DOI:
10.1039/C7RA13536J
(Paper)
RSC Adv., 2018,
8, 5760-5767
Conjugated polymers containing tetraphenylethylene in the backbones and side-chains for highly sensitive TNT detection
Received
21st December 2017
, Accepted 27th January 2018
First published on 2nd February 2018
Abstract
The novel polytriphenylamines derivatives PTPA1-TPE and PTPA2-TPE with TPE as side groups and bis(tert-butyl)-TPE in the backbones have been successfully synthesized via Yamamoto-type coupling under microwave heating. The incorporation of TPE units in the backbones and side-chains endows the polymers with a distinct AIE effect and high fluorescence quantum yields in the solid state. Meanwhile, nitroaromatic explosive detection based on dispersed, electron-rich PTPA1-TPE and PTPA2-TPE nanoaggregates in THF/water was carried out. Both of them show high sensitivity towards the TNT analyte, with a maximum Stern–Volmer quenching constant Ksv of up to 8.3 × 104 M−1. In addition, the paper strip test demonstrates the promising practical application of the polymers as solid state sensors towards PL-based nitroaromatic explosive detection.
Introduction
According to the United Nations, there are still several hundred millions of landmines in more than 60 countries and regions around the world. The question of how to detect explosives effectively has become a global challenge. The explosives include aromatic compounds, such as 2,4,6-trinitrotoluene (TNT), 2,4-dinitrotoluene (DNT), picric acid (PA) and others. Among them, TNT is the most commonly available nitroaromatic explosive.1–3 TNT can not only cause many insecure conditions to society and humans, but it is also a kind of environmental pollutant due to its high toxicity. TNT can penetrate into the human body through the skin, or the respiratory and digestive tracts.4,5 Therefore, the realization of fast and effective detection schemes for the TNT explosive is of great importance, especially at crowded places, e.g. airports, train stations or docks, etc. There are many methods/instruments that have been developed for detecting TNT molecules, such as ones based on X-ray diffusion,6,7 surface acoustic waves,8 neutron analysis,9 gas chromatography,10,11 surface-enhanced Raman spectroscopy,12,13 ion mobility14,15 and electrochemical sensing,16,17etc. However, they are either low-sensitive, or expensive, or require large apparatus or complicated operation procedures, which make them not suitable for the practical in-field application at the above-mentioned occasions. Photoluminescence (PL)-based sensors18–20 towards TNT explosives, possessing high sensitivity, selectivity, stability, low-cost and portability, has attracted a lot of attention around the world. Among them, especially conjugated polymer-based PL sensors are widely investigated because of the occurrence of a molecular wire effect, leading to an amplified response signal by the polymers.21–24 This behavior is different from that of small molecules, which only show a linear response. So far, many kinds of conjugated polymers have been developed for TNT detection, with structural modification in the polymer backbone or in the side-chains. Based on the TNT sensing mechanism of fluorescence quenching that is mainly caused by photo-induced electron transfer from electron-rich conjugated polymers to electron-deficient TNT analytes, the electron-donating property of the conjugated polymer25–29 (e.g. polycarbazoles and polytriphenylamines) is essential for the high sensitivity detection of TNT. Moreover, constructing suitable transport paths (e.g. by adjusting the aggregate structures of the polymers) for diffusion and adsorption of TNT molecules is crucial for realizing fast and sensitive TNT sensing as well.24,30–33 Through a proper modification of the conjugated polymers one can enhance the interaction between polymer and TNT, thus leading to very sensitive fluorescence sensing towards TNT.
As we know, most conventional conjugated polymers display so-called aggregation-caused quenching (ACQ),34,35 which limits their applications in the aggregated state, such as in organic light emitting devices (OLEDs), organic solid lasers, chemo/biosensors and many others. Since the novel phenomenon of aggregation induced emission (AIE)33,36–39 was solidified by Tang and co-workers, AIE-active materials have been widely introduced to the above mentioned application fields, especially for the application in nitroaromatics detection, for their high PL quantum yields in the aggregated state. For example, Zheng's group reported several AIE-active TPE-based macrocycles, which demonstrated a high sensitivity and selectivity towards TNT sensing via different mechanisms, in water and in the solid state.40–42 Stang and coworkers developed a collection of multi-TPE Pt(II) metallacycles for sensing nitroaromatic analytes with high quenching constants of up to 2 × 106 M−1 for PA.43 Bhunia and Janiak et al. synthesized highly photoluminescent, triazine-based microporous frameworks (PCTF-8) containing TPE units for a sensitive detection of picric acid.44 Tang and coworkers reported a series of AIE-active, non-conjugated polymers containing TPE units, and applied them for the detection of nitroaromatic compounds.45–47 Despite the large number of known AIE-active small molecules for sensing applications, the examples of conjugated, AIE-active polymers for the detection of nitroaromatic explosives are still rare.
Here, we designed two novel AIE-active conjugated polymers, with AIE-active tetraphenylethylene (TPE) moieties both in the backbones and side-chains. Simultaneously, polytriphenylamines are incorporated into the polymer main-chains, to endow the electron-donating property of the obtained polymers, thus offering strong binding between the polymers and TNT analytes. Noteworthy the TPE structural unit which is incorporated into the polymer backbones contain two tert-butyl groups. There are three advantages of introducing bis(tert-butyl)-TPE main-chain units: (1) the presence of the tert-butyl substituents will improve the solubility of the polymers, to improve the solution processability; (2) the bis(tert-butyl)-TPE unit should be more effective in suppressing unwanted π–π stacking in the aggregated state due to its bulky structure; and (3) the bulky TPE side-chains together with the bis(tert-butyl)-TPE in the backbones may create micropores as transport channels for analyte molecules in the aggregated state, which will be beneficial for a high sensitivity detection of TNT molecules.
Experimental
Characterizations of the materials
NMR spectra were recorded on a Bruker AVANCE 400 or AVANCE III 600 NMR spectrometer. 1H and 13C NMR spectra were measured with tetramethylsilane (TMS) as internal standard. Gel permeation chromatography (GPC) measurements were carried out on a PSS/Agilent SECurity GPC system equipped with polystyrene gel columns using chloroform as eluent. APLI (Atmospheric Pressure Laser Ionization) measurements were carried out on Bruker Daltronik Bremen with micrOTOF. Elemental analyses were performed on a Vario EL II (CHNS) instrument. UV-visible absorption spectra were recorded on a Jasco V-670 spectrometer, and PL spectra on a Varian CARY Eclipse F2500. The PL quantum yields of polymer solid powder were measured with an integrating sphere.
Synthesis
All reagents were obtained from commercial suppliers and were used without further purification. All reactions were carried out under argon atmosphere by standard and Schlenk techniques. The solvents were used as commercial p.a. quality. Monomer 1 and 2 were synthesized according to the reported literature.22
4,4′-(2,2-Bis(4-(tert-butyl)phenyl)ethene-1,1-diyl)bis(bromobenzene) (3)
To a solution of bis[4-(tert-butyl)phenyl]methane (3.03 g, 10.82 mmol) in dry THF (20 mL) a 2.8 M solution of n-BuLi in hexane (3.86 mL, 10.82 mmol) was added at 0 °C under an argon atmosphere. The resulting orange-red solution was stirred for 1 h at that temperature. To this solution, 4,4′-dibromobenzophenone (3.07 g, 9.02 mmol) in THF (25 mL) was added. Next, the reaction mixture was allowed to warm up to room temperature overnight. The reaction was stopped by addition of saturated aqueous NH4Cl solution. The aqueous layer was extracted with chloroform, and the combined organic layers were washed with a saturated brine solution and dried over anhydrous MgSO4. The solvents were removed by evaporation and the resulting crude alcohol (containing excess diphenylmethane) was subjected to acid-catalyzed dehydration as follows: the crude product was dissolved in toluene (50 mL) containing p-toluenesulphonic acid (0.45 g, 2.34 mmol) in a 100 mL flask, and the mixture was refluxed at 110 °C overnight. The toluene layer was washed with aqueous NaHCO3 solution, dried over MgSO4 and the solvent removed to afford the crude tetraphenylethylene derivative. The product was purified by silica gel chromatography (eluent: dichloromethane/hexane = 1/9) to give desired compound as a white solid in 30% yield (1.63 g). 1H NMR (600 MHz, CDCl3) δ (ppm) 7.23 (d, J = 8.6 Hz, 4H), 7.15 (d, J = 8.5 Hz, 4H), 6.93 (d, J = 8.5 Hz, 4H), 6.88 (d, J = 8.6 Hz, 4H), 1.29 (s, 18H). 13C NMR (150 MHz, CDCl3) δ (ppm) 149.80, 142.70, 142.24, 140.03, 137.35, 133.01, 130.83, 130.80, 124.63, 120.35, 34.47, 31.28. MS (APLI): m/z calcd 602.10; found 602.08. Elemental anal. calcd: C, 67.78%; H, 5.69%. Found: C, 68.14%; H, 5.66%.
PTPA1-TPE
A solution of monomer 3 (200 mg, 0.33 mmol), monomer 1 (218 mg, 0.33 mmol), Ni(COD)2 (457 mg, 1.66 mmol), 2,2′-bipyridine (Bpy) (259 mg, 1.66 mmol) and 1,5-cyclooctadiene (COD) (180 mg, 1.66 mmol) in THF (5 mL) was reacted under microwave heating at 120 °C for 12 min. The reaction was quenched by adding water and the mixture extracted with chloroform. The collected organic phases were washed with aqueous 2 M HCl, aqueous NaHCO3 solution, saturated aqueous EDTA solution and brine, and finally dried over MgSO4. Afterwards, the solvent was removed under vacuum. The resulting solid powder was dissolved in a small amount of chloroform and precipitated into 500 mL of methanol to afford the target polymer as light-green solid. Subsequent Soxhlet extractions were carried out with methanol, acetone, ethyl acetate and chloroform, respectively. After reprecipitation of the chloroform-soluble fraction into methanol, the light-green polymer was obtained in 80% yield (248 mg). 1H NMR (400 MHz, C2D2Cl4, 60 °C) δ (ppm) 7.58–7.30 (m, 8H), 7.27–6.99 (m, 31H), 6.99–6.82 (m, 4H), 1.42–1.19 (m, 18H). Elem. anal. calcd for PTPA1-TPE: C, 91.97%; H, 6.54%; N, 1.49%. Found: C, 91.72%; H, 6.53%; N, 1.52%. Mn 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700, Mw 189000, and Mw/Mn 3.34 (GPC, PS calibration).
700, Mw 189000, and Mw/Mn 3.34 (GPC, PS calibration).
PTPA2-TPE
A solution of monomer 3 (200 mg, 0.33 mmol), monomer 2 (244 mg, 0.33 mmol), Ni(COD)2 (457 mg, 1.66 mmol), BPy (259 mg, 1.66 mmol) and COD (180 mg, 1.66 mmol) in THF (5 mL) was reacted under microwave heating at 120 °C for 12 min. The workup procedure was similar to that described for the preparation of PTPA1-TPE. The light-green polymer was obtained in 87% yield (293 mg). 1H NMR (400 MHz, C2D2Cl4, 60 °C) δ (ppm) 7.62–7.44 (m, 6H), 7.44–7.28 (m, 6H), 7.28–6.89 (m, 35H), 1.28 (s, 18H). Elem. anal. calcd for PTPA2-TPE: C, 92.18%; H, 6.45%; N, 1.38%. Found: C, 91.89%; H, 6.39%; N, 1.39%. Mn 32700, Mw 165000, and Mw/Mn 5.03 (GPC, PS calibration).
Results and discussion
Synthesis and characterization
The synthetic routes to the monomers and polymers PTPA1-TPE and PTPA2-TPE are depicted in Scheme 1. The triphenylethylene- and TPE-substituted dibromotriphenylamine monomers 1 and 2 were generated in a Suzuki-type coupling of tribromotriphenylamine with triphenylethylene- or TPE-boronic ester. The dibromo-TPE monomer 3 was obtained by treating 4-bromobenzophenone with bis(4-(tert-butyl)phenyl)methyl lithium followed by an acid-catalyzed dehydration. The random copolymers PTPA1-TPE and PTPA2-TPE were synthesized via Yamamoto-type couplings of 1:1 mixtures of monomer 3 and 1 or 2, respectively, with bis(1,5-cyclooctadiene)nickel(0) (Ni(COD)2) as coupling reagent in a mixture of THF, 1,5-cyclooctadiene (COD) and 2,2′-bipyridine (Bpy) under microwave heating. Applying the microwave method we could obtain the targeted polymers in a short reaction time (12 min). The chemical structure of the monomers 1–3 was confirmed by NMR spectroscopy, mass spectrometry and elemental analysis. The chemical structure of the obtained polymers was finally confirmed by combining NMR spectroscopy, elemental analysis, GPC and optical spectroscopy. The exact composition ratio x:y in the resulting random copolymers was estimated from the carbon and nitrogen contents of the elemental analyses: x:y is calculated to be 0.95 or 0.97 for PTPA1-TPE or PTPA2-TPE, respectively. Therefore, the incorporation ratios of two co-monomers into PTPA1-TPE and PTPA2-TPE are in good accordance with the feed ratios. The obtained high molecular weight copolymers (PTPA1-TPE: Mn = 56700; PTPA2-TPE: Mn = 32700) show good solubility in common solvents, such as chloroform, dichloromethane, THF, DMF or toluene.
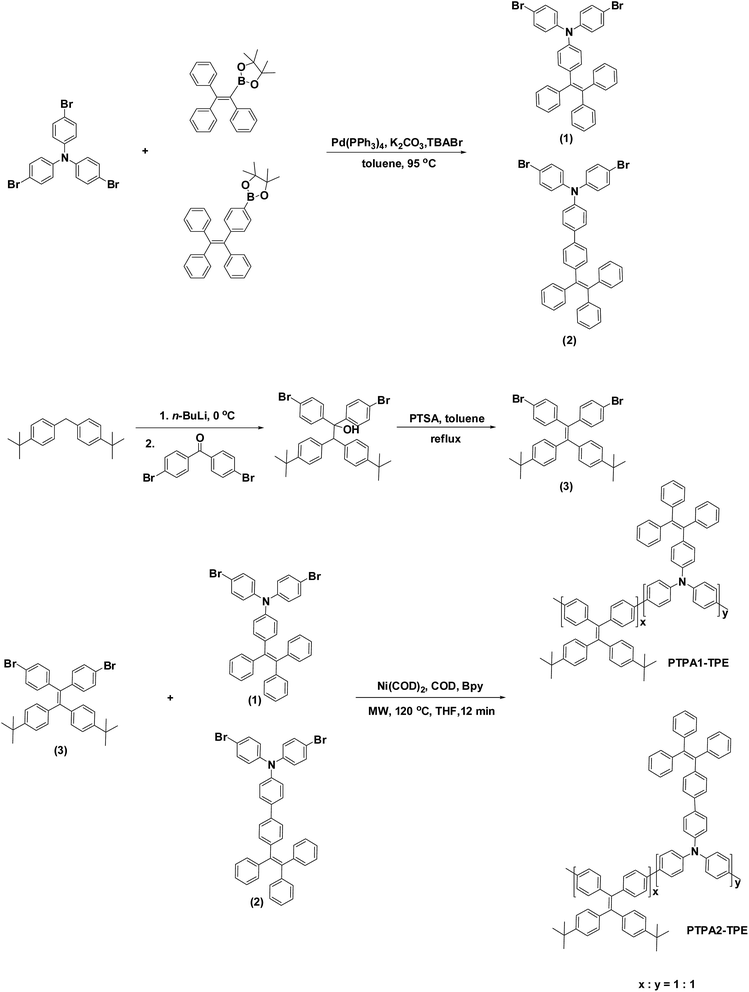 |
| | Scheme 1 Synthesis scheme towards monomers and copolymers PTPA1-TPE and PTPA2-TPE. | |
Photophysical properties
Fig. 1 shows the absorption and PL spectra of the two PTPA-TPE copolymers in dilute THF solution and as thin films. The absorption spectra of PTPA1-TPE and PTPA2-TPE are quite similar, with absorption maxima at ca. 372 nm both in solution and as thin films. The almost identical absorption of the polymers is attributed to their similar chemical structures, with only one additional phenyl ring for PTPA2-TPE. The PL spectra of PTPA1-TPE and PTPA2-TPE reflect a green-emission, with PL maxima of the films centered at 516 nm and 512 nm, respectively. The slight blue shift from PTPA1-TPE (516 nm) to PTPA2-TPE (512 nm) is attributed to the presence of biphenyl linkers in PTPA2-TPE, which decrease the conjugative interaction between the polymer backbone and TPE side groups. The optical data of the polymers are summarized in Table 1. The PL quantum yields (PLQYs) of PTPA1-TPE and PTPA2-TPE in dilute THF solution have been determined to be 0.4% and 1.4%, respectively, using quinine sulfate as standard. Weakly emitting in dilute solution, both polymers, however, show a much stronger PL emission in the solid state. The PLQYs of the solid powders are 20% and 37% for PTPA1-TPE and PTPA2-TPE, respectively, corresponding to 50 and 26 times PL increase, respectively, if compared to their PLQYs in dilute solution. These results demonstrate the occurrence of distinct AIE phenomena. The good AIE effects are attributed to the introduction of TPE units both in the backbones and side-chains of the copolymers.
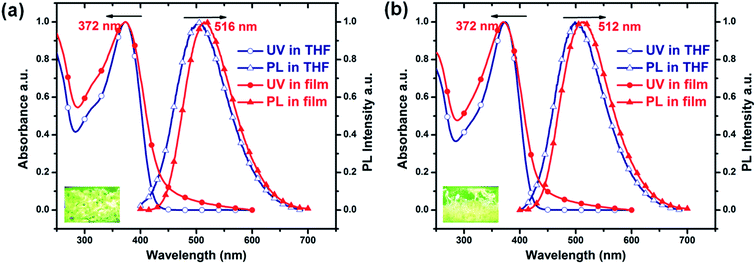 |
| | Fig. 1 Normalized absorption and PL spectra of (a) PTPA1-TPE and (b) PTPA2-TPE in dilute THF solution (10−5 M) and as spin-coated films (measured at room temperature). The insets in (a) and (b) show fluorescent images of PTPA1-TPE and PTPA2-TPE powders under UV irradiation, respectively. | |
Table 1 Optical data of PTPA1-TPE and PTPA2-TPE
| Polymers |
λ
UV/nm in THF |
λ
PL/nm in THF |
λ
UV/nm in film |
λ
PL/nm in film |
η
PL (%) in THF |
η
PL (%) in powder |
| PTPA1-TPE |
372 |
507 |
372 |
516 |
0.4 |
20 |
| PTPA2-TPE |
373 |
502 |
372 |
512 |
1.4 |
37 |
To further investigate the AIE phenomenon of the copolymers, a series of PL spectra were recorded in the solvent mixture of THF/water (Fig. 2). Since water is a non-solvent for the copolymers, aggregation is induced starting from a certain water content. The PL intensity of all copolymers increases with increasing water fraction, again confirming the occurrence of AIE effects. The maximum increase of PL intensity is 42 and 30 times; in 9:1 (v/v) water/THF for PTPA1-TPE or 8:2 (v/v) water/THF for PTPA2-TPE, respectively, if compared to non-aggregated THF solutions. The introduction of bis(tert-butyl)-TPE units into the backbones of the copolymers leads to well-resolved AIE effects that are coupled to an improved solubility of the copolymers, thus simplifying their solution processing.
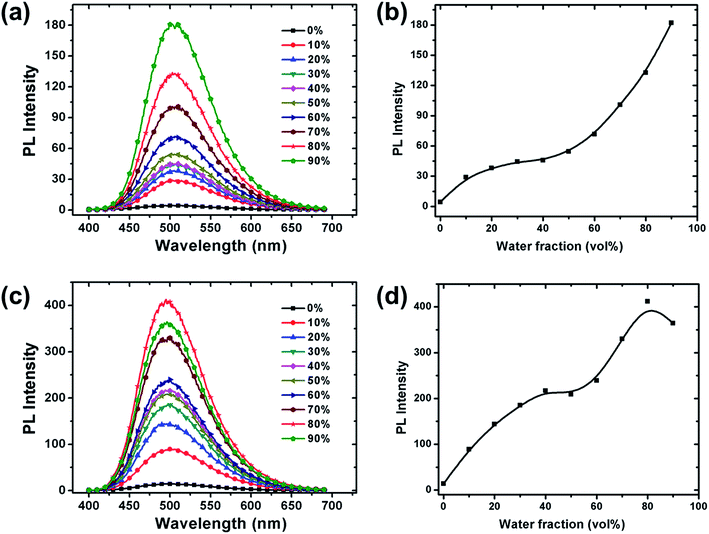 |
| | Fig. 2 PL spectra of (a) PTPA1-TPE and (b) PTPA2-TPE in THF/water mixtures with different water content; PL intensity with increasing water fraction for (c) PTPA1-TPE and (d) PTPA2-TPE, respectively (polymer concentration: 10−5 M). | |
Explosive detection
To explore the potential of the obtained polymers for explosive detection, we chose TNT as prototypical nitroaromatic analyte, since it is a very frequently used component of explosives. The polymers PTPA1-TPE and PTPA2-TPE both possess an electron-rich backbone, which should be beneficial for the interaction with electron-deficient TNT. Moreover, both polymers show a distinct AIE effect coupled with high PLQYs in the aggregated state. Both is beneficial for the high sensitivity PL detection of TNT; in combination with the loosely packed structure of the polymer aggregates, due to the presence of bulky TPE moieties in the side-chains and bulky bis(tert-butyl) substituted TPE units in the backbones. Based on the above statement, the polymers PTPA1-TPE and PTPA2-TPE are expected to be sensitive towards TNT.
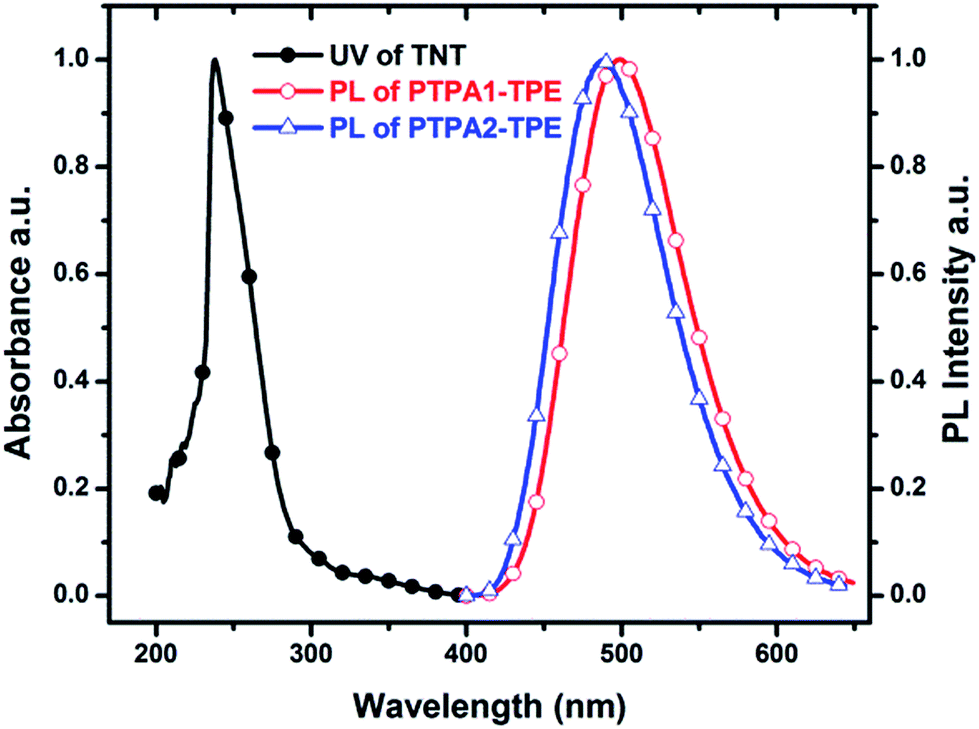
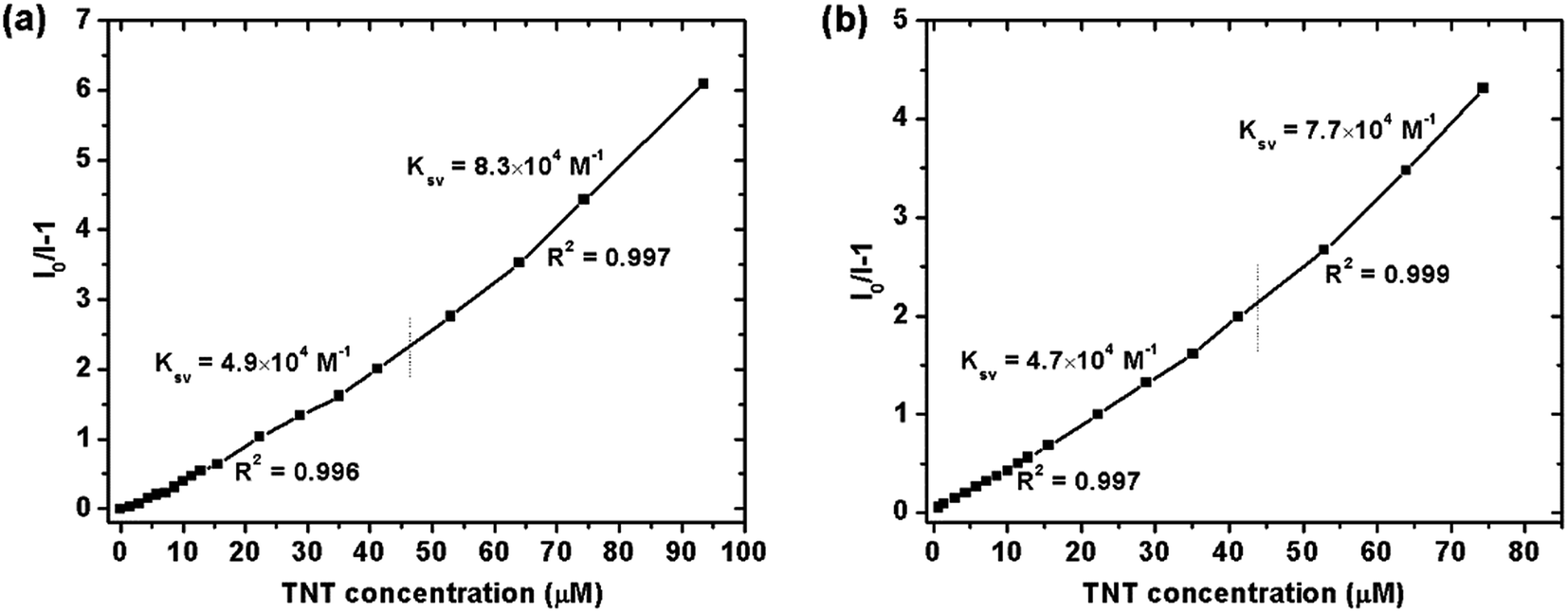
For the detection of TNT, dispersed nanoaggregates of the polymers are prepared in 9:1 (v/v) water/THF mixtures, with concentration of 10−5 M. A series of PL spectra of the polymers were recorded during addition of TNT molecules, as shown in Fig. 3. The PL intensities of PTPA1-TPE and PTPA2-TPE decrease gradually with increasing TNT concentration, thus demonstrating the sensitive interaction of the polymers with the analyte TNT. With a TNT concentration of 93 μM, the fluorescence of both polymers can be quenched by more than 85%. The limit of detection was calculated to be 0.8 μM or 0.5 μM for PTPA1-TPE or PTPA2-TPE, respectively.48 During the PL quenching process, there is no significant change of the PL spectra, thus meaning, that no different emissive species are formed. The quenching mechanism for our polymers towards TNT could be an electron transfer between the exited state of PTPA1-TPE or PTPA2-TPE and TNT, since there is no spectral overlap between the absorption spectrum of TNT and PL spectra of the polymers, a necessary prerequisite for Förster-type energy transfer (Fig. 4). The Stern–Volmer plots of relative PL intensities (I0/I − 1) of PTPA1-TPE and PTPA2-TPE vs. TNT concentration are shown in Fig. 5. Different from small molecule-based PL quenchers as sensitive materials, the Stern–Volmer plots here display upward-bend curves, which illustrates an amplified quenching effect at high TNT concentration. This amplified quenching effect may be attributed to the loosely packed, three-dimensional morphology of the polymer nanoaggregates in 9:1 (v/v) water/THF, thus leading to the formation of transport channels for TNT diffusion. The Stern–Volmer curves have been divided into two linear subparts, for TNT concentrations of 0–41 μM and for TNT concentrations of >41 μM, both with correlation coefficients R2 of >0.996 of the individual subparts. Both PTPA1-TPE and PTPA2-TPE indicate highly sensitive quenching towards TNT, with maximum Ksv values calculated from the Stern–Volmer curves of 8.3 × 104 M−1 for PTPA1-TPE and 7.7 × 104 M−1 for PTPA2-TPE, respectively. These values are among the best in the published literature.23,29,49,50
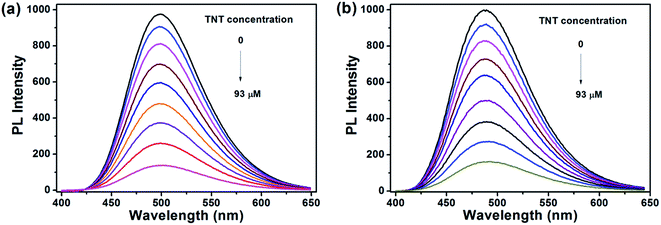 |
| | Fig. 3 PL spectra of (a) PTPA1-TPE and (b) PTPA2-TPE in 9:1 (v/v) water/THF solution containing different amounts of TNT. Polymer concentration: 10−5 M. | |
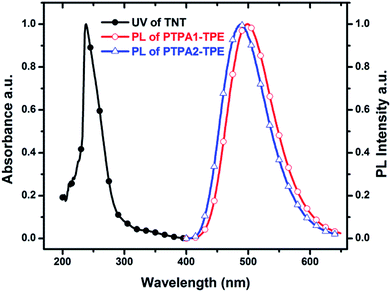 |
| | Fig. 4 Normalized absorption spectrum of TNT in THF solution and PL spectra of PTPA1-TPE and PTPA2-TPE nanoaggregates in 9:1 (v/v) water/THF dispersion. | |
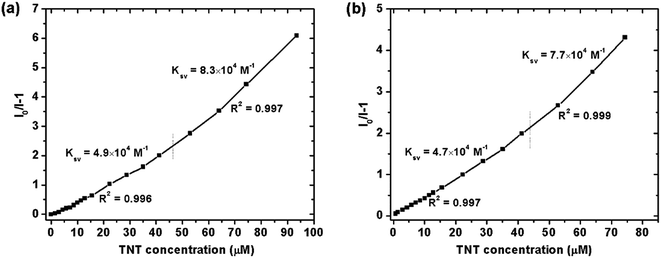 |
| | Fig. 5 Stern–Volmer plots of the relative PL intensities of (a) PTPA1-TPE and (b) PTPA2-TPE (I0/I − 1) vs. TNT concentration (I: PL intensity, I0: PL intensity at zero TNT concentration). | |
The response of dispersed PTPA1-TPE nanoaggregates towards other aromatic analytes have been also tested, as shown in Fig. 6. For the nitroaromatic compounds 2,4-dinitrotoluene (DNT) and nitrobenzene (NB), the PL of PTPA1-TPE is also quenched to some extent, with similar Ksv quenching constants that are, however, lower than the Ksv towards TNT (Fig. 6a) due to their weaker electron-accepting character. Moreover, toluene was also tested as analyte. However, the PL of a PTPA1-TPE dispersion remains almost unchanged upon toluene addition (Fig. 6b) confirming that the PL quenching of the polymer dispersion is restricted to electron-accepting analytes.
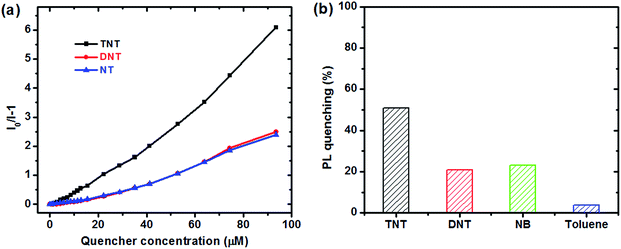 |
| | Fig. 6 (a) Stern–Volmer plots of the relative PL intensities of PTPA1-TPE (I0/I − 1) vs. concentration of the analytes TNT, DNT and NB (I: PL intensity, I0: PL intensity at zero analyte concentration). (b) PL quenching of PTPA1-TPE for analyte concentrations of 22 μM. | |
For the practical detection of nitroaromatic explosives, the availability of solid state sensors will be very important. Towards this aid, paper strips with deposits of both polymers were prepared by drop-coating polymer solutions (10−4 M in THF) onto the filter papers, followed by drying them under air stream. The original PL emission of the paper strips is bright green, as shown in Fig. 7. When TNT solutions with concentrations of 0.001, 0.005, 0.01, 0.05, 0.1 and 0.2 mg mL−1, respectively, are dropped onto polymer-impregnated paper strips, the PL from both TPA1-TPE or TPA2-TPE is quenched progressively. PL quenching for both types of paper strips towards TNT is already found for a concentration of 0.001 mg mL−1, representing micromolar TNT concentrations. In addition, our paper strip tests were extended to the detection of TNT vapor. Hereby, the strips were placed above solid TNT for 5 min at room temperature. As consequence, the PL of the strips is distinctly quenched, as shown in Fig. 7. The results demonstrate a certain potential of the polymers TPA1-TPE and TPA2-TPE for the fabrication of solid state sensor devices for sufficiently sensitive TNT detection.
 |
| | Fig. 7 Paper test strips impregnated with PTPA1-TPE or PTPA2-TPE before and after dropping TNT solutions in THF onto them (concentration: 0.001, 0.005, 0.01, 0.05, 0.1 and 0.2 mg mL−1, respectively), and after storage above TNT vapor for 5 min. | |
Conclusions
Two polytriphenylamines derivatives with TPE side groups and bis(tert-butyl)-TPE units in the backbones have been successfully synthesized via Yamamoto-type coupling under microwave heating. TPE modification of polymer backbones and side-chains of PTPA1-TPE and PTPA2-TPE facilitate a good solubility of the polymers coupled with a distinct AIE effect and high solid state PLQYs. For the application in nitroaromatic explosive detection, PL sensing based on PTPA1-TPE and PTPA2-TPE nanoaggregates in 9:1 (v/v) water/THF was tested. Both nanoaggregate dispersions exhibit a sensitive and amplified PL quenching towards TNT, with a maximum Stern–Volmer quenching constant Ksv of up to 8.3 × 104 M−1. In addition, paper strips tests also demonstrated promising potential for low-cost practical application as solid state sensors for nitroaromatic explosive detection.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to thank the financial support from Scientific Research Projects of Jilin Provincial Department of Education, China.
Notes and references
- Y. Salinas, R. Martinez-Manez, M. D. Marcos, F. Sancenon, A. M. Castero, M. Parra and S. Gil, Chem. Soc. Rev., 2012, 41, 1261 RSC.
- H. Ma, F. Li, Z. Zhang and M. Zhang, Sens. Actuators, B, 2017, 244, 1080 CrossRef CAS.
- A. Kumar and P. S. Chae, Sens. Actuators, B, 2017, 240, 1 CrossRef CAS.
- X. Sun, Y. Wang and Y. Lei, Chem. Soc. Rev., 2015, 44, 8019 RSC.
- R. S. Aparna, J. S. A. Devi, P. Sachidanandan and S. George, Sens. Actuators, B, 2018, 254, 811 CrossRef CAS.
- A. M. Soldate and R. M. Noyes, Anal. Chem., 1947, 19, 442 CrossRef CAS.
- R. Luggar, M. Farquharson, J. Horrocks and R. Lacey, X-Ray Spectrom., 1998, 27, 87 CrossRef CAS.
- E. J. House, R. A. McGill, V. K. Nguyen, R. Chung and D. W. Weir, Proc. SPIE, 2000, 4038, 504 CrossRef.
- G. Vourvopoulos and P. C. Womble, Talanta, 2001, 54, 459 CrossRef CAS.
- M. Elsner, M. A. Jochmann, T. B. Hofstetter, D. Hunkeler, A. Bernstein, T. C. Schmidt and A. Schimmelmann, Anal. Bioanal. Chem., 2012, 403, 2471 CrossRef CAS.
- M. E. Walsh, Talanta, 2001, 54, 427 CrossRef CAS.
- A. K. M. Jamil, A. Sivanesan, E. L. Izake, G. A. Ayoko and P. M. Fredericks, Sens. Actuators, B, 2015, 221, 273 CrossRef CAS.
- J. M. Sylvia, J. A. Janni, J. D. Klein and K. M. Spencer, Anal. Chem., 2000, 72, 5834 CrossRef CAS.
- M. Najarro, M. E. D. Morris, M. E. Staymates, R. Fletcher and G. Gillen, Analyst, 2012, 137, 2614 RSC.
- D. S. Moore, Rev. Sci. Instrum., 2004, 75, 2499 CrossRef CAS.
- M. Krausa and K. J. Schorb, J. Electroanal. Chem., 1999, 461, 10 CrossRef CAS.
- E. S. Forzani, D. Lu, M. J. Leright, A. D. Aguilar, F. Tsow, R. A. Iglesias, Q. Zhang, J. Lu, J. Li and N. Tao, J. Am. Chem. Soc., 2009, 131, 1390 CrossRef CAS.
- N. Venkatramaiah, S. Kumar and S. Patil, Chem. Commun., 2012, 48, 5007 RSC.
- K. R. Ghosh, S. K. Saha and Z. Y. Wang, Polym. Chem., 2014, 5, 5638 RSC.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537 CrossRef CAS.
- Q. Zhou and T. M. Swager, J. Am. Chem. Soc., 1995, 117, 12593 CrossRef CAS.
- W. Dong, Y. Pan, M. Fritsch and U. Scherf, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1753 CrossRef CAS.
- X.-S. Ma, D.-H. Wang, Y.-Z. Cui, F.-R. Tao, Y.-T. Wang and T.-D. Li, Sens. Actuators, B, 2017, 251, 851 CrossRef CAS.
- H. Nie, Y. Lv, L. Yao, Y. Pan, Y. Zhao, P. Li, G. Sun, Y. Ma and M. Zhang, J. Hazard. Mater., 2014, 264, 474 CrossRef CAS.
- W. Dong, J. Pina, Y. Pan, E. Preis, J. S. Seixas de Melo and U. Scherf, Polymer, 2015, 76, 173 CrossRef CAS.
- P. E. Shaw and P. L. Burn, Phys. Chem. Chem. Phys., 2017, 19, 29714 RSC.
- A. Räupke, A. Palma-Cando, E. Shkura, P. Teckhausen, A. Polywka, P. Görrn, U. Scherf and T. Riedl, Sci. Rep., 2016, 6, 29118 CrossRef.
- B. Xu, X. Wu, H. Li, H. Tong and L. Wang, Macromolecules, 2011, 44, 5089 CrossRef CAS.
- T. L. Duniho, B. J. Laughlin, A. A. Buelt, W. F. Baker, C. A. Conrad and R. C. Smith, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1487 CrossRef CAS.
- Y. Wu, B. He, C. Quan, C. Zheng, H. Deng, R. Hu, Z. Zhao, F. Huang, A. Qin and B. Z. Tang, Macromol. Rapid Commun., 2017, 38, 1700070 CrossRef.
- Z. Zhao, T. Jiang, Y. Guo, L. Ding, B. He, Z. Chang, J. W. Y. Lam, J. Liu, C. Y. K. Chan, P. Lu, L. Xu, H. Qiu and B. Z. Tang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2265 CrossRef CAS.
- H. Zhou, X. Wang, T. T. Lin, J. Song, B. Z. Tang and J. Xu, Polym. Chem., 2016, 7, 6309 RSC.
- W. Dong, T. Fei, A. Palma-Cando and U. Scherf, Polym. Chem., 2014, 5, 4048 RSC.
- R. Jakubiak, C. J. Collison, W. C. Wan and L. Rothberg, J. Phys. Chem. A, 1999, 103, 2394 CrossRef CAS.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS.
- J. Shi, Y. Wu, B. Tong, J. Zhi and Y. Dong, Sens. Actuators, B, 2014, 197, 334 CrossRef CAS.
- Y. Zhang, J. Xia, X. Feng, B. Tong, J. Shi, J. Zhi, Y. Dong and Y. Wei, Sens. Actuators, B, 2012, 161, 587 CrossRef CAS.
- R. Hu, Y. Kang and B. Z. Tang, Polym. J., 2016, 48, 359 CrossRef CAS.
- H.-T. Feng and Y.-S. Zheng, Chem.–Eur. J., 2014, 20, 195 CrossRef CAS PubMed.
- H.-T. Feng, J.-H. Wang and Y.-S. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 20067 CrossRef CAS.
- J.-H. Wang, H.-T. Feng and Y.-S. Zheng, Chem. Commun., 2014, 50, 11407 RSC.
- X. Yan, H. Wang, C. E. Hauke, T. R. Cook, M. Wang, M. L. Saha, Z. Zhou, M. Zhang, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2015, 137, 15276 CrossRef CAS.
- A. Bhunia, D. Esquivel, S. Dey, R. Fernández-Terán, Y. Goto, S. Inagaki, P. Van Der Voort and C. Janiak, J. Mater. Chem. A, 2016, 4, 13450 RSC.
- R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494 RSC.
- Y. Zhang, G. Chen, Y. Lin, L. Zhao, W. Z. Yuan, P. Lu, C. K. W. Jim, Y. Zhang and B. Z. Tang, Polym. Chem., 2015, 6, 97 RSC.
- H. Li, H. Wu, E. Zhao, J. Li, J. Z. Sun, A. Qin and B. Z. Tang, Macromolecules, 2013, 46, 3907 CrossRef CAS.
- M. Shortreed, R. Kopelman, M. Kuhn and B. Hoyland, Anal. Chem., 1996, 68, 1414 CrossRef CAS.
- S. Mondal, A. Jana, R. Bera and N. Das, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3725 CrossRef CAS.
- M. F. Almassio, M. J. Romagnoli, P. G. Del Rosso, A. B. Schvval and R. O. Garay, Polymer, 2017, 113, 167 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Teng
Fei
*a,
Teng
Fei