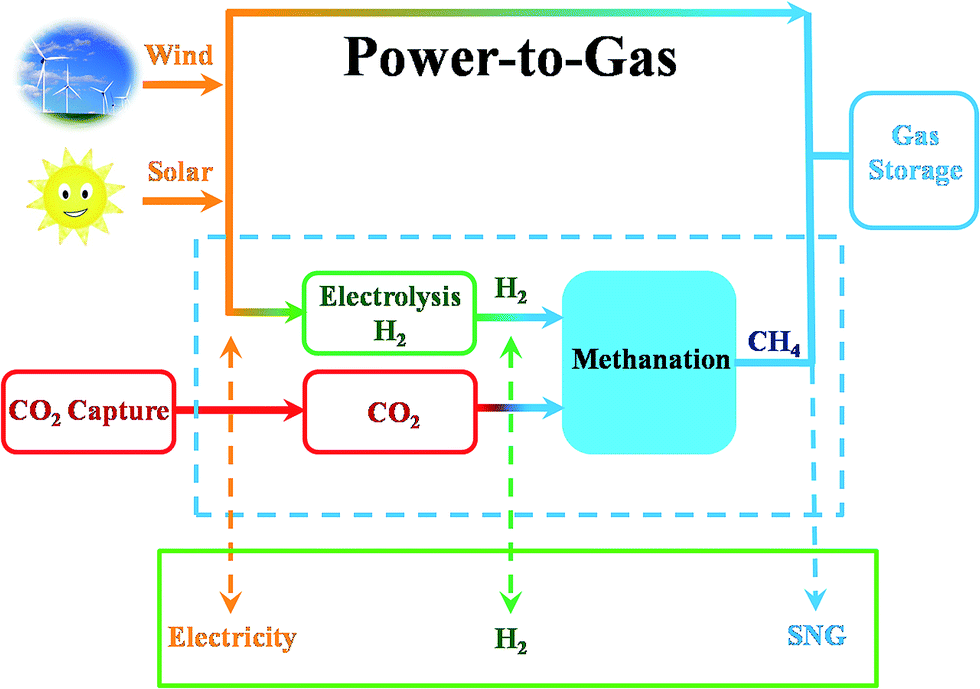 | ||
| Fig. 2 Schematic illustration of CO2-based sustainable production of chemicals and fuels. | ||
CO2 methanation was first reported by the French chemist Paul Sabatier in 1902.36 Due to the increasing demand for mitigating global warming and storing surplus renewable power, this ancient art has attracted renewed attention. The Sabatier reaction is an advantageous way to store renewable energy such as wind and solar power, to transfer biogas effectively to biomethane, and to convert CO2 to chemical feedstocks and fuels.37,38 CO2 methanation is exothermic with high equilibrium conversion between 25 °C and 400 °C as shown in Fig. 3.39,40 CO2 methanation can reach 99% CH4 selectivity through use of appropriate catalysts, avoid the subsequent product separation and overcome the difficulty of dispersed product distribution. Therefore, such a thermodynamic feature makes CO2 methanation more significant in terms of energy efficiency and economic viability.
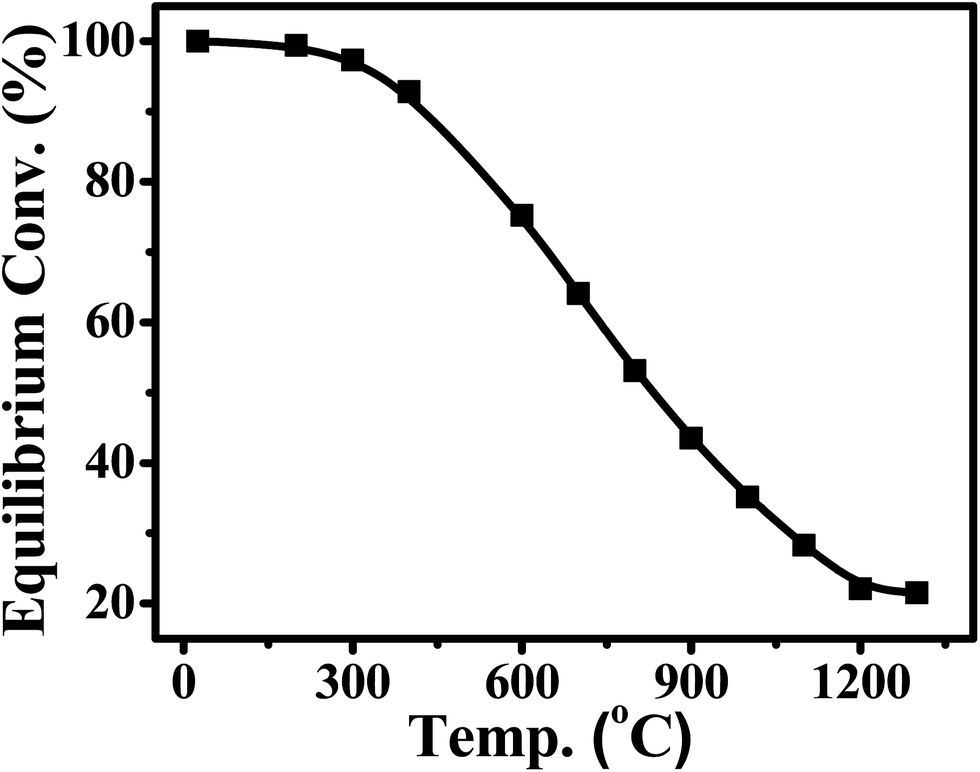 | ||
| Fig. 3 Equilibrium conversion of CO2 in methanation at different temperatures (plotted using the data from the literature).39,40 | ||
2.1 Metal-based heterogeneous catalysts
CO2 methanation can be catalyzed by transition metals such as Co,41–44 Ni,7,45 Ru,46,47 Rh,48 and Pd.49,50 Based on previously published results, the activity performance of various metal-based catalysts decreases in the following order: Ru > Rh > Ni > Co > Pt > Pd.34 Co- and Ni-based catalysts are preferred because of their low cost compared with the noble metals (Ru, Rh, Pd). Ni-based catalysts are the most commonly used for industrial purposes due to their high activity, high CH4 selectivity, and easy availability. The catalytic performances of some representative catalysts are summarized in Table 1, as well as the preparation methods and reaction conditions.| Catalyst | Preparation method | Reactor | GHSV [mL h−1 g−1] | P [MPa] | T [°C] | XCO2 [%] | SCH4 [%] | Stability tests | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| NiO–MgO@SiO2 | Co-precipitation method | Fixed-bed quartz reactor | 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
0.1 | 300 | 80 | 97 | Stable after 100 h | 94 |
| NiWMgOx | Precipitation | Fix-bed quartz tube micro-reactor | 40000 |
0.1 | 300 | 83 | 99 | Stable after 100 h | 74 |
| 15% Ni 2% La-hydrotalcite | Co-precipitation method | Tubular quartz reactor | 12000 |
— | 250 | 46.5 | 99 | Stable after 5 h | 75 |
| 10Ni/Ce–ZrO2 | Ammonia evaporation method | Fixed-bed quartz reactor | 20000 |
— | 275 | 55 | 99.8 | Stable after 70 h | 88 |
| Ni/CeO2 | Excess impregnation | Fixed-bed quartz reactor | 22000 |
0.1 | 340 | 91.1 | 100 | Decreased by 18% after 700 min | 64 |
| 14% Ni 7% Ce/USY | Impregnation | Flow tubular reactor in Pyrex | 43000 |
0.1 | 400 | 68.3 | 95.1 | Stable after 10 h | 95 |
| 10% Ni/MOF-5 | Impregnation | Fixed-bed quartz reactor | 2000 | 0.1 | 320 | 75.1 | 100 | Stable after 100 h | 84 |
| 20% Ni/55% γ-Al2O3–15% ZrO2–15% TiO2–15% CeO2 | Impregnation | Fix-bed quartz reactor | 20000 |
0.1 | 300 | 85 | 98 | Stable after 400 min | 79 |
| 80% Ni–Al hydrotalcite | Co-precipitation method | Fix-bed quartz reactor | 20000 |
0.1 | 300 | 86 | 98 | Stable after 25 h | 80 |
| 2.5% Ce–10% Ni/Al2O3 | Ultrasonic impregnation | Fixed-bed reactor | 7200 | 0.1 | 400 | 74 | 98 | — | 17 |
| 12% Ni/Al2O3 (3DFD structure) | Impregnation, coated on 3DFD structures | Fix-bed quartz tubular reactor | 1500 | 0.1 | 350 | 85 | 98 | Stable after 53 h | 90 |
| 10% Ni/TiO2 | Dielectric barrier discharge plasma | Fix-bed quartz tubular reactor | 60000 |
0.1 | 350 | 73.2 | 99 | — | 91 |
| RQ Ni | Rapid quenching | Capacity hastelloy autoclave | — | 3 | 200 | 60 | 99 | Five successive cycles without reactivation | 78 |
| 10Ni3Pr/Al2O3 | Evaporation induced self-assembly | Fix-bed quartz tubular reactor | 15000 |
0.1 | 400 | 76 | 98 | Stable after 50 h | 76 |
| 0.03% Pt–20% Co–80% Al2O3 | Double flame spray pyrolysis | U-shaped tube reactor | 36000 |
0.1 | 400 | 70 | 98 | — | 89 |
| Co/(0.01)PC-600 | ZIF-67-derived carbonization | Fix-bed reactor | 72000 |
3 | 270 | 59 | 99 | Stable after 420 min | 85 |
| Pt@CSN | Water-soaking-assisted phase-transformation method | Fix-bed reactor | 4800 | 3 | 320 | 41.8 | 95 | — | 87 |
| 10Co/ZrO2 | Impregnation | Fix-bed reactor | 3600 | 3 | 400 | 92.5 | 99 | Stable after 300 h | 62 |
| 20% Co/KIT-6 | Impregnation | Fix-bed quartz tubular reactor | 22000 |
0.1 | 260 | 46 | 99 | — | 42 |
| 2.5% Ru/P25 | Impregnation | Continuous flow fixed-bed reactor | 6000 | 0.1 | 200 | 27.4 | 100 | — | 67 |
| Ru/CeO2 | Single-step flame spray pyrolysis | Fix-bed reactor | 7640 | — | 300 | 83 | 99 | — | 66 |
Zhou et al.63 prepared a series of CeO2-supported Ni-based catalysts with various textural properties by hard-template method, soft-template method, and precipitation method, and examined their activity performance in CO2 methanation. Among them, they found that the one prepared by the hard-template method exhibited a higher CO2 methanation activity, and attributed such superiority to the mesoporous structure and high specific surface area. Furthermore, in situ FT-IR and in situ XPS results illustrate that the surface oxygen vacancies on the CeO2 support were capable of activating the chemisorbed CO2 and subsequently forming the CO intermediate.64 Martin et al.65 investigated Rh, Pd, and Ni catalysts supported on different substrates (Al2O3, CeO2, SiO2, and zeolites) for CO2 methanation. Rh/Al2O3 and Rh/CeO2 exhibited the highest CO2 conversion, but differed in mechanism. In situ DRIFTS interferograms showed that the linear Rh–CO species was evident on Rh/Al2O3, suggesting CO2 dissociation, while the CO was formed through formate and carbonate intermediate species on Rh/CeO2. These advantageous results indicate that the surface oxygen vacancies on the CeO2 substrate enabled the interaction with CO2, and promoted the CO2 hydrogenation. Li et al.62 prepared Co/ZrO2 catalysts for CO2 methanation, as well as Co/Al2O3 catalysts for comparison. The Co/ZrO2 catalysts displayed a higher CO2 methanation activity with a practically stable performance even after 300 h on stream, while the Co/Al2O3, in contrast, deactivated rapidly within the same period of time. The Co–Zr interface was observed on the samples in reduced form, which enabled the redistribution of active Co on the ZrO2 support due to the special metal–support interaction (Fig. 4). The special Co–Zr interface is crucial for the superior CO2 methanation activity. Dreyer et al.66 have investigated the influence of metal oxide support reducibility on Ru-based catalysts for CO2 methanation. They pointed out that the intermediate CO should have an appropriate coverage and strong adsorption, which ensures the occurrence of H2 dissociation. The reducible CeO2 support is the most suitable to support Ru for CO2 methanation compared with the irreducible Al2O3 which gives a quasi-saturated CO adsorption and limits the co-adsorption of H2 and reducible ZnO which has a weak CO adsorption and leads to the reverse water–gas shift (RWGS) reaction.
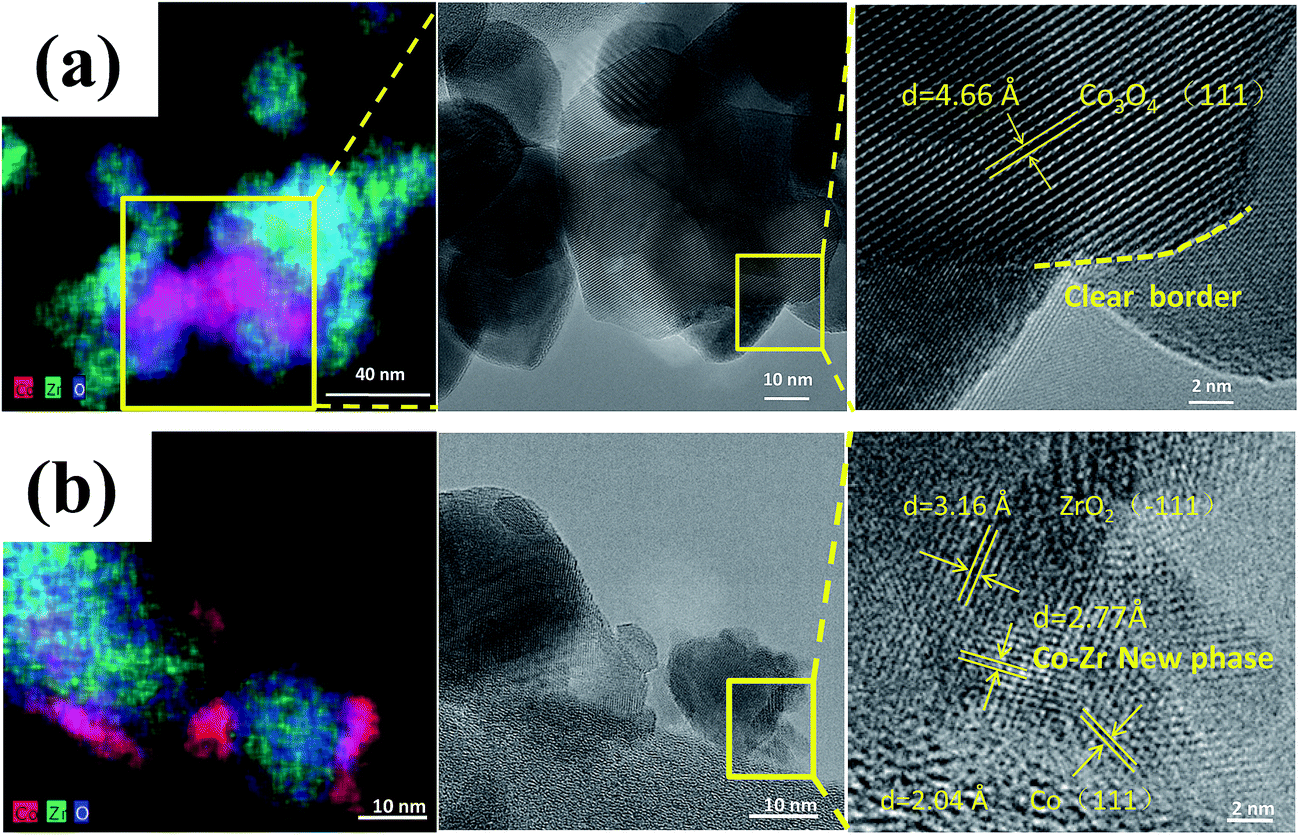 | ||
| Fig. 4 STEM-EDS maps and corresponding TEM images of (a) calcined catalyst precursors Co3O4/ZrO2 and (b) reduced catalyst Co/ZrO2. Reprinted with permission from ref. 62. Copyright 2017 Elsevier. | ||
In addition, metal oxide supports with the same chemical composition and different crystal phase also have an influence on the chemical state of the supported metal. Kim et al.67 synthesized monodispersed 2 nm RuO2 nanoparticle colloidal suspension, and impregnated it onto TiO2 with different crystal phases for CO2 methanation. The activity and product selectivity were strongly dependent on the composition of different crystal phases of TiO2, wherein P25, with 20% of anatase and the rest of rutile, exhibited the highest CO2 conversion and CH4 selectivity. Inspired by these results, they further developed a fundamental understanding of the composition structure–activity performance relationship.68 The phenomenon that RuO2 nanoparticles tended to migrate towards rutile TiO2 during the CO2 methanation process when rutile and anatase TiO2 co-existed was evidenced by the stabilization of RuO2 on rutile TiO2 based on characterization results. Such rutile-favored migration led to the formation of highly dispersed Ru in the reduced form, thereby exhibiting a superior activity (Fig. 5). Lin et al.69 also observed a similar phenomenon on Ni/TiO2 catalysts with different TiO2 crystal phases for both CO and CO2 methanation. Chen et al.56 found that PtCo bimetallic catalysts were capable of shifting the selectivity from CO to CH4 by altering the oxide supports from TiO2 to ZrO2, respectively. In other words, PtCo/ZrO2 tends to favor CH4 formation compared with PtCo/TiO2. Both XPS and DFT calculations were carried out to elucidate the origins for CO formation on PtCo/TiO2 and CH4 formation on PtCo/ZrO2. Experimentally, both *HCOO and *HOCO were identified as reaction intermediates on both PtCo/TiO2 and PtCo/ZrO2, whereas *CH3O was only evidenced on PtCo/ZrO2. DFT results illustrate that the CO desorption energy is much lower than that of its hydrogenation to *CHO on PtCo/TiO2. Therefore, the chemisorbed CO favored desorption energetically rather than the subsequent hydrogenation, thereby leading to a selective production of CO. On the other hand, CO formation was hindered on PtCo/ZrO2 catalyst, and CH4 was formed. Apparently, the interaction between metal and support plays an important role in product selectivity.
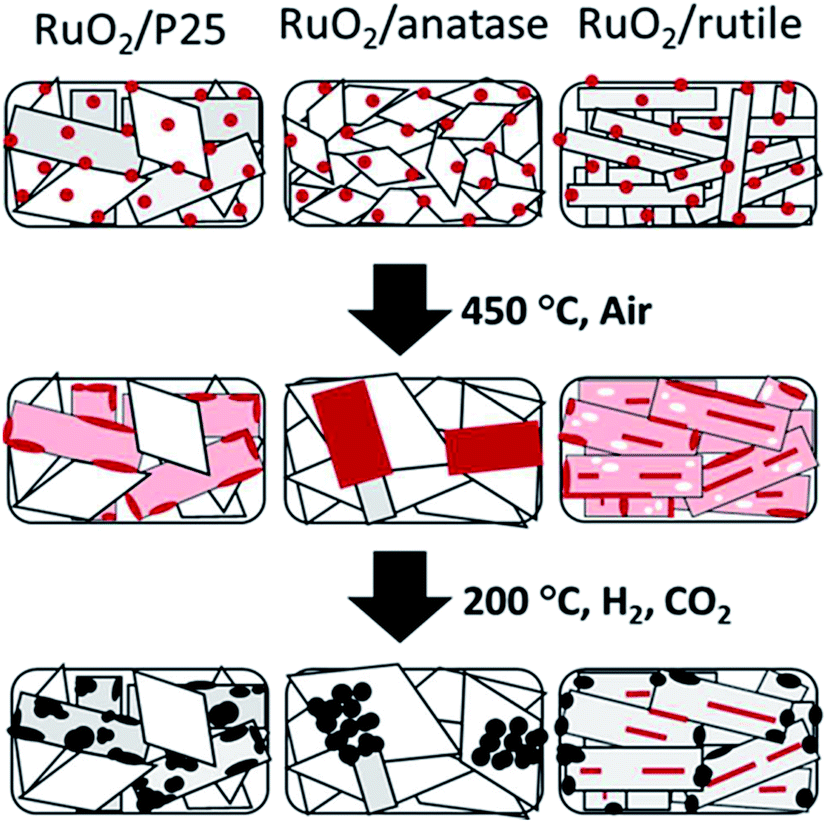 | ||
| Fig. 5 Schematic illustration of the shape evolution of RuO2/TiO2 catalysts: after RuO2 nanoparticle deposition, after thermal annealing at 450 °C, and after reduction and methanation. Red indicates RuO2, pink indicates thin RuO2 layer, white indicates Ru depleted area, and black indicates metallic Ru. Reprinted with permission from ref. 67. Copyright 2013 RSC. | ||
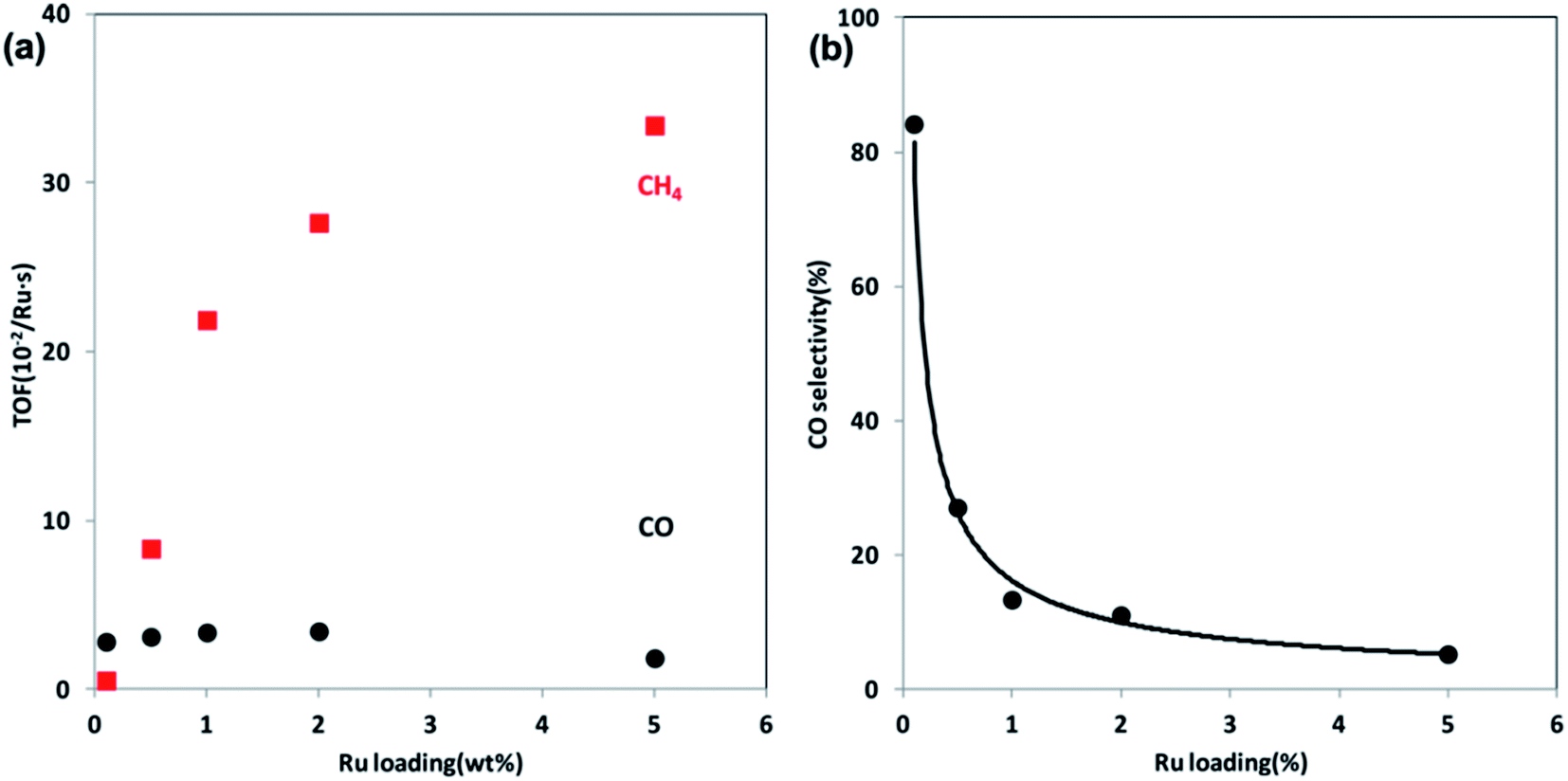 | ||
| Fig. 6 (a) TOFs of CO and CH4 formation at steady state at 300 °C over Ru/Al2O3 catalysts as a function of Ru loading. (b) CO selectivity as a function of Ru loading at 300 °C. Reprinted with permission from ref. 47. Copyright 2013 ACS. | ||
Seen from the results above, both noble and non-noble metal-based catalysts can be applied to CO2 methanation, and the experimental results indicate that, within a certain range of metal particle size, the atom-scale structured catalysts tend to favor the RWGS reaction, while the larger metal particles facilitate CH4 formation.50 To unveil the underlying reasons behind the size-dependent effect, Ma et al.73 prepared Ir/CeO2 catalysts with various Ir loadings using a ligand-free method, and tested them in CO2 methanation. The catalysts with low Ir loading presented partially oxidized Ir species, and displayed catalytic selectivity for CO production. On the other hand, more metallic Ir species appeared to emerge when increasing the Ir loading level, leading to a preference for desired CH4 formation. Their results suggest that the chemical state of Ir could be finely tuned by altering the loading of the metal. Actually, the particle size effect is the chemical state effect. The metal loading essentially affects the active metal state on the supports and further affects the reaction routes.
Similarly, Wierzbicki et al.75 increased the surface basicity by adding 2 wt% lanthanum to Ni–Mg–Al hydrotalcite-derived catalysts, which remarkably enhanced the CO2 methanation activity. In addition, a series of rare earth-doped (La, Ce, Sm, and Pr) Ni-based mesoporous materials were facilely fabricated by the one-pot evaporation-induced self-assembly strategy for low-temperature CO2 methanation.76 The rare earth-doped catalysts with enhanced surface basicity displayed two or three times higher catalytic activities than the pristine MA-10Ni catalyst in the low-temperature region (200–250 °C). Generally speaking, catalyst basicity improvement increases the CO2 adsorption and activation by the second metal addition.
Centi and co-workers have developed γ-Al2O3–ZrO2–TiO2–CeO2 composite oxide-supported Ni catalysts79 and Ni–Al hydrotalcite80 catalysts for CO2 methanation. A better performance of the catalysts was achieved because of the improvements in the reducibility of active metal Ni.
Metal oxides have relatively low surface areas without featuring pore structures, thereby limiting the intimate contact between reagents and active sites, and even leading to mass transfer limitation. To resolve such issues, some high BET surface supports have been explored. MOFs are a class of crystalline, nanoporous materials that offer such tailorability through large accessible surface areas, tunable pore functionalities, and reactive open metal sites.81–83 Zhen et al.84 prepared Ni-based catalysts using MOF-5 (surface area of 2961 m2 g−1) as support, and obtained a high Ni dispersion (41.8%). Such 10Ni/MOF-5 catalyst with highly dispersed Ni showed a higher activity than the benchmark Ni/SiO2, and presented a superior stability after 100 h on stream for low-temperature CO2 methanation. Li et al.85 prepared ZIF-67-derived Co-based porous carbon catalysts, and achieved both excellent catalytic performance and good stability in comparison to the traditional Al2O3-supported counterpart. This catalyst even exhibited prominent activity performance in CO2 methanation at low temperatures compared with the other Co-based catalysts supported on either metal oxides or zeolites.41,42,86 Meanwhile, Zeng et al.87 have proposed a general synthesis route of ZIF-67-derived bifunctional nanoreactors via a water-soaking-assisted phase-transformation method for CO2 hydrogenation. They point out that CO2 converts to CO on the Pt active sites and CO methanation to CH4 occurs on the Co active sites. The Pt@CSN (cobalt silicate nanoparticles) bifunctional nanoreactors increase the CO2 conversion and CH4 selectivity obviously through prolonging the intermediate retention time on the catalyst surface and enhancing the probability for CO to further convert to CH4. These achievements provide insight into adapting these advancements toward the industrial utilization of CO2 in terms of economic and sustainable viability. However, MOFs are unfavorable for high-temperature reaction because of their instability under the hydrothermal reaction conditions, especially given that CO2 hydrogenation usually requires high temperature. From another point of view, developing low-temperature methanation catalysts with high activity is also a promising way for CO2 conversion from the energy conservation perspective.
In addition to the traditional homogeneous catalyst preparation methods, such as impregnation method and co-precipitation method, more and more innovation techniques have been employed to prepare catalytic materials to remedy the defects existing in traditional methods. Kawi and co-workers developed Ni/Ce–ZrO2 catalysts by the ammonia evaporation (AE) method, and remarkably improved the Ni reducibility (total H2 uptake = 3.37 mmol g−1) compared with the impregnation (total H2 uptake = 3.32 mmol g−1) and deposition–precipitation (total H2 uptake = 2.06 mmol g−1) methods.88 The Ni/Ce–ZrO2-AE possessed more oxygen vacancies, and a strengthened metal–support interaction, thereby contributing to the high activity and stability for low-temperature CO2 methanation. Schubert et al.89 used the double flame spray pyrolysis technique to control the Pt content to as low as 0.03 wt%, and improved the catalytic performance of PtCo–Al2O3 significantly. Protasova et al.90 manufactured macro-porous catalytic supports by an innovative and highly reproducible robocasting technique, three dimensional fiber deposition (3DFD), and the supports were coated with Ni/Al2O3 suspension to achieve sufficient catalytic coating. The catalysts coated on 3DFD supports had improved mass and heat transfer properties, and prevented metal sintering efficiently. These advantages were conducive to maintain the stability of the catalysts. Liu et al.91 attained a better catalytic performance of Ni/TiO2 catalysts prepared by dielectric barrier discharge plasma, depending on which more active Ni(111) facets were selectively exposed for CO2 methanation.92
To accord with practical applications, researchers have recently paid attention to the integration of carbon dioxide capture and utilization processes by incorporating together both CO2 capture system and catalytic CO2 conversion system. Farrauto8,93 and co-workers devoted their effort to exploring dual functional catalysts which enable CO2 capture from an emission source, and conversion of it to synthetic natural gas in the same reactor at the same temperature (320 °C). In this process, the catalyst composition comprised 5% Ru 10% CaO/Al2O3, wherein the components of CaO and Ru were responsible for CO2 adsorption and conversion, respectively. Of note, this new approach utilized flue gas sensible heat, and needed no additional heat input, which made it more attractive in mitigating the current energy shortage. Grunwaldt et al.33 explored Ni-based catalysts under dynamic reaction conditions, especially under a fluctuating supply of renewable H2. They found the oxidation of Ni particles occurred after the removal of H2 from the gas stream, and a lower catalytic performance was observed consequently. Apparently, the Ni/CaO–Al2O3 catalyst was unadapted to the dynamic reaction conditions. Such an issue impeded its implementation in real industry, which also made the search for efficient ways for renewable H2 supply more important.
2.2 Mechanisms of CO2 methanation
CO2 methanation can be catalyzed through either CO route or formate route, which is determined by the properties of different active metals and supports. The CH4 selectivity is likely determined by the competition between the hydrogenation and C–O bond scission reactions of the *HxCO intermediates. To achieve high CH4 selectivity, the binding of *HxCO species should be strong enough to facilitate C–O bond cleavage.61Duan et al.96 investigated the effect of oxygen vacancies on the catalytic performance of Rh/CeO2 catalysts in CO2 methanation, and developed an understanding of its role in the proposed mechanism. The existence of Ce3+, surface hydroxyl, and oxygen vacancies on Ru/CeO2 was evidenced from operando XANES, IR, and Raman analyses. Steady-state isotope transient kinetic analysis (SSITKA)-type in situ DRIFTS was employed to detect the surface intermediates and track their transformation during the reaction process on both Ru/CeO2 and Ru/Al2O3, wherein the latter was introduced as reference, as it barely had any oxygen vacancies. On the Ru/Al2O3 catalyst, the carbonyl species, originating from the chemisorbed CO*, was observed until 250 °C, and CH4 generation occurred within the same temperature range. In contrast, formate and methanol corresponding bands emerged for Ru/CeO2, in which the former was identified as a key intermediate via this route, and methanol-to-methane transformation was the rate-determining step at a much lower temperature (150 °C). In this work, oxygen vacancies played a crucial role in CO2 activation and formate formation. Sharma et al.97 prepared Ru-substituted Ce0.95Ru0.05O2 catalyst for CO2 methanation, and interpreted a plausible reaction mechanism by combining the characterization results (TPR and DRIFTS) and DFT calculation. In this case, surface CO* species, rather than formate, was more likely to act as a key intermediate for CH4 production, wherein the reaction proceeded through the following steps: CO2 → CO → OCH2 → OCH3 → CH4. Note that this proposed reaction pathway differed from that of the supported metal catalyst (Ru/CeO2), which proceeded via CO2 → CO → HCOO− → C → CH4.
Ren et al.92 investigated the mechanisms of CO2 methanation on Ni(111) surfaces by DFT through three routes with and without CO formation (Fig. 7). Considering the energy barriers and reaction energies for these different routes, the CO route was more favorable energetically for CO2 methanation on Ni(111) surface: CO2 → CO + O → C + O + 4H → CH2 + 2H → CH3 + H → CH4. Salmeron et al.98 also concluded that the methanation reaction proceeded via CO intermediate on Ni(111) surface as evidenced by ambient pressure X-ray photoelectron spectroscopy. Meanwhile, they reported that the Ni(110) seemed to convert CO much more easily to atomic carbon than Ni(111). Henriques et al.18 also investigated the mechanism of USY zeolites-supported Ni catalysts for CO2 methanation, and reported results also supporting the CO route. On the other hand, Gonzalez et al.99 identified the surface species on Ni/ZrO2 catalysts and bare ZrO2 during CO2 methanation using DRIFTS, and they proposed a different scenario with the formate route as the favorable one, as displayed in Fig. 8. In this mechanism, chemisorbed CO2 reacted with surface hydroxyl groups of ZrO2 to give bicarbonate species that can be reversibly converted to carbonate species. H2 was dissociated on the surface of Ni particles, which may migrate to the reducible metal oxide support by a spillover process, resulting in the formation of surface hydroxyl groups, metal–H species, and formate species. Next, the formate species took part in further hydrogenation to form CH4.
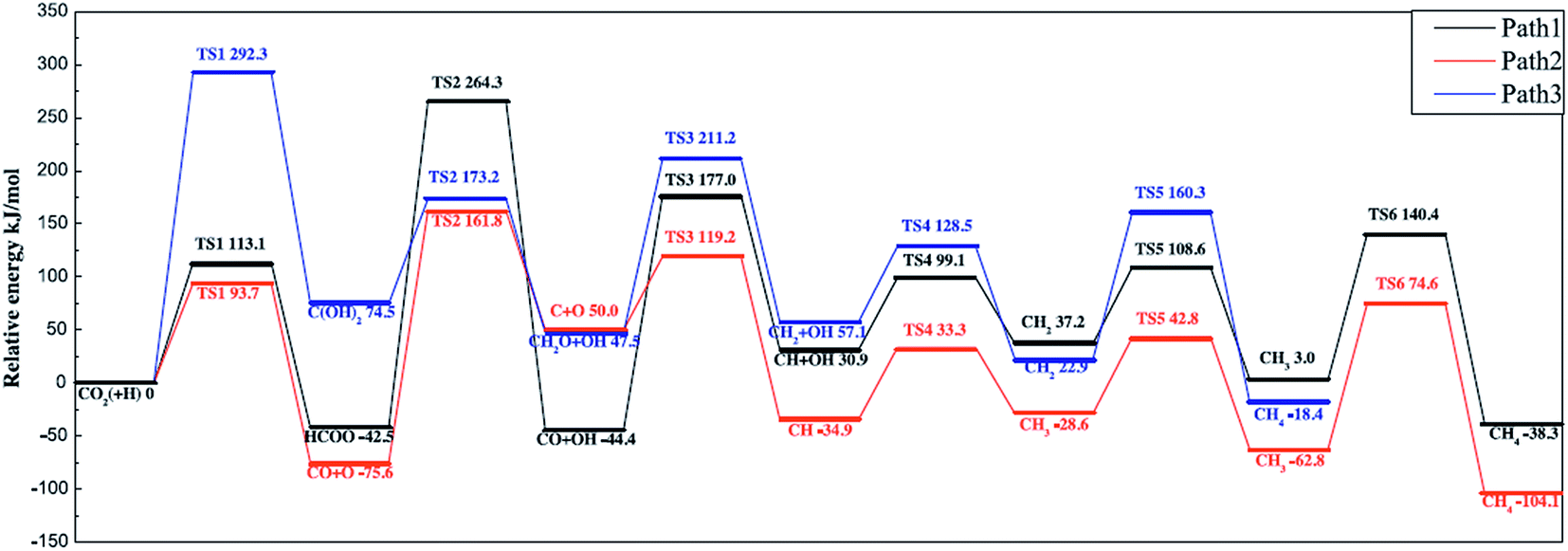 | ||
| Fig. 7 Potential energy diagram of three mechanisms of CO2 methanation. Reprinted with permission from ref. 92. Copyright 2015 Elsevier. | ||
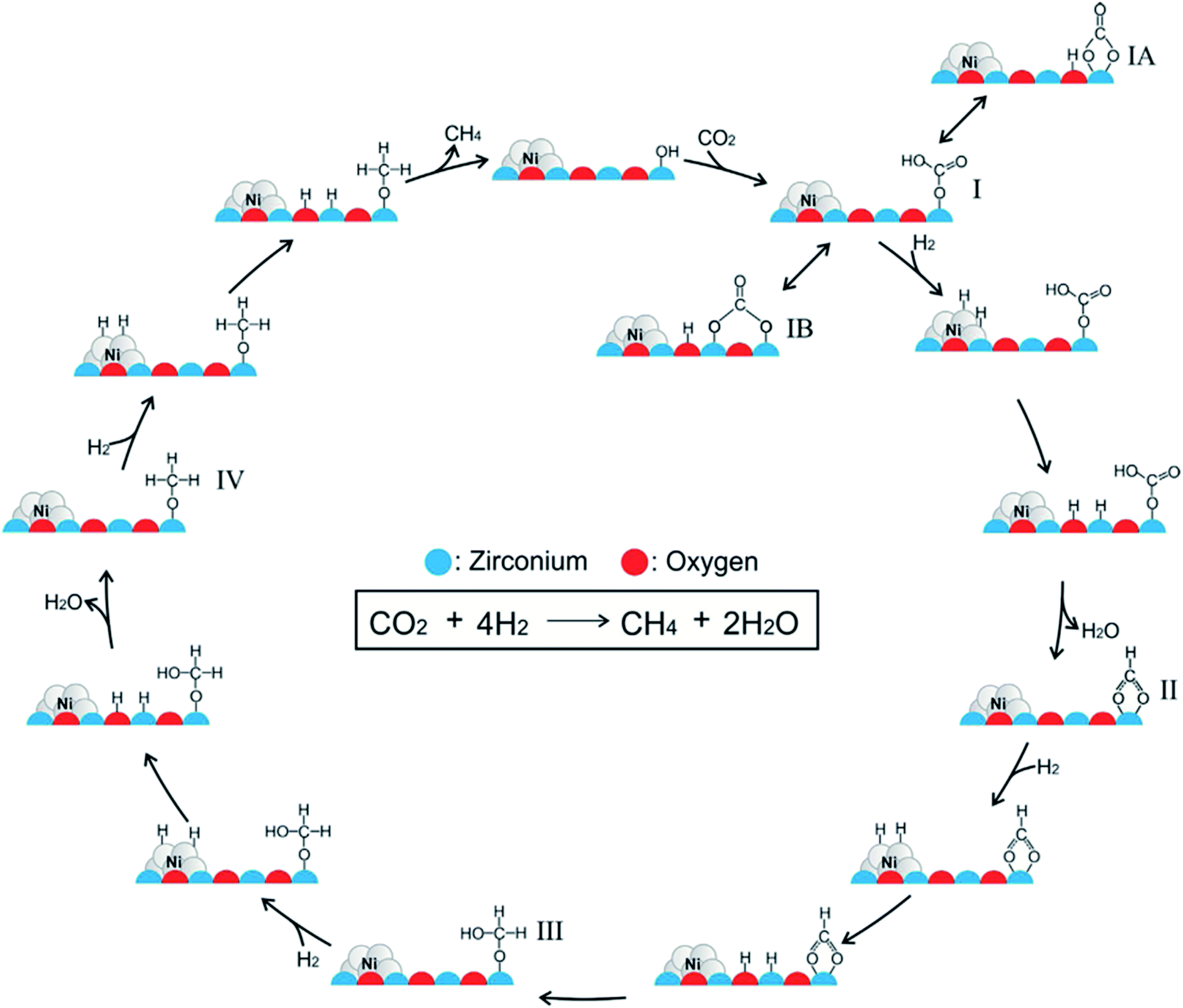 | ||
| Fig. 8 Mechanism of CO2 methanation on ZrO2-supported Ni catalysts. Reprinted with permission from ref. 99. Copyright 2017 Elsevier. | ||
In contrast to other reports,18,92 the Ni/ZrO2 catalysts barely presented vibrational bands of carbonyl species on Ni surface. Instead, carbonate and bicarbonate species were identified on both Ni/ZrO2 and bare ZrO2, and even their subsequent transition to formate species was evidenced. Clearly, the incorporation of Ni and ZrO2 was characteristic of bifunctionality for CO2 methanation, in which the former metal sites were responsible for providing hydrogen through dissociation, while the latter support accounted for the CO2 stabilization and activation. In other words, CO2 methanation can be catalyzed through the formate route rather than the CO route on ZrO2-supported Ni catalysts.
In sum, the reaction pathway of CO2 methanation varied depending on the catalytic system used, and strongly depended on the selection of active metals and supports and their interactions.
2.3 Deactivation of CO2 methanation catalysts
Deactivation of metal catalysts is a big challenge in CO2 methanation. The deactivation of methanation catalysts can be divided into two types: (a) chemical deactivation and (b) physical deactivation.The chemical deactivation of CO2 methanation catalysts is mainly directed toward the decrease of active sites caused by the formation of spinel structure. Li et al.62 prepared Co/ZrO2 catalysts for CO2 methanation, as well as Co/Al2O3 catalysts for comparison. The Co/ZrO2 catalysts displayed a higher CO2 methanation activity with a practically stable performance even after 300 h on stream, while the Co/Al2O3, in contrast, deactivated rapidly within the same period of time. The deactivation of the Co/Al2O3 catalyst was further studied through thermogravimetric analysis and hydrothermal (H2O) treatment verification tests. Extra H2O was pumped into the reaction system which led to a large amount of CoAl2O4 being formed and accelerated the Co/Al2O3 catalyst deactivation. Thus, the product H2O promotes the formation of the inactive phase CoAl2O4, leading to the rapid deactivation of Co/Al2O3 catalysts. Carbon deposition is one of the reasons for deactivation; however, the main reason for deactivation is the formation of inactive phase CoAl2O4 spinel structure.
Physical deactivation is caused by carbon deposition and active metal sintering. Kesavana et al.100 synthesized Ni/YSZ catalysts by different methods. On the Ni/YSZ catalyst obtained by impregnation method, graphitic filaments are formed on Ni0 particles exposing flat surfaces, whereas thin layers of carbon are formed on Ni0 particles with spherical shape. Ni/YSZ(EDTA) catalyst showed remarkable stability and operando XAS showed that Ni/YSZ(EDTA) catalyst did not undergo deactivation by Ni0 → Ni2+ oxidation using high CO2:H2 ratio. Carbon deposition on the catalyst can be avoided by adding steam or increasing the H2/CO2 ratio because hydrogen reacts with the carbon deposits and prevents catalyst deactivation. To mitigate metal sintering, common strategies are increasing metal dispersion through strong metal–support interaction,62 adding catalyst promoters,101 and developing advanced synthesis methods.89 Li et al.85 prepared MOF-derived Co-based porous carbon catalysts in which the active Co particles are separated by the graphite-like carbon avoiding metal sintering effectively. A special catalyst structure can also resist metal sintering effectively. Li et al.94 prepared NiO–MgO@SiO2 core–shell structured catalysts, and activity performance was successfully retained after 100 h on stream. In summary, particles with appropriate size are beneficial for CH4 formation.
3. CO2 hydrogenation to C2+ hydrocarbons
CO2 hydrogenation to C2+ hydrocarbons is of great importance because the long-chain hydrocarbons possess higher energy density, and could be used as fuels and chemicals for a wide range of applications. Utilizing CO as the carbon source for hydrocarbon synthesis through Fischer–Tropsch synthesis has been widespread in industry, and continuing studies to further improve the activity, tune the product distribution, and develop deep understandings of catalytic composition-performance-physicochemical relationships are still ongoing worldwide.102,103 Substituting CO with CO2 as carbon source makes the reaction thermodynamically more difficult (see the reaction enthalpy below).104 Besides, not only will the challenge originate from the chemical inertness of CO2, but also from the competition with CO2 methanation.73 The relatively small amount of CO2 adsorbed species compared with dissociated H* on a catalyst surface leads to a low C/H ratio, which favors the fast hydrogenation of surface-adsorbed intermediates and the formation of methane and prevents chain growth.12,56,105 Despite the difficulties, the transformation of CO2 to value-added chemicals still receives great attention worldwide because of the significance in providing sustainable alternatives to solve urgent issues such as those of energy and the environment. This section will discuss the most recent advances in CO2 hydrogenation to hydrocarbons. Similarly, some representative catalysts for CO2 hydrogenation to C2+ hydrocarbons are presented in Table 2.| Catalyst | Preparation method | GHSV [mL h−1 g−1] | P [MPa] | T [°C] | XCO2 [%] | SCO [%] | SCH4 [%] | SC2–C4 [%] | SC5+ [%] | O/P | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
a Calculated from equation 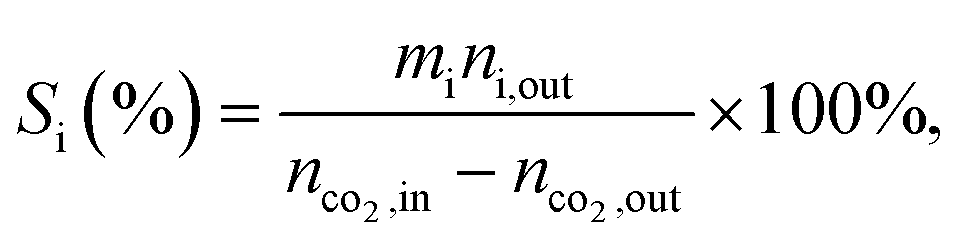 where nco2,in and nco2,out represent the molar concentration of CO2 in the feed and effluent, respectively; ni,out represents the molar concentration of product i in the effluent; and mi represents the number of carbon atoms in product i. Products include CO and hydrocarbons.b Calculated from equation: where nco2,in and nco2,out represent the molar concentration of CO2 in the feed and effluent, respectively; ni,out represents the molar concentration of product i in the effluent; and mi represents the number of carbon atoms in product i. Products include CO and hydrocarbons.b Calculated from equation: 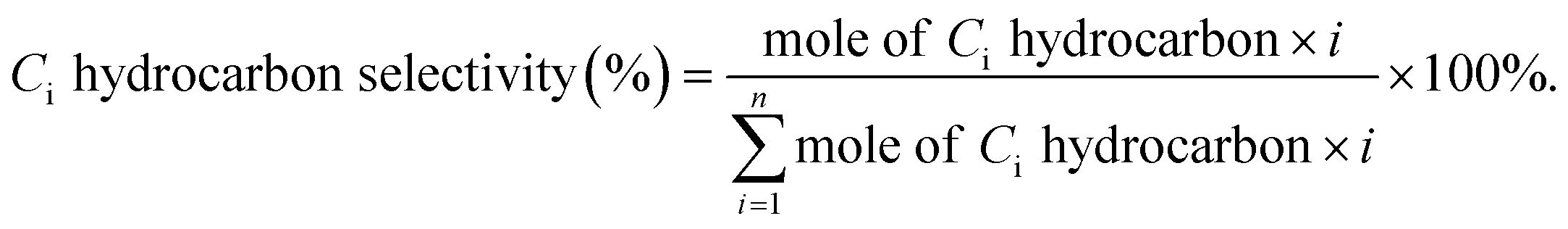 |
|||||||||||
| Fe2O3-CT600 | Precipitation method | 1140 | 0.15 | 350 | 40.0 | 15a | 12a | 37a | 36a | 2.7 | 112 |
| 20% Fe/cube-CeO2 | Incipient wetness impregnation | 200 | — | 390 | 18.9 | 73.5a | 75.5b | 22.2b | 1.9b | 4.1 | 115 |
| Fe–Co(0.17)/K(1.0)/Al2O3 | Incipient wetness impregnation | 3600 | 1.1 | 300 | 31.0 | 18a | 13a | 69a | — | 116 | |
| 10K13Fe2Co100ZrO2 | Electro-spinning method | 3600 | 3 | 400 | 42.3 | 21.9a | 25.7a | 34a | 18.4a | 4.2 | 117 |
| CeO2–Pt@mSiO2–Co | Hydrothermal and impregnation | 50400 |
0.6 | 250 | 2.0 | 78.0 | 60.0b | 40.0b | 0b | — | 118 |
| Delafossite-CuFeO2 | Hydrothermal method | 1800 | 1 | 300 | 16.7 | 31.4a | 2.4b | 32.7b | 64.9b | 7.7 | 119 |
| 0.05MnFe | One-step sol–gel process | 6000 | 0.1 | 340 | 30 | 7.7a | 29.3b | 67.1b | 0.37 | 120 | |
| ZnGa2O4/SAPO-34 | Physical mixing | 5400 | 2 | 370 | 13 | 46a | 1b | 97b | 2b | 7.8 | 121 |
| ZnZrO/SAPO-34 | Physical mixing | 3600 | 2 | 380 | 12.6 | 47a | 3b | 94b | 3b | 5.7 | 122 |
| In2O3/HZSM-5 | Granule stacking | 9000 | 3 | 340 | 13.1 | 45a | 1b | 13.1b | 78.6b | — | 26 |
| In–Zr/SAPO-34 | Physical mixing | 9000 | 3 | 400 | 35 | 75a | 5b | 93b | 3b | 6.1 | 123 |
| Na–Fe3O4/HZSM-5 | Granule mixin | 4000 | 3 | 320 | 33.6 | 14.2a | 7.9b | 18.4b | 73.7b | — | 105 |
| Fe–Zn–Zr@HZSM-5-Hbeta | Claddingmethod | 3000 | 8 | 340 | 14.9 | 38.6a | 1.5a | 71.7a | 26.8a | — | 124 |
| Cu–Zn–Al/modified-HB | Physical mixing | 1500 | 0.98 | 300 | 27.6 | 53.4a | 0.7a | 43.2a | 2.3a | — | 125 |
3.1 Modified Fischer–Tropsch synthesis (FTS) route
Primarily, CO2 hydrogenation to hydrocarbons could proceed through both direct and indirect pathways. The direct way is straightforward conversion of CO2 to hydrocarbons106 (eqn (1)) or going through the RWGS reaction and FTS reaction (eqn (2) and (3)).107,108 In addition to these proposed reaction pathways, some scientists have also attempted to use methanol as a bridge and building unit to synthesize long-chain hydrocarbons via CO2 hydrogenation, and made a major breakthrough most recently.96 This newly developed reaction pathway is another alternative option for CO2 hydrogenation to hydrocarbons through an indirect way.| CO2 HYD, nCO2 + 3nH2 = CnH2n + 2nH2O, ΔRH573 K = −128 kJ mol−1 | (1) |
| RWGS, CO2 + H2 = CO + H2O, ΔRH573 K = +38 kJ mol−1 | (2) |
| FTS, 2nCO + (3n + 2)H2 = 2CnH2n+2 + 2nH2O, ΔRH573 K = −166 kJ mol−1 | (3) |
Since converting CO2 to hydrocarbons directly makes the reaction kinetically more difficult,104 some scientists have turned to the modified FTS route. This section will mainly focus on the major progress made in CO2 hydrogenation to C2+ hydrocarbons through the modified FTS route. Currently, FTS is mainly catalyzed by Fe-based catalysts because the metal Fe possesses the catalytic characteristic of improving C–C coupling, which was proposed as a rate-determining step.109 In 1978, Dwyer et al.110 found that the presence of CO2 impacted the product distribution of FTS on Fe-based catalysts, and such an inspired finding exploited a new field for the development of active catalysts for hydrocarbon synthesis from CO2, an alternative carbon source with even greater proportion in the atmosphere. Computational results demonstrate that the kinetics of FTS is not comparable to that of RWGS, which makes it another challenge other than carbon chain growth.111 Even so, the similarity between CO and CO2 hydrogenation motivated people to apply Fe catalysts, which exhibited good performance in FTS, to CO2 hydrogenation at an early stage, and more researchers then endeavored to apply modified Fe catalysts with desired features. Albrecht et al.112 prepared dopant-free bulk Fe2O3 by a cellulose-template (CT) synthesis method, and applied it in CO2 hydrogenation to hydrocarbons. The catalysts selectively catalyze CO2 to C2–C4 hydrocarbons (selectivity = 37%) with an olefin to paraffin (O/P) ratio of 2.7. Iron carbide, as high as 81 wt%, was detected on the spent Fe2O3-CT600 catalysts, which was considered as active sites for the FTS.113,114 In comparison to FTS, this CT-supported Fe2O3 catalyst yielded comparable C2–C4 hydrocarbons, which could be attributed to the improved reducibility and in situ formation of iron carbide promoted by the CT-synthesized catalyst precursor.
Owing to their tailorable pore structure and featured physicochemical properties, MOFs and their subclass zeolitic imidazolate frameworks (ZIFs) have attracted considerable attention in a diversity of energy-related applications such as CO2 capture and even CO2 activation.130–132 Driven by their unique characteristic, researchers employed this group of materials as supports to prepare heterogeneous catalysts for CO2 hydrogenation to hydrocarbons.84,133 Some metal-based MOFs and ZIFs, which already were proven to be active for hydrocarbon synthesis from either CO2 or CO hydrogenation, were selectively chosen for activity tests in an attempt to collect first-hand data. Guo and coworkers proposed and carried out a series of tests using these novel catalysts, and obtained interesting results. In an early work, they employed MIL-53(Al) and ZIF-8 as supports for preparing α-Fe2O3 catalysts by a solid grinding method.133 Preliminary results indicated that ZIF-8-supported catalysts exhibited a higher CO2 conversion than MIL-53-supported ones because the acidity on the MIL-53 impeded CO2 adsorption, an important step for heterogeneous catalysis.
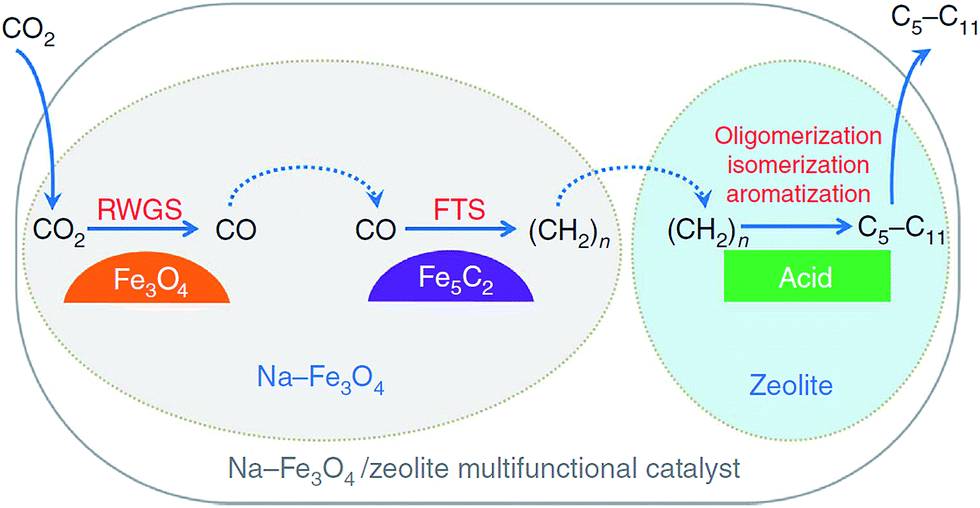 | ||
| Fig. 9 Reaction scheme for CO2 hydrogenation to gasoline-range hydrocarbons through modified FTS route. Reprinted with permission from ref. 105. Copyright 2017 Nature. | ||
Yang et al.118 prepared CeO2–Pt@mSiO2–Co core–shell catalysts for converting CO2 to C2–C4 hydrocarbons. The two interfaces of Pt–CeO2 and Co–SiO2 were intentionally created depending on the unique core–shell structure, wherein the former accounted for converting CO2 to CO through RWGS, while the latter accounted for the subsequent hydrogenation to C2–C4 through FTS. Notably, this novel catalyst yielded 60% of C2–C4 hydrocarbons in total carbon-containing products except CO (Fig. 10).
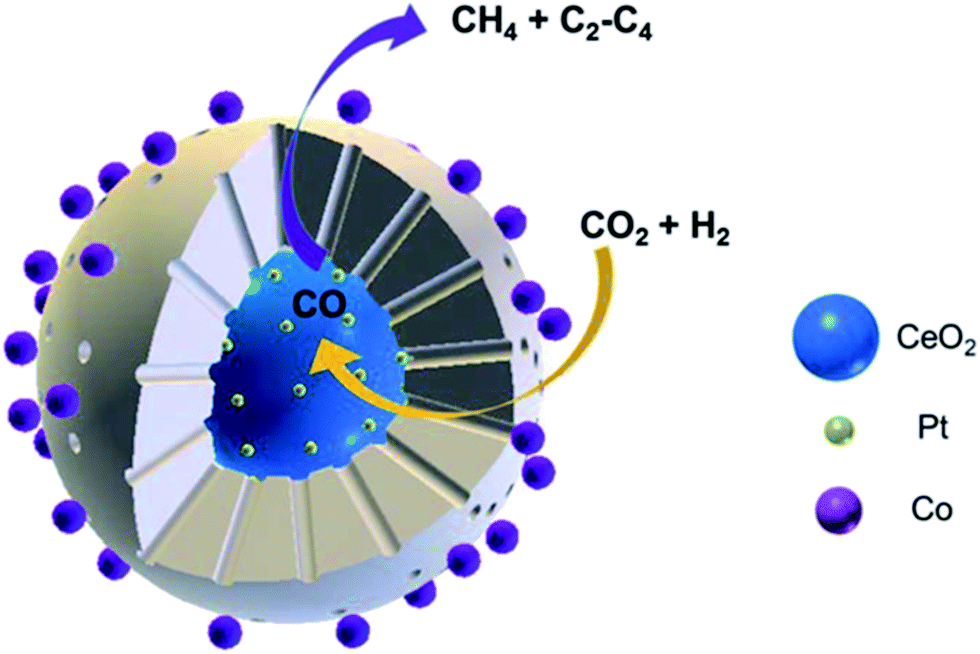 | ||
| Fig. 10 Schematic diagram of CO2 hydrogenation on CeO2–Pt@mSiO2–Co core–shell catalysts. Reprinted with permission from ref. 118. Copyright 2017 ACS. | ||
Another promising strategy to improve the yield of higher hydrocarbons is the application of bimetallic synergy. Satthawong and coworkers137 conducted comprehensive screening tests over Fe-M/Al2O3 (M = Co, Ni, Cu, and Pd) catalysts with fixed M/(M + Fe) atomic ratios at 0.1 at per at and their K-promoted counterparts. The combination of Fe with either Co, Cu, or Pd led to a significant improvement of chain-growth possibility and bimetallic promoting effect on C2+ hydrocarbon formation, while the Fe–Ni(0.1) catalysts, on the contrary, selectively produced undesired CH4. Interestingly, the combination of Fe and Co, Cu or Pd enhanced the catalyst activity obviously compared with their monometallic counterparts, indicating a strong synergetic effect and intimate proximity existed between the combined metal components. Also, the K addition further increased the CO2 conversion and the C2–C4 olefin production. In following work, the Fe–Co bimetallic catalysts with a wide range of Co/(Fe + Co) atomic ratios (i.e., 0.0–1.0 at per at) were selectively chosen for a systematic examination to unveil the synergetic regime of this bimetallic combination and the function of promoter in terms of adsorption properties.116,138 Evidently, doping Fe with appropriate amount of Co (e.g., Co/(Co + Fe) = 0.17 mol mol−1) can maximize the promotion of C2+ hydrocarbon production. Inspired by such significant bimetallic synergetic effect, Li et al.117 synthesized Fe–Co–Zr polymetallic fibers for CO2 hydrogenation, and obtained 27.5% C2–C4 olefins with the addition of K.
3.2 Methanol-mediated route
In addition to these proposed reaction pathways, some scientists also attempted to use methanol as a bridge and building unit to synthesize long-chain hydrocarbons via CO2 hydrogenation, and made a major breakthrough most recently.96 This newly developed reaction pathway is another alternative option for CO2 hydrogenation to hydrocarbons through an indirect way.The products of FTS generally follow a statistical hydrocarbon distribution, which is known as the Anderson–Schulz–Flory (ASF) distribution.139 In the ideal case, the chain-growth probability (α) is independent of carbon chain length. Therefore the product distribution is determined by the chain-growth probability (α). For example, if CO2 hydrogenation is through the modified FTS route, ASF distribution of FTS products limits the maximum selectivities of C5–C11 (gasoline range) and C12–C20 (diesel range) hydrocarbons to roughly 45% and 30%, respectively.140,141 To break the limitation of ASF distribution and get more gasoline or lower olefins, the methanol-mediated route is an ideal path. Enlightened by the superior selectivity to methanol (ca. 100%) on In2O3-based catalysts from CO2 hydrogenation,142 Sun et al.26 used In2O3/HZSM-5 composite to selectively produce 78.6% C5–C11 hydrocarbons with a high octane number through the methanol route, and broke the ASF restraint. In the proposed reaction pathway, CO2 first was converted to methanol on the In2O3 surface, which was further transformed to hydrocarbons on HZSM-5 via the hydrocarbon-pool mechanism. DFT calculation indicates that the CO2 was first chemisorbed on the oxygen vacancies on the In2O3, and the active site was not the metallic phase. Fujiwara et al.125 prepared composite catalysts obtained by the physical mixing of Cu–Zn–Al oxide and HB zeolite that was modified with 1,4-bis(hydroxydimethylsilyl)benzene, which were very effective for the production of C2+ hydrocarbons, and reached ca. 12.6% at 300 °C and 0.98 MPa. They proposed a reaction scheme of CO2 hydrogenation over the composite catalyst as illustrated in Fig. 11. The methanol was synthesized on the Cu–Zn–Al oxide catalyst, and methanol was converted to C2+ hydrocarbons on the zeolite, which proceeded simultaneously in a single catalytic bed. The preservation of the strong acid sites of the modified HB zeolite with hydrophobic surface improved the second-step CH3OH conversion activity.143
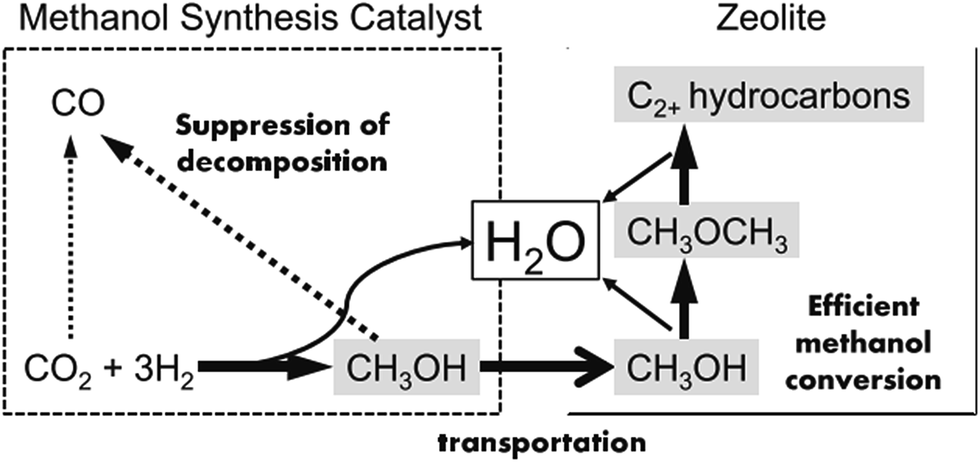 | ||
| Fig. 11 Reaction steps of CO2 hydrogenation over composite catalyst (favorable paths are shown in bold lines). Reprinted with permission from ref. 125. Copyright 2015 Elsevier. | ||
There are some differences between CO2 hydrogenation and CO hydrogenation to hydrocarbons through the methanol-mediated route, such as the molecular polarity,105 number of C–O bonds and adsorption capacity of reactants.144 However, there are also some similarities, such as the subsequent reaction on the zeolites when methanol is formed. So, the design of bifunctional catalysts for CO2 hydrogenation can be inspired by the syngas conversion catalysts. Recently, the products of syngas conversion over the bifunctional catalysts have broken through the traditional ASF distribution and giving the desired products selectively. Wang et al.145,146 prepared mesoporous H-ZSM-5-supported cobalt nanoparticles for conversion of syngas to hydrocarbons. The C5–C11 selectivity can reach as high as ca. 70% which was due to the hydrocracking/isomerization of the higher hydrocarbons on the Brønsted acidic sites of H-ZSM-5. A series of core–shell catalysts (Fe–Zn–Zr@zeolites) were synthesized by Wang et al.124 to adjust product distribution, especially in an attempt to improve isoalkane content by the confinement effect. Over 80% isoalkanes among all hydrocarbons were produced on Fe–Zn–Zr@HZSM5-Hbeta catalyst.
Wang and co-workers also integrated the methanol synthesis and methanol-to-olefins reactions with a bifunctional catalyst, Zr–Zn (2:1)/SAPO-34. The C2–C4 olefin selectivity can reach 74% with a CO conversion of 11% at 673 K, thus breaking the limitation of ASF distribution.103 Furthermore, Wang et al. synthesized ZnGa2O4/SAPO-34 for CO2 hydrogenation to C2–C4 olefins with a selectivity of 86% using the oxygen vacancies on ZnGa2O4 to activate CO2 molecules.121 Additionally, the importance of oxygen vacancies was also evidenced by Sun and co-workers. They prepared a bifunctional catalyst composed of indium–zirconium composite oxide and SAPO-34 zeolite which offered C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) –C4 selectivity as high as 80% at more than 35% CO2 conversion.123 DFT calculations revealed that In2O3 was a unique catalyst in CO2 activation and hydrogenation to methanol with its surface oxygen vacancies and that the reaction followed a mechanism comprising the cyclic creation and annihilation of oxygen vacancies.147 These results indicated that the incorporation of Zr into In2O3 created new kinds of vacancies with high concentration, which progressively enhanced the reaction rate evidenced by DFT calculations. It is worth noting that no obvious deactivation is observed over 150 h, indicating a promising potential for industrial application. Bao et al.148 presented a process to eliminate the ASF distribution for synthesis gas to light olefins that was enabled by a bifunctional catalyst with two types of active sites. The partially reduced oxide surface (ZnCrOx) activated CO and H2, and C–C coupling was subsequently manipulated within the confined acidic pores of SAPO zeolites. They suggested that the appropriate distance of the two active sites was beneficial for C2–C4 formation. The C2–C4 selectivity could be optimized as high as 80%, and the C2–C4 selectivity was 94%, which broke the theoretical limit of only 58% for C2–C4 hydrocarbons as well. Li et al.122 also fabricated a tandem catalyst, ZnZrO/SAPO-34, for CO2 conversion with a selectivity for lower olefins as high as 80–90% among hydrocarbon products. It is proposed that CO2 and H2 were activated on ZnZrO and the C–C bond formation was performed on SAPO through DRIFT characterization. CHxO was considered as intermediate species that included not only methanol. This tandem catalyst showed a resistance to thermal and sulfur treatments (H2S and SO2), suggesting promising potential application in industry.
–C4 selectivity as high as 80% at more than 35% CO2 conversion.123 DFT calculations revealed that In2O3 was a unique catalyst in CO2 activation and hydrogenation to methanol with its surface oxygen vacancies and that the reaction followed a mechanism comprising the cyclic creation and annihilation of oxygen vacancies.147 These results indicated that the incorporation of Zr into In2O3 created new kinds of vacancies with high concentration, which progressively enhanced the reaction rate evidenced by DFT calculations. It is worth noting that no obvious deactivation is observed over 150 h, indicating a promising potential for industrial application. Bao et al.148 presented a process to eliminate the ASF distribution for synthesis gas to light olefins that was enabled by a bifunctional catalyst with two types of active sites. The partially reduced oxide surface (ZnCrOx) activated CO and H2, and C–C coupling was subsequently manipulated within the confined acidic pores of SAPO zeolites. They suggested that the appropriate distance of the two active sites was beneficial for C2–C4 formation. The C2–C4 selectivity could be optimized as high as 80%, and the C2–C4 selectivity was 94%, which broke the theoretical limit of only 58% for C2–C4 hydrocarbons as well. Li et al.122 also fabricated a tandem catalyst, ZnZrO/SAPO-34, for CO2 conversion with a selectivity for lower olefins as high as 80–90% among hydrocarbon products. It is proposed that CO2 and H2 were activated on ZnZrO and the C–C bond formation was performed on SAPO through DRIFT characterization. CHxO was considered as intermediate species that included not only methanol. This tandem catalyst showed a resistance to thermal and sulfur treatments (H2S and SO2), suggesting promising potential application in industry.
The current results have demonstrated that the preparation of bifunctional catalysts combining metal oxides and zeolites is an effective way to control product selectivity for C1 conversion, and the appropriate hydrogenation ability of the two components in such bifunctional catalyst is crucial for adjusting product selectivity.
In summary, CO2 hydrogenation to C2+ hydrocarbons can be catalyzed through modified FTS route or methanol-mediated route to promote hydrocarbon chain growth. For the modified FTS metal-based catalysts, appropriate active metal should be chosen, such as Fe, to get the best hydrogenation capacity. The support basicity and oxygen vacancies should also be improved to increase CO2 adsorption and activation.62 In addition, adding promoters to adjust the surface C/H ratio and reduction capacity of the active metal is another approach to promote hydrocarbon chain growth.116,120,135 For the methanol-mediated route, bifunctional catalysts combining metal oxides and zeolites are crucial for obtaining a higher selectivity of long-chain hydrocarbons. Acid sites are important for the conversion of methanol to hydrocarbons and the channel diameter can influence product selectivities due to the shape-selectivity characteristic.143 SPAO-34 with 8-ring pore structure is beneficial for C2–C4 formation148 and ZSM-5 with 10-ring pore structure will lead to C5–C11 formation.105 Therefore, tuning acid strength and pore size plays a significant role in the formation of C2+ hydrocarbons and it is a promising direction to promote chain growth.
3.3 Mechanisms of C–C coupling
There is an essential and very large difference between CO2 methanation and CO2 hydrogenation to C2+ hydrocarbons, which is the C–C coupling barrier for C2+ hydrocarbon formation. In light of the hydrocarbon formation mechanism, the key point for producing long-chain hydrocarbons is controlling the active H/C to an appropriate ratio, wherein too much surface H* will lead to excessive hydrogenation and methane formation, while the opposite condition will offset the hydrogenation ability and reduce the activity of CO2 conversion. Satthawong and co-workers added different amounts of potassium to Fe–Co bimetallic catalysts to tune the product selectivity. The CO2 adsorption was promoted and the H2 adsorption was suppressed with increasing K content through CO2-TPD and H2-TPD; the C5+ selectivity also increased with more potassium addition. However, when the K/Fe atomic ratio increased from 0.5 to 1, the CO2 conversion decreased. They attributed the selectivity of the product to the type and concentration of chemisorbed hydrogen and carbon dioxide on the catalyst surface.116 Apparently, tuning the surface H/C ratios to manipulate and optimize the product distribution appears to be decisive in the synthesis of C2+ hydrocarbons via CO2 hydrogenation.The crucial step of hydrocarbon synthesis by CO2 hydrogenation is first C–C bond formation and C–O bond cleavage. Many computational studies have been conducted to investigate the C–C coupling step over various catalysts.149–157 Different metals showed diverse catalytic performances. Co- and Fe-based catalysts are widely used to catalyze CO2 or CO hydrogenation to hydrocarbons.158–163 Cu with unfavorable ability for C–O bond cleavage usually converted CO2 or CO to C2+ hydrocarbons164 or alcohols.28,150,165–168
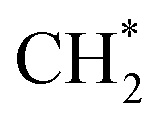 and
and 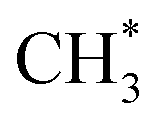 hydrogenation.155 Compared with the CO* insertion mechanism,
hydrogenation.155 Compared with the CO* insertion mechanism, 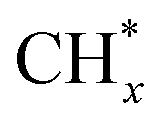 coupling appeared to be a possible C2 hydrocarbon synthesis route on the χ-Fe5C2(510) surface. The chemisorbed CO* dissociated to become C* on the χ-Fe5C2(510) surface, and was hydrogenated to CH* species in the following step. C* + CH* and CH* + CH* were the most likely coupling pathways, and were characterized with carbide mechanism.
coupling appeared to be a possible C2 hydrocarbon synthesis route on the χ-Fe5C2(510) surface. The chemisorbed CO* dissociated to become C* on the χ-Fe5C2(510) surface, and was hydrogenated to CH* species in the following step. C* + CH* and CH* + CH* were the most likely coupling pathways, and were characterized with carbide mechanism.Recently Nie et al.109 studied C–C coupling over Fe–Cu bimetallic catalysts (Fig. 12). CH* was proposed as the most likely monomer over both pure Fe(100) and Cu-doped Fe(100) surfaces, though CH* formation was quite different over the two surfaces. On the Fe(100) facet, CO2 was directly dissociated to form CO* then to CH* through subsequent hydrogenation. Differently, the intermediate CO* was transformed to HCOO* then to CH* through hydrogenation and dissociation in sequence on the Cu-doped Fe(100) facet. The pure Fe(100) favored CH4 synthesis with a low barrier of CH* hydrogenation, while Cu promoted C2H4 synthesis with a low barrier CH* + CH*. The 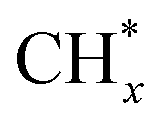 coupling pathway was also proposed as a plausible mechanism for C2 hydrocarbon synthesis over other iron facets.169,170 Clearly, the appropriate catalysts for CO2 hydrogenation to hydrocarbons do not always go through the CO route (modified FTS route), which offers an alternative to break the restraint of the ASF distribution and the equilibrium conversion of CO2 to methanol.
coupling pathway was also proposed as a plausible mechanism for C2 hydrocarbon synthesis over other iron facets.169,170 Clearly, the appropriate catalysts for CO2 hydrogenation to hydrocarbons do not always go through the CO route (modified FTS route), which offers an alternative to break the restraint of the ASF distribution and the equilibrium conversion of CO2 to methanol.
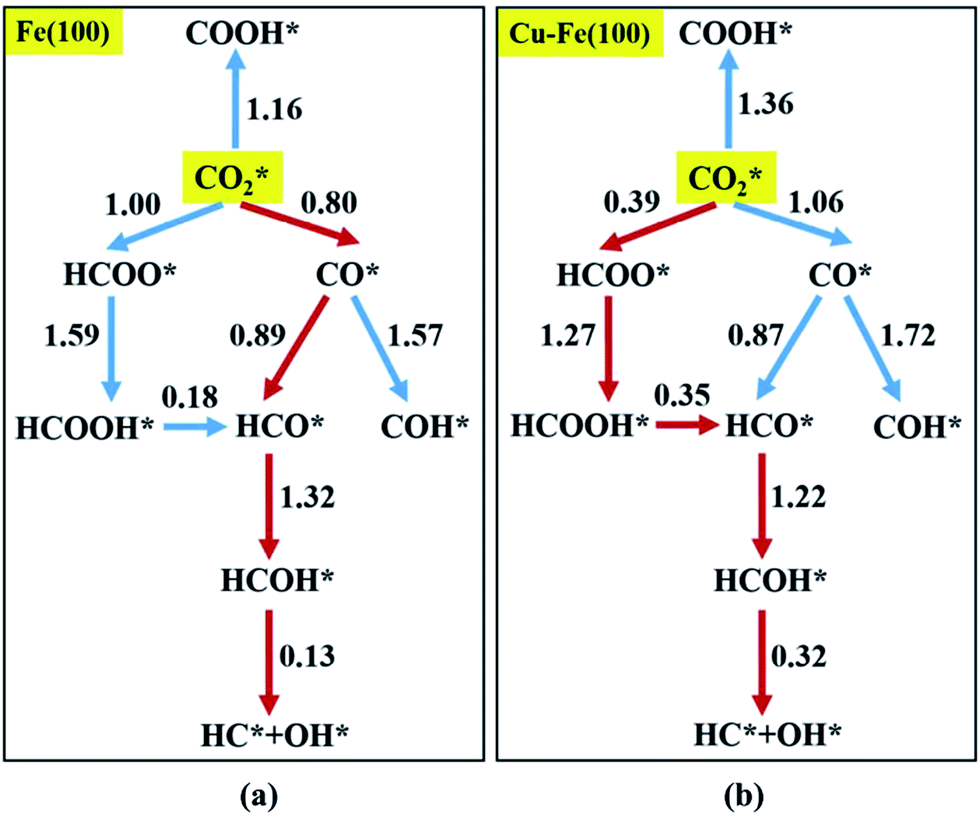 | ||
| Fig. 12 Reaction networks examined to identify energetically favorable C1 species from CO2 hydrogenation on (a) Fe(100) and (b) Cu–Fe(100) surface at 4/9 ML Cu coverage. Activation barriers are given in eV (the networks connected with red arrows represent the preferred path for CO2 conversion to CH*). Reprinted with permission from ref. 109. Copyright 2017 ACS. | ||
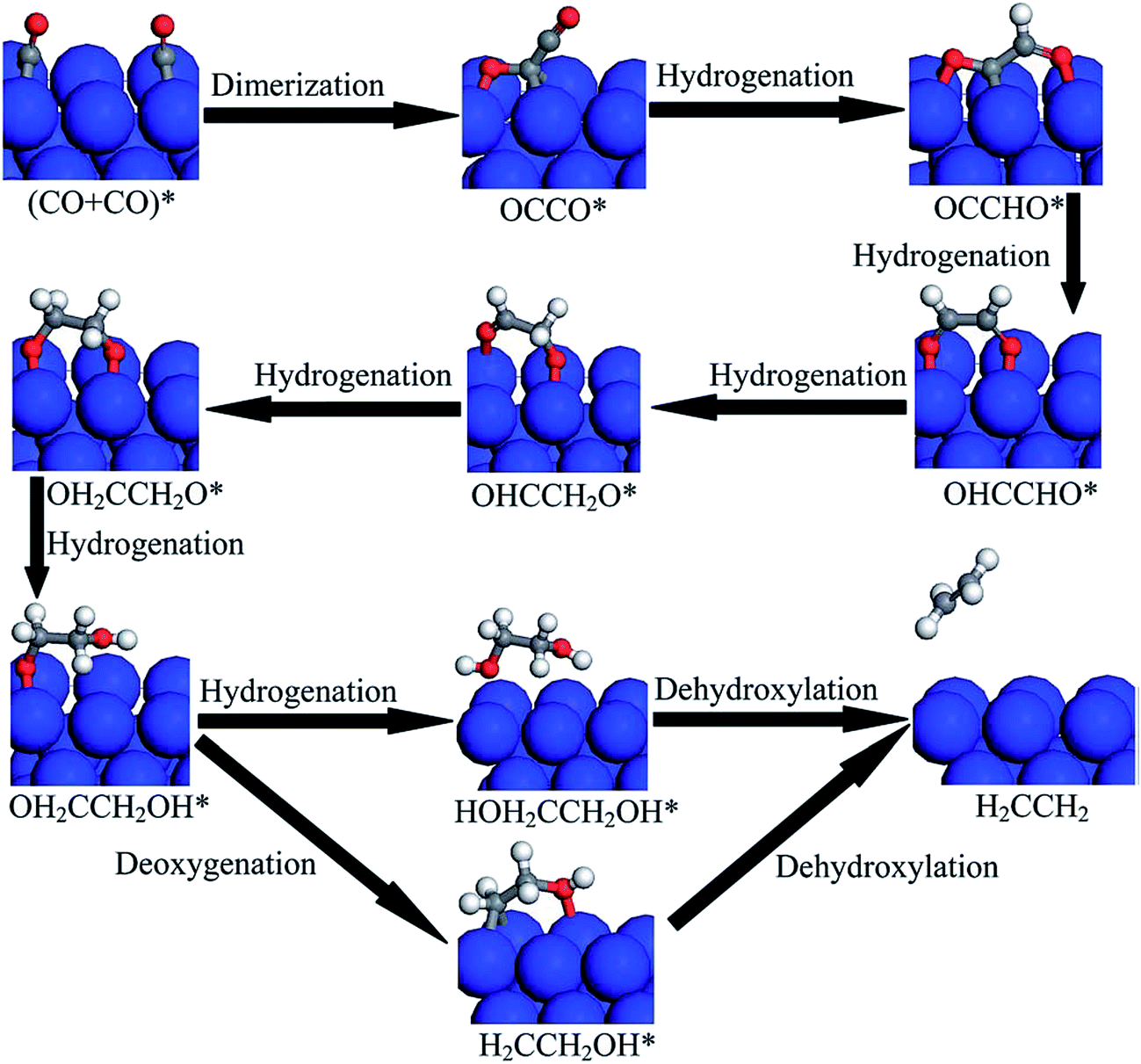 | ||
| Fig. 13 Proposed reduction pathways for the production of C2H4 in the reduction mechanism of CO dimer on Cu(100). Reprinted with permission from ref. 150. Copyright 2015 RSC. | ||
Compared with the CO* dimerization mechanism, CO* coupled with CHx also contributed to hydrocarbon formation over Cu-based catalysts. Wang et al.152–154 investigated the effect of Cu on higher alcohol and hydrocarbon formation. The higher alcohol formation was facilitated by lowering the barrier of CO* coupling with 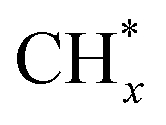 using copper as the promoter over Co-based catalysts.153 The C2 oxygenate was the main product over CuΣ5(310) surface by CO* coupling with
using copper as the promoter over Co-based catalysts.153 The C2 oxygenate was the main product over CuΣ5(310) surface by CO* coupling with 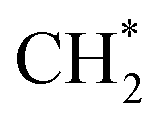 which could not occur over pure Cu(111) and Cu(100) facets.152 The coverage of
which could not occur over pure Cu(111) and Cu(100) facets.152 The coverage of 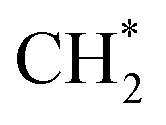 has an essential role in this mechanism. Cu exhibited much better catalytic performance for the association reaction than Co and Ni, while Co benefited the dissociation reaction.154,171 With the assistance of Co and the CuΣ5(310) surface's special active sites, the formation of
has an essential role in this mechanism. Cu exhibited much better catalytic performance for the association reaction than Co and Ni, while Co benefited the dissociation reaction.154,171 With the assistance of Co and the CuΣ5(310) surface's special active sites, the formation of 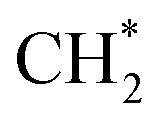 was accelerated, and the CO* coupling mechanism was favored. Co and Ni were capable of catalyzing CO to CH* and further to
was accelerated, and the CO* coupling mechanism was favored. Co and Ni were capable of catalyzing CO to CH* and further to 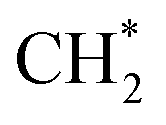 as the favorable monomer. Due to the low barrier of CO* insertion into
as the favorable monomer. Due to the low barrier of CO* insertion into 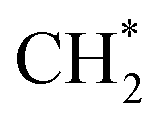 over Co(111), the Co-based catalysts favored chain growth. On the other hand, Ni-based catalysts were used for CO or CO2 methanation with higher barrier of CO* insertion and lower barrier of
over Co(111), the Co-based catalysts favored chain growth. On the other hand, Ni-based catalysts were used for CO or CO2 methanation with higher barrier of CO* insertion and lower barrier of 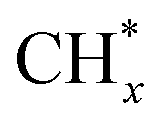 hydrogenation.92,154,172,173 Zhang et al.171 investigated the CO hydrogenation over Co-decorated Cu alloy catalyst, and stated that the Co–Cu(211) surface was conducive to ethanol formation rather than methane or methanol, and the C–C coupling was accomplished by interacting CO* with
hydrogenation.92,154,172,173 Zhang et al.171 investigated the CO hydrogenation over Co-decorated Cu alloy catalyst, and stated that the Co–Cu(211) surface was conducive to ethanol formation rather than methane or methanol, and the C–C coupling was accomplished by interacting CO* with 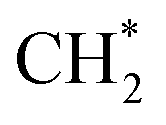 and
and 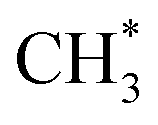 .
.
Zuo et al.156 explored ethanol synthesis by syngas over alloy-like CoCu(111) surface, and they found that CO* + 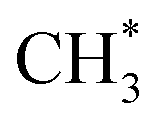 was the most likely pathway of coupling. The above computational studies demonstrate that CO*, as the main intermediate or the reactant during CO2 hydrogenation, was able to interact with surface CO* or
was the most likely pathway of coupling. The above computational studies demonstrate that CO*, as the main intermediate or the reactant during CO2 hydrogenation, was able to interact with surface CO* or 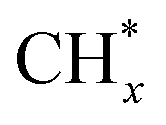 species over Cu- and Co-based catalysts, through which C–C coupling was available for the formation of long-chain products.
species over Cu- and Co-based catalysts, through which C–C coupling was available for the formation of long-chain products.
3.4 Deactivation of catalysts for CO2 hydrogenation to hydrocarbons
Lee et al.174 investigated the reasons for deactivation of Fe–K/γ-Al2O3 for CO2 hydrogenation to hydrocarbons through XPS, HR-TEM, TPO, and Mössbauer spectroscopy. The reasons for deactivation are different at different positions in the reactor. As time progressed, the Fe2O3 was reduced to active phase χ-Fe5C3 and finally the χ-Fe5C3 was transformed to θ-FeC3, which is not an active species for CO2 hydrogenation. Hence, in the inlet reactor region, the deactivation pathway was phase transformation. Conversely, the main factor at the outlet part of the reactor was coke deposition.Li et al.117 observed the remarkable metal sintering on supported FeCo/ZrO2 catalysts which was responsible for the rapid deactivation of activity. In contrast, Fe–Co–Zr polymetallic fibers obtained by a one-step electrospinning technique showed stable activity over the reaction period. Co and Fe were dispersed in proximity to ZrO2, but separately from each other, which, in turn, helped reduce the possibility of sintering. Active metals encapsulated in hollow zeolite175 or confined in nanotubes176 were also applied to resist metal sintering and increase catalyst stability, which are good references for CO2 hydrogenation catalysts.
4. Conclusion and prospects
Environmental issues have pushed the necessity to reduce CO2 emissions caused by the use of fossil fuels. Many efforts have been made to develop catalysts and understand the reaction mechanisms. Heterogeneous thermocatalysis is a promising direction for application in CO2 conversion. The catalyst performance can be affected by many factors, such as metal–support interaction, metal particle size and promoters. Ni-based catalysts are mainly used in CH4 production from CO2 hydrogenation. In addition, Co, Ru, Ir and Rh are also applied for CO2 methanation. Fe is an active metal for CO2 hydrogenation to C2+ hydrocarbons through modified FTS route or methanol-mediated route. Fe–metal bimetallic catalysts have shown markedly improved performance. The preparation of bifunctional catalysts combining metal oxides and zeolites is an effective way to control the product selectivity for C1 conversion. Some experiments and DFT calculations have given the encouraging result that CO2 conversion can be catalyzed through the formate intermediate route which is neither the CO route nor the methanol route, which will not be limited by the ASF distribution and the equilibrium conversion of CO2 to methanol. The crucial mechanisms of the initial C–C bond formation and C–O bond cleavage are different between Fe-based catalysts and Cu-based catalysts in DFT calculations.In general, future research directions for CO2 hydrogenation are proposed as follows:
1. To adjust the catalyst surface H/C ratio and facilitate C–C coupling and generate high-value-added products.
2. To improve the support basicity and oxygen vacancies and increase the CO2 adsorption and activation.
3. To explore more novel catalytic materials and improve the catalyst stability.
4. To explore more active catalysts for low-temperature and energy-saving CO2 hydrogenation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2016YFB0600902-5).References
- B. Kahn, Earth's CO2 Passes the 400 PPM Threshold—Maybe Permanently, Climate Central, 2016 Search PubMed.
- A Roadmap for moving to a competitive low carbon economy in 2050, European Commission, 2011 Search PubMed.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- I. Dimitriou, P. García-Gutiérrez, R. H. Elder, R. M. Cuéllar-Franca, A. Azapagic and R. W. K. Allen, Energy Environ. Sci., 2015, 8, 1775–1789 CAS.
- X. Ma, X. Wang and C. Song, J. Am. Chem. Soc., 2009, 131, 5777–5783 CrossRef CAS PubMed.
- P. J. Lunde and F. L. Kester, Ind. Eng. Chem. Process Des. Dev., 1974, 13, 27–33 CAS.
- G. Du, S. Lim, Y. Yang, C. Wang, L. Pfefferle and G. L. Haller, J. Catal., 2007, 249, 370–379 CrossRef CAS.
- M. S. Duyar, M. A. A. Treviño and R. J. Farrauto, Appl. Catal., B, 2015, 168–169, 370–376 CrossRef CAS.
- Y. Yoon, A. S. Hall and Y. Surendranath, Angew. Chem., Int. Ed., 2016, 128, 1–6 CrossRef.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- P. G. Jessop, F. Joó and C. C. Tai, Coord. Chem. Rev., 2004, 248, 2425–2442 CrossRef CAS.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- J. Ma, N. Sun, X. Zhang, N. Zhao, F. Xiao, W. Wei and Y. Sun, Catal. Today, 2009, 148, 221–231 CrossRef CAS.
- S. Saeidi, N. A. S. Amin and M. R. Rahimpour, J. CO2 Util., 2014, 5, 66–81 CrossRef CAS.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef CAS.
- G. Centi and S. Perathoner, Greenhouse Gases: Sci. Technol., 2011, 1, 21–35 CrossRef CAS.
- L. Zhou, Q. Wang, L. Ma, J. Chen, J. Ma and Z. Zi, Catal. Lett., 2015, 145, 612–619 CrossRef CAS.
- A. Westermann, B. Azambre, M. C. Bacariza, I. Graca, M. F. Ribeiro, J. M. Lopes and C. Henriques, Appl. Catal., B, 2015, 174, 120–125 CrossRef.
- D. C. Upham, A. R. Derk, S. Sharma, H. Metiu and E. W. McFarland, Catal. Sci. Technol., 2015, 5, 1783–1791 CAS.
- N. Shimoda, D. Shoji, K. Tani, M. Fujiwara, K. Urasaki, R. Kikuchi and S. Satokawa, Appl. Catal., B, 2015, 174, 486–495 CrossRef.
- R. Razzaq, C. Li, M. Usman, K. Suzuki and S. Zhang, Chem. Eng. J., 2015, 262, 1090–1098 CrossRef CAS.
- S. K. Beaumont, S. Alayoglu, C. Specht, W. D. Michalak, V. V. Pushkarev, J. Guo, N. Kruse and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 9898–9901 CrossRef CAS PubMed.
- F. Wang, C. Li, X. Zhang, M. Wei, D. G. Evans and X. Duan, J. Catal., 2015, 329, 177–186 CrossRef CAS.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 CAS.
- C. G. Visconti, M. Martinelli, L. Falbo, A. Infantes-Molina, L. Lietti, P. Forzatti, G. Iaquaniello, E. Palo, B. Picutti and F. Brignoli, Appl. Catal., B, 2017, 200, 530–542 CrossRef CAS.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Nat. Chem., 2017, 9, 1019–1024 CrossRef CAS PubMed.
- H. Song, N. Zhang, C. Zhong, Z. Liu, M. Xiao and H. Gai, New J. Chem., 2017, 41, 9170–9177 RSC.
- K. Larmier, W. C. Liao, S. Tada, E. Lam, R. Verel, A. Bansode, A. Urakawa, A. Comas-Vives and C. Coperet, Angew. Chem., Int. Ed., 2017, 56, 1–7 CrossRef.
- X. Jiang, N. Koizumi, X. Guo and C. Song, Appl. Catal., B, 2015, 170–171, 173–185 CrossRef CAS.
- S. Bai, Q. Shao, P. Wang, Q. Dai, X. Wang and X. Huang, J. Am. Chem. Soc., 2017, 139, 6827–6830 CrossRef CAS PubMed.
- K. Mazloomi, N. Sulaiman and H. Moayedi, Int. J. Electrochem. Sci., 2012, 7, 3314–3326 CAS.
- M. S. Duyar, A. Ramachandran, C. Wang and R. J. Farrauto, J. CO2 Util., 2015, 12, 27–33 CrossRef CAS.
- B. Mutz, H. W. P. Carvalho, S. Mangold, W. Kleist and J. D. Grunwaldt, J. Catal., 2015, 327, 48–53 CrossRef CAS.
- M. Younas, L. L. Kong, M. J. K. Bashir, H. Nadeem, A. Shehzad and S. Sethupathi, Energy Fuels, 2016, 30, 8815–8831 CrossRef CAS.
- S. Rönsch, J. Schneider, S. Matthischke, M. Schlüter, M. Götz, J. Lefebvre, P. Prabhakaran and S. Bajohr, Fuel, 2016, 166, 276–296 CrossRef.
- M. Y. K. Hashimoto and S. Meguro, Corros. Sci., 2002, 44, 371–386 CrossRef.
- A. D. Tomsett, T. Haginwara, A. Miyamoto and T. Inui, Appl. Catal., 1986, 26, 391 CrossRef CAS.
- K. R. Thampi, J. Kiwi and M. Gratzel, Nature, 1987, 327, 506 CrossRef CAS.
- J. Gao, Y. Wang, Y. Ping, D. Hu, G. Xu, F. Gu and F. Su, RSC Adv., 2012, 2, 2358 RSC.
- F. Koschany, D. Schlereth and O. Hinrichsen, Appl. Catal., B, 2016, 181, 504–516 CrossRef CAS.
- J. Janlamool, P. Praserthdam and B. Jongsomjit, J. Nat. Gas Chem., 2011, 20, 558–564 CrossRef CAS.
- Z. Guilin, W. Tian, X. Hongmei and Z. Xuxu, Int. J. Hydrogen Energy, 2013, 38, 10012–10018 CrossRef.
- G. L. Zhou, T. Wu, H. B. Zhang, H. M. Xie and Y. C. Feng, Chem. Eng. Commun., 2014, 201, 233–240 CrossRef CAS.
- H. H. Shin, L. Lu, Z. Yang, C. J. Kiely and S. McIntosh, ACS Catal., 2016, 6, 2811–2818 CrossRef CAS.
- I. Rossetti, C. Biffi, C. L. Bianchi, V. Nichele, M. Signoretto, F. Menegazzo, E. Finocchio, G. Ramis and A. Di Michele, Appl. Catal., B, 2012, 117–118, 384–396 CrossRef CAS.
- D. Theleritis, S. Souentie, A. Siokou, A. Katsaounis and C. G. Vayenas, ACS Catal., 2012, 2, 770–780 CrossRef CAS.
- J. H. Kwak, L. Kovarik and J. Szanyi, ACS Catal., 2013, 3, 2449–2455 CrossRef CAS.
- A. Karelovic and P. Ruiz, Appl. Catal., B, 2012, 113–114, 237–249 CrossRef CAS.
- J. N. Park and E. W. McFarland, J. Catal., 2009, 266, 92–97 CrossRef CAS.
- J. H. Kwak, L. Kovarik and J. Szanyi, ACS Catal., 2013, 3, 2094–2100 CrossRef CAS.
- J. Liu, D. Cui, J. Yu, F. Su and G. Xu, Chin. J. Chem. Eng., 2015, 23, 86–92 CrossRef CAS.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono, R. R. Mukti, Y. H. Taufiq-Yap and M. R. Sazegar, Appl. Catal., B, 2014, 147, 359–368 CrossRef CAS.
- Z. Q. Wang, Z. N. Xu, S. Y. Peng, M. J. Zhang, G. Lu, Q. S. Chen, Y. Chen and G. C. Guo, ACS Catal., 2015, 5, 4255–4259 CrossRef CAS.
- G. R. Johnson and A. T. Bell, ACS Catal., 2016, 6, 100–114 CrossRef CAS.
- G. Zeng, J. Qiu, Z. Li, P. Pavaskar and S. B. Cronin, ACS Catal., 2014, 4, 3512–3516 CrossRef CAS.
- S. Kattel, W. Yu, X. Yang, B. Yan, Y. Huang, W. Wan, P. Liu and J. G. Chen, Angew. Chem., Int. Ed., 2016, 55, 1–7 CrossRef PubMed.
- Y. Liu, K. Fang, J. Chen and Y. Sun, Green Chem., 2007, 9, 611 RSC.
- S. Sokolov, E. V. Kondratenko, M. M. Pohl, A. Barkschat and U. Rodemerck, Appl. Catal., B, 2012, 113–114, 19–30 CrossRef CAS.
- V. Nichele, M. Signoretto, F. Menegazzo, A. Gallo, V. Dal Santo, G. Cruciani and G. Cerrato, Appl. Catal., B, 2012, 111–112, 225–232 CrossRef CAS.
- A. M. Abdel-Mageed, D. Widmann, S. E. Olesen, I. Chorkendorff, J. Biskupek and R. J. Behm, ACS Catal., 2015, 5, 6753–6763 CrossRef CAS.
- S. Kattel, P. Liu and J. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS PubMed.
- W. Li, X. Nie, X. Jiang, A. Zhang, F. Ding, M. Liu, Z. Liu, X. Guo and C. Song, Appl. Catal., B, 2018, 220, 397–408 CrossRef CAS.
- G. Zhou, H. Liu, K. Cui, H. Xie, Z. Jiao, G. Zhang, K. Xiong and X. Zheng, Int. J. Hydrogen Energy, 2017, 42, 16108–16117 CrossRef CAS.
- G. Zhou, H. Liu, K. Cui, A. Jia, G. Hu, Z. Jiao, Y. Liu and X. Zhang, Appl. Surf. Sci., 2016, 383, 248–252 CrossRef CAS.
- N. M. Martin, P. Velin, M. Skoglundh, M. Bauer and P. A. Carlsson, Catal. Sci. Technol., 2017, 7, 1086–1094 CAS.
- J. A. H. Dreyer, P. Li, L. Zhang, G. K. Beh, R. Zhang, P. H. L. Sit and W. Y. Teoh, Appl. Catal., B, 2017, 219, 715–726 CrossRef CAS.
- A. Kim, C. Sanchez, G. Patriarche, O. Ersen, S. Moldovan, A. Wisnet, C. Sassoye and D. P. Debecker, Catal. Sci. Technol., 2016, 6, 8117–8128 CAS.
- A. Kim, D. P. Debecker, F. Devred, V. Dubois, C. Sanchez and C. Sassoye, Appl. Catal., B, 2018, 220, 615–625 CrossRef CAS.
- Y. Lin, Y. Zhu, X. Pan and X. Bao, Catal. Sci. Technol., 2017, 7, 2813–2818 CAS.
- H. C. Wu, Y. C. Chang, J. H. Wu, J. H. Lin, I. K. Lin and C. S. Chen, Catal. Sci. Technol., 2015, 5, 4154–4163 CAS.
- V. Iablokov, S. K. Beaumont, S. Alayoglu, V. V. Pushkarev, C. Specht, J. Gao, A. P. Alivisatos, N. Kruse and G. A. Somorjai, Nano Lett., 2012, 12, 3091–3096 CrossRef CAS PubMed.
- J. C. Matsubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137, 3076–3084 CrossRef CAS PubMed.
- S. Li, Y. Xu, Y. Chen, W. Li, L. Lin, M. Li, Y. Deng, X. Wang, B. Ge, C. Yang, S. Yao, J. Xie, Y. Li, X. Liu and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 1–6 CrossRef.
- Y. Yan, Y. Dai, H. He, Y. Yu and Y. Yang, Appl. Catal., B, 2016, 196, 108–116 CrossRef CAS.
- D. Wierzbicki, R. Debek, M. Motak, T. Grzybek, M. E. Gálvez and P. Da Costa, Catal. Commun., 2016, 83, 5–8 CrossRef CAS.
- L. Xu, F. Wang, M. Chen, D. Nie, X. Lian, Z. Lu, H. Chen, K. Zhang and P. Ge, Int. J. Hydrogen Energy, 2017, 42, 15523–15539 CrossRef CAS.
- M. P. Andersson, F. Abild-Pedersen, I. N. Remediakis, T. Bligaard, G. Jones, J. Engbæk, O. Lytken, S. Horch, J. H. Nielsen, J. Sehested, J. R. Rostrup-Nielsen, J. K. Nørskov and I. Chorkendorff, J. Catal., 2008, 255, 6–19 CrossRef CAS.
- H. Wang, K. Xu, X. Yao, D. Ye, T. Pei, H. Hu, M. Qiao, Z. H. Li, X. Zhang and B. Zong, ACS Catal., 2018, 8, 1207–1211 CrossRef CAS.
- S. Abate, C. Mebrahtu, E. Giglio, F. Deorsola, S. Bensaid, S. Perathoner, R. Pirone and G. Centi, Ind. Eng. Chem. Res., 2016, 55, 4451–4460 CrossRef CAS.
- S. Abate, K. Barbera, E. Giglio, F. Deorsola, S. Bensaid, S. Perathoner, R. Pirone and G. Centi, Ind. Eng. Chem. Res., 2016, 55, 8299–8308 CrossRef CAS.
- T. Kajiwara, M. Higuchi, D. Watanabe, H. Higashimura, T. Yamada and H. Kitagawa, Chem.–Eur. J., 2014, 20, 15611–15617 CrossRef CAS PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- B. Chen, S. Xiang and G. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS PubMed.
- W. Zhen, B. Li, G. Lu and J. Ma, Chem. Commun., 2015, 51, 1728–1731 RSC.
- W. Li, A. Zhang, X. Jiang, C. Chen, Z. Liu, C. Song and X. Guo, ACS Sustainable Chem. Eng., 2017, 5, 7824–7831 CrossRef CAS.
- N. Srisawad, W. Chaitree, O. Mekasuwandumrong, A. Shotipruk, B. Jongsomjit and J. Panpranot, React. Kinet., Mech. Catal., 2012, 107, 179–188 CrossRef CAS.
- G. Zhan and H. C. Zeng, ACS Catal., 2017, 7, 7509–7519 CrossRef CAS.
- J. Ashok, M. L. Ang and S. Kawi, Catal. Today, 2017, 281, 304–311 CrossRef CAS.
- M. Schubert, S. Pokhrel, A. Thomé, V. Zielasek, T. M. Gesing, F. Roessner, L. Mädler and M. Bäumer, Catal. Sci. Technol., 2016, 6, 7449–7460 CAS.
- S. Danaci, L. Protasova, J. Lefevere, L. Bedel, R. Guilet and P. Marty, Catal. Today, 2016, 273, 234–243 CrossRef CAS.
- R. Zhou, N. Rui, Z. Fan and C. J. Liu, Int. J. Hydrogen Energy, 2016, 41, 22017–22025 CrossRef CAS.
- J. Ren, H. Guo, J. Yang, Z. Qin, J. Lin and Z. Li, Appl. Surf. Sci., 2015, 351, 504–516 CrossRef CAS.
- Q. Zheng, R. Farrauto and A. Chau Nguyen, Ind. Eng. Chem. Res., 2016, 55, 6768–6776 CrossRef CAS.
- Y. Li, G. Lu and J. Ma, RSC Adv., 2014, 4, 17420 RSC.
- I. Graça, L. V. González, M. C. Bacariza, A. Fernandes, C. Henriques, J. M. Lopes and M. F. Ribeiro, Appl. Catal., B, 2014, 147, 101–110 CrossRef.
- F. Wang, S. He, H. Chen, B. Wang, L. Zheng, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2016, 138, 6298–6305 CrossRef CAS PubMed.
- S. Sharma, K. B. Sravan Kumar, Y. M. Chandnani, V. S. Phani Kumar, B. P. Gangwar, A. Singhal and P. A. Deshpande, J. Phys. Chem. C, 2016, 120, 14101–14112 CAS.
- C. Heine, B. A. Lechner, H. Bluhm and M. Salmeron, J. Am. Chem. Soc., 2016, 138, 13246–13252 CrossRef CAS PubMed.
- A. Solis-Garcia, J. F. Louvier-Hernandez, A. Almendarez-Camarillo and J. C. Fierro-Gonzalez, Appl. Catal., B, 2017, 218, 611–620 CrossRef CAS.
- J. K. Kesavana, I. Luisetto, S. Tuti, C. Meneghini, G. Iucci, C. Battocchio, S. Mobilio, S. Casciardi and R. Sisto, J. CO2 Util., 2018, 23, 200–211 CrossRef.
- H. Li, J. Ren, X. Qin, Z. Qin, J. Lin and Z. Li, RSC Adv., 2015, 5, 96504–96517 RSC.
- A. A. Muleja, Y. Yao, D. Glasser and D. Hildebrandt, Ind. Eng. Chem. Res., 2017, 56, 469–478 CrossRef CAS.
- K. Cheng, B. Gu, X. Liu, J. Kang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 4725–4728 CrossRef CAS PubMed.
- Y. Zheng, W. Zhang, Y. Li, J. Chen, B. Yu, J. Wang, L. Zhang and J. Zhang, Nano Energy, 2017, 40, 512–539 CrossRef CAS.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Nat. Commun., 2017, 8, 15174 CrossRef PubMed.
- G. Pekridis, K. Kalimeri, N. Kaklidis, E. Vakouftsi, E. F. liopoulou, C. Athanasiou and G. E. Marnellos, Catal. Today, 2007, 127, 337–346 CrossRef CAS.
- P. Kangvansura, L. M. Chew, W. Saengsui, P. Santawaja, Y. Poo-arporn, M. Muhler, H. Schulz and A. Worayingyong, Catal. Today, 2016, 275, 59–65 CrossRef CAS.
- L. M. Chew, P. Kangvansura, H. Ruland, H. J. Schulte, C. Somsen, W. Xia, G. Eggeler, A. Worayingyong and M. Muhler, Appl. Catal., A, 2014, 482, 163–170 CrossRef CAS.
- X. Nie, H. Wang, M. J. Janik, Y. Chen, X. Guo and C. Song, J. Phys. Chem. C, 2017, 121, 13164–13174 CAS.
- D. J. Dwyer and G. A. Somorjai, J. Catal., 1978, 52, 291–301 CrossRef CAS.
- H. D. Willauer, R. Ananth, M. T. Olsen, D. M. Drab, D. R. Hardy and F. W. Williams, J. CO2 Util., 2013, 3–4, 56–64 CrossRef CAS.
- M. Albrecht, U. Rodemerck, M. Schneider, M. Bröring, D. Baabe and E. V. Kondratenko, Appl. Catal., B, 2017, 204, 119–126 CrossRef CAS.
- G. B. Yu, B. Sun, Y. Pei, S. Xie, S. Yan, M. Qiao, K. Fan, X. Zhang and B. Zong, J. Am. Chem. Soc., 2010, 132, 935–937 CrossRef CAS PubMed.
- F. C. Emiel de Smit, A. M. Beale, O. V. Safonova, P. S. Wouter van Beek and B. M. Weckhuysen, J. Am. Chem. Soc., 2010, 132, 14928–14941 CrossRef PubMed.
- L. Torrente-Murciano, R. S. Chapman, A. Narvaez-Dinamarca, D. Mattia and M. D. Jones, Phys. Chem. Chem. Phys., 2016, 18, 15496–15500 RSC.
- R. Satthawong, N. Koizumi, C. Song and P. Prasassarakich, Catal. Today, 2015, 251, 34–40 CrossRef CAS.
- W. Li, A. Zhang, X. Jiang, M. J. Janik, J. Qiu, Z. Liu, X. Guo and C. Song, J. CO2 Util., 2018, 23, 219–225 CrossRef CAS.
- C. Xie, C. Chen, Y. Yu, J. Su, Y. Li, G. A. Somorjai and P. Yang, Nano Lett., 2017, 17, 3798–3802 CrossRef CAS PubMed.
- Y. H. Choi, Y. J. Jang, H. Park, W. Y. Kim, Y. H. Lee, S. H. Choi and J. S. Lee, Appl. Catal., B, 2017, 202, 605–610 CrossRef CAS.
- M. Al-Dossary, A. A. Ismail, J. L. G. Fierro, H. Bouzid and S. A. Al-Sayari, Appl. Catal., B, 2015, 165, 651–660 CrossRef CAS.
- X. Liu, M. Wang, C. Zhou, W. Zhou, K. Cheng, J. Kang, Q. Zhang, W. Deng and Y. Wang, Chem. Commun., 2018, 54, 140–143 RSC.
- Z. Li, J. Wang, Y. Qu, H. Liu, C. Tang, S. Miao, Z. Feng, H. An and C. Li, ACS Catal., 2017, 7, 8544–8548 CrossRef CAS.
- P. Gao, S. Dang, S. Li, X. Bu, Z. Liu, M. Qiu, C. Yang, H. Wang, L. Zhong, Y. Han, Q. Liu, W. Wei and Y. Sun, ACS Catal., 2017, 8, 571–578 CrossRef.
- X. Wang, G. Yang, J. Zhang, S. Chen, Y. Wu, Q. Zhang, J. Wang, Y. Han and Y. Tan, Chem. Commun., 2016, 52, 7352–7355 RSC.
- M. Fujiwara, T. Satake, K. Shiokawa and H. Sakurai, Appl. Catal., B, 2015, 179, 37–43 CrossRef CAS.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B. Kniep, M. Tovar, R. Fischer, J. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- F. Ding, A. Zhang, M. Liu, Y. Zuo, K. Li, X. Guo and C. Song, Ind. Eng. Chem. Res., 2014, 53, 17563–17569 CrossRef CAS.
- K. Zhao, W. Wang and Z. Li, J. CO2 Util., 2016, 16, 236–244 CrossRef CAS.
- J. Wang, Z. You, Q. Zhang, W. Deng and Y. Wang, Catal. Today, 2013, 215, 186–193 CrossRef CAS.
- X. Liu, Y. Jia, Y. Zhang, G. Ren, R. Feng, S. Zhang, M. Zaworotko and X. Bu, Inorg. Chem. Front., 2016, 3, 1510–1515 RSC.
- B. Mousavi, S. Chaemchuen, B. Moosavi, Z. Luo, N. Gholampour and F. Verpoort, New J. Chem., 2016, 40, 5170–5176 RSC.
- J. Ye and J. Johnson, ACS Catal., 2015, 5, 2921–2928 CrossRef CAS.
- S. Hu, M. Liu, F. Ding, C. Song, G. Zhang and X. Guo, J. CO2 Util., 2016, 15, 89–95 CrossRef CAS.
- J. Wei, J. Sun, Z. Wen, C. Fang, Q. Ge and H. Xu, Catal. Sci. Technol., 2016, 6, 4786–4793 CAS.
- T. Riedel, M. Claeys, H. Schulz, G. Schaub, S. Nam, K. Jun, M. Choi, G. Kishan and K. Lee, Appl. Catal., A, 1999, 186, 201–213 CrossRef CAS.
- C. G. Visconti, M. Martinelli, L. Falbo, L. Fratalocchi and L. Lietti, Catal. Today, 2016, 277, 161–170 CrossRef CAS.
- R. Satthawong, N. Koizumi, C. Song and P. Prasassarakich, Top. Catal., 2014, 57, 588–594 CrossRef CAS.
- R. Satthawong, N. Koizumi, C. Song and P. Prasassarakich, J. CO2 Util., 2013, 3–4, 102–106 CrossRef CAS.
- Q. Zhang, J. Kang and Y. Wang, ChemCatChem, 2010, 2, 1030–1058 CrossRef CAS.
- U. Rodemerck, M. Holeňa, E. Wagner, Q. Smejkal, A. Barkschat and M. Baerns, ChemCatChem, 2013, 5, 1948–1955 CrossRef CAS.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Energy Environ. Sci., 2010, 3, 884 CAS.
- O. Martin, A. J. Martín, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferré and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef CAS PubMed.
- M. Fujiwara, H. Sakurai, K. Shiokawa and Y. Iizuka, Catal. Today, 2015, 242, 255–260 CrossRef CAS.
- M. Kogler, E. M. Köck, B. Klötzer, L. Perfler and S. Penner, J. Phys. Chem. C, 2016, 120, 3882–3898 CAS.
- K. Cheng, L. Zhang, J. Kang, X. Peng, Q. Zhang and Y. Wang, Chem.–Eur. J., 2015, 21, 1928–1937 CrossRef CAS PubMed.
- J. Kang, K. Cheng, L. Zhang, Q. Zhang, J. Ding, W. Hua, Y. Lou, Q. Zhai and Y. Wang, Angew. Chem., Int. Ed., 2011, 50, 5200–5203 CrossRef CAS PubMed.
- J. Ye, C. Liu, D. Mei and Q. Ge, ACS Catal., 2013, 3, 1296–1306 CrossRef CAS.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS PubMed.
- L. Ou, J. Mol. Model., 2016, 22, 246 CrossRef PubMed.
- L. Ou, W. Long, Y. Chen and J. Jin, RSC Adv., 2015, 5, 96281–96289 RSC.
- H. Xiao, T. Cheng and W. A. Goddard, J. Am. Chem. Soc., 2017, 139, 130–136 CrossRef CAS PubMed.
- J. Wang, Q. Sun, S. Chan and H. Su, Appl. Catal., A, 2016, 509, 97–104 CrossRef CAS.
- J. Wang, X. Zhang, Q. Sun, S. Chan and H. Su, Catal. Commun., 2015, 61, 57–61 CrossRef CAS.
- J. Wang, Y. Kawazoe, Q. Sun, S. Chan and H. Su, Surf. Sci., 2016, 645, 30–40 CrossRef CAS.
- T. H. Pham, Y. Qi, J. Yang, X. Duan, G. Qian, X. Zhou, D. Chen and W. Yuan, ACS Catal., 2015, 5, 2203–2208 CrossRef CAS.
- Z. J. Zuo, F. Peng and W. Huang, Sci. Rep., 2016, 6, 34670 CrossRef CAS PubMed.
- T. H. Pham, X. Duan, G. Qian, X. Zhou and D. Chen, J. Phys. Chem. C, 2014, 118, 10170–10176 CAS.
- M. K. Gnanamani, G. Jacobs, H. H. Hamdeh, W. D. Shafer, F. Liu, S. D. Hopps, G. A. Thomas and B. H. Davis, ACS Catal., 2016, 6, 913–927 CrossRef CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS PubMed.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 CAS.
- P. Helden, J. Berg, M. Petersen, W. Rensburg, I. Ciobica and J. van de Loosdrecht, Faraday Discuss., 2017, 197, 117–151 RSC.
- D. Chakrabarti, A. de Klerk, V. Prasad, M. Gnanamani, W. Shafer, G. Jacobs, D. Sparks and B. Davis, Ind. Eng. Chem. Res., 2015, 54, 1189–1196 CrossRef CAS.
- M. Landau, N. Meiri, N. Utsis, R. Vidruk Nehemya and M. Herskowitz, Ind. Eng. Chem. Res., 2017, 56, 13334–13355 CrossRef CAS.
- M. Bersani, K. Gupta, A. K. Mishra, R. Lanza, S. F. R. Taylor, H. U. Islam, N. Hollingsworth, C. Hardacre, N. H. de Leeuw and J. A. Darr, ACS Catal., 2016, 6, 5823–5833 CrossRef CAS.
- S. Kattel, B. Yan, Y. Yang, J. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS PubMed.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS PubMed.
- X. Nie, W. Luo, M. J. Janik and A. Asthagiri, J. Catal., 2014, 312, 108–122 CrossRef CAS.
- W. Luo, X. Nie, M. J. Janik and A. Asthagiri, ACS Catal., 2016, 6, 219–229 CrossRef CAS.
- H. J. Li, C. C. Chang and J. J. Ho, J. Phys. Chem. C, 2011, 115, 11045–11055 CAS.
- J. Cheng, P. Hu, P. Ellis, S. French, G. Kelly and C. M. Lok, J. Phys. Chem. C, 2008, 112, 6082–6086 CAS.
- R. Zhang, F. Liu and B. Wang, Catal. Sci. Technol., 2016, 6, 8036–8054 CAS.
- M. Roiaz, E. Monachino, C. Dri, M. Greiner, A. Knop-Gericke, R. Schlogl, G. Comelli and E. Vesselli, J. Am. Chem. Soc., 2016, 138, 4146–4154 CrossRef CAS PubMed.
- C. Zhi, R. Zhang and B. Wang, Mol. Catal., 2017, 438, 1–14 CrossRef CAS.
- S. C. Lee, J. S. Kim, W. C. Shin, M. J. Choi and S. J. Choung, J. Mol. Catal. A: Chem., 2009, 301, 98–105 CrossRef CAS.
- C. Dai, S. Zhang, A. Zhang, C. Song and X. Guo, J. Mater. Chem. A, 2015, 3, 16461–16468 CAS.
- J. Kang, S. Zhang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2009, 48, 2565–2568 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |