
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient photocatalysis with graphene oxide/Ag/Ag2S–TiO2 nanocomposites under visible light irradiation†
Shuang Shuang a,
Ruitao Lvb,
Xiaoyang Cuia,
Zheng Xieac,
Jian Zhengd and
Zhengjun Zhang*b
a,
Ruitao Lvb,
Xiaoyang Cuia,
Zheng Xieac,
Jian Zhengd and
Zhengjun Zhang*b
aState Key Laboratory of New Ceramics and Fine Processing, School of Materials Science and Engineering, Tsinghua University, Beijing 100084, China
bKey Laboratory of Advanced Materials (MOE), School of Materials Science and Engineering, Tsinghua University, Beijing 100084, China. E-mail: zjzhang@tsinghua.edu.cn
cHigh-Tech Institute of Xi'an, Xi'an 710025, China
dDepartment of Chemistry, University of Oslo, Sem Sælands Vei 26, 0371 Oslo, Norway
First published on 5th February 2018
Abstract
Lack of visible light response and low quantum yield hinder the practical application of TiO2 as a high-performance photocatalyst. Herein, we present a rational design of TiO2 nanorod arrays (NRAs) decorated with Ag/Ag2S nanoparticles (NPs) synthesized through successive ion layer adsorption and reaction (SILAR) and covered by graphene oxide (GO) at room temperature. Ag/Ag2S NPs with uniform sizes are well-dispersed on the TiO2 nanorods (NRs) as evidenced by electron microscopic analyses. The photocatalyst GO/Ag/Ag2S decorated TiO2 NRAs shows much higher visible light absorption response, which leads to remarkably enhanced photocatalytic activities on both dye degradation and photoelectrochemical (PEC) performance. Its photocatalytic reaction efficiency is 600% higher than that of pure TiO2 sample under visible light. This remarkable enhancement can be attributed to a synergy of electron-sink function and surface plasmon resonance (SPR) of Ag NPs, band matching of Ag2S NPs, and rapid charge carrier transport by GO, which significantly improves charge separation of the photoexcited TiO2. The photocurrent density of GO/Ag/Ag2S–TiO2 NRAs reached to maximum (i.e. 6.77 mA cm−2 vs. 0 V). Our study proves that the rational design of composite nanostructures enhances the photocatalytic activity under visible light, and efficiently utilizes the complete solar spectrum for pollutant degradation.
Introduction
Heterogeneous photocatalysis of pollutants over semiconductor materials has recently emerged as an efficient method for purifying water and air. Among different semiconductors, TiO2, usually as n-type semiconductor, is the most widely used photocatalyst due to its excellent photocatalytic activity, non-toxicity and good stability.1,2 However, the wide band gap (3.2 eV) of TiO2 seriously limits its utilization under visible light which occupies a large portion of the solar spectrum. Till now, different methods such as metal decoration,3,4 metallic ion doping,5 non-metal doping,6,7 and dye sensitization8,9 have been proposed to modify and narrow its band gap, and enhance its photocatalytic activity under visible light radiation.10–12Decorating TiO2 with metal, as one of the promising methods to develop highly efficient visible light photocatalysts, is becoming attractive. The deposition of metal on TiO2, as one of the ways to combine metal with TiO2, can greatly improve its photo-efficiency through the Schottky barrier conduction band (CB) electron trapping and consequent longer electron–hole pair lifetime.3,13 Hu et al.14 reported a highly efficient Pt-decorated TiO2 which demonstrates enhanced photocatalytic activity for NOx oxidation under both UV and visible light irradiation. On the other hand, some noble metals such as Ag and Au, exhibit strong UV-vis absorption due to their plasmon resonance, produced by the collective oscillations of surface electrons.15
In other ways, coupling TiO2 with different semiconductors is one of common ways to utilize the merit of various semiconductors and enhance the performance of photocatalysts.16 Some typical semiconductors like Ag2S,17 GO,18 WOx,19 AgVOx20 and et al. Ag2S as a typical n-type semiconductor, has low energy bandgap (about 1.0 eV) which enables it to absorb visible and near infrared (NIR) light without the necessity of further doping process. Moreover, quantum dots (QDs) of Ag2S has attracted increasing research interest due to their low toxicity, narrow bandgap, and high absorption coefficient.21 So, it has been demonstrated that Ag2S QDs could perform as a potential excellent visible and NIR photocatalyst.22
Graphene oxide (GO) having sufficient reactive oxygen functional groups, is a good candidate for supporting metal or metal oxide particles. The existence of p-conjugation structure and oxygen groups increases the photosensitivity of GO under visible light irradiation, and make it hydrophilic with good electronic performance. The electron energy gap of GO can be adjusted from 3.5 eV to 2.5 eV,23,24 depending directly on the oxidization degree of the graphene and the species of the oxygen groups. However, solo GO exhibits weak photocatalytical activity. Combining with other semiconductors could improve their property and prepare excellent photocatalysts. The pioneering work of Zhang et al.25 on chemically bonded TiO2 (P25)–graphene composite photocatalyst was firstly reported. They discovered high photocatalytical performance of the nanocomposite containing GO and TiO2 (P25). Afterwards, there are many researches on the preparation of graphene–TiO2 composite26–28 and its wide application such as photocatalytic bactericide,29 hydrogen evolution,30 or dye-sensitized solar cell.31 Such kind of composites owns at least two advantages: (1) controllable reduction of the GO nanosheets incorporated in the composition by using UV irradiation and (2) enhanced photocatalytic activity of TiO2 thin films for higher efficiency of solar light irradiation. However, all the previous report about photocatalysts were usually powder and amorphous, which was difficult to handle and restricted its usage in practical applications.
In this work, we report a novel synthesis of TiO2 NRAs on Si, quartz and F-doped SnO2 (FTO) to form self-standing structures. The photocatalyst is much easier to recycle and extends its application. Besides, we tried to introduce Ag, Ag2S and GO together according to the effects described in above description and prepare a unique photocatalyst with high activity. Based on our recent work on GO synthesis,32 a rational design of GO/Ag/Ag2S–TiO2 nanocomposite is proposed for highly efficient photocatalysis under visible light irradiation. In this regard, the effect of different combination among Ag NPs, Ag2S NPs, GO and TiO2 NRAs on degradation efficiency and photoelectrocatalytic property are discussed briefly.
Experimental methods
Synthesis of TiO2 NRAs
The inclined TiO2 NRAs were deposited by glancing angle deposition technique (GLAD) technique on different substrates and annealed as previously reported.33 Before the deposition, the chamber was evacuated to a vacuum level above 1 × 10−8 Torr. During deposition, the vapor flux incident angle was set to ∼86° off the surface normal to the substrates. And the deposition rate (∼0.75 nm s−1) and height of the NRs were monitored by a quartz crystal microbalance. Ti films were oxidized in a tube furnace in order to obtain TiO2 NRAs. The Ti films were heated up to 400 °C for 2 h at a ramp of 5 °C min−1 under oxygen environment to obtain crystalline nanostructures for high photocatalytic activity.Ag2S NPs deposition on TiO2 NRAs
Ag2S NPs were deposited on TiO2 NRs through successive ion layer adsorption and reaction (SILAR) method with slight modification as described in our previous work.15 Firstly, TiO2 NRAs were immersed in 0.1 mg mL−1 AgNO3 solution for 30 s, followed by rinsing with DI water and then immersed in Na2S solution (12 mg mL−1) for 30 s. After this, they were rinsed in DI water for several times. This SILAR process was repeated for several cycles until the desired quantity of decorated nanocrystallites was achieved.Ag nanoparticles deposition on TiO2 NRAs
Ag NPs were deposited using the same method. The NRs were immersed in 0.1 mg mL−1 AgNO3 solution for 60 s, followed by rinsing with DI water and then immersed in NaBH4 solution (1.0 mg mL−1) for another 60 s, after which the resultant was rinsed with DI water for several times. This SILAR process was repeated for several cycles until the desired quantity of metallic nanocrystallites was achieved.Ag/Ag2S NPs deposition on TiO2 NRAs
The TiO2 NRAs were alternately coated with Ag and Ag2S NPs respectively for specific times. To determine the optimal loading amounts of Ag and Ag2S NPs decoration, the orthogonal experimental design was used, and results were shown in Table S1† (5 μM MO under visible light).Combination with GO
GO was prepared via modified Hummers method. Typically, 5 g natural graphite flakes (Sigma Aldrich) and 3.0 g NaNO3 was added to a 1000 mL beaker containing 150 mL concentrated sulphuric acid in an ice-bath and stirred for 1 h. Then 20 g KMnO4 was gradually added while stirring, keeping the temperature below 30 °C. The mixture was stirred at room temperature for 24 h. Next, the beaker was placed in an ice bath and 300 mL DI water slowly added to the beaker, keeping the reaction temperature below 98 °C. After that, 30 mL H2O2 (30%) was added to the end reaction. Once H2O2 was added, the colour of the suspension turned bright yellow. The suspension was stirred for 30 min and then centrifuged at 8000 rpm to remove large flakes, then washed with 10% HCl solution, followed by washing with DI water and a three-week dialysis. Then Ag/Ag2S–TiO2 NRs with substrates were immersed in this solution for few seconds. The samples were dried in a muffle furnace at 50 °C for 2 hours to obtain the GO/Ag/Ag2S–TiO2 NRAs photocatalyst. GO–TiO2 NRAs, GO/Ag–TiO2 NRAs, GO/Ag2S–TiO2 NRAs were prepared in the similar way. The typical procedure were illustrated in Fig. 1(a).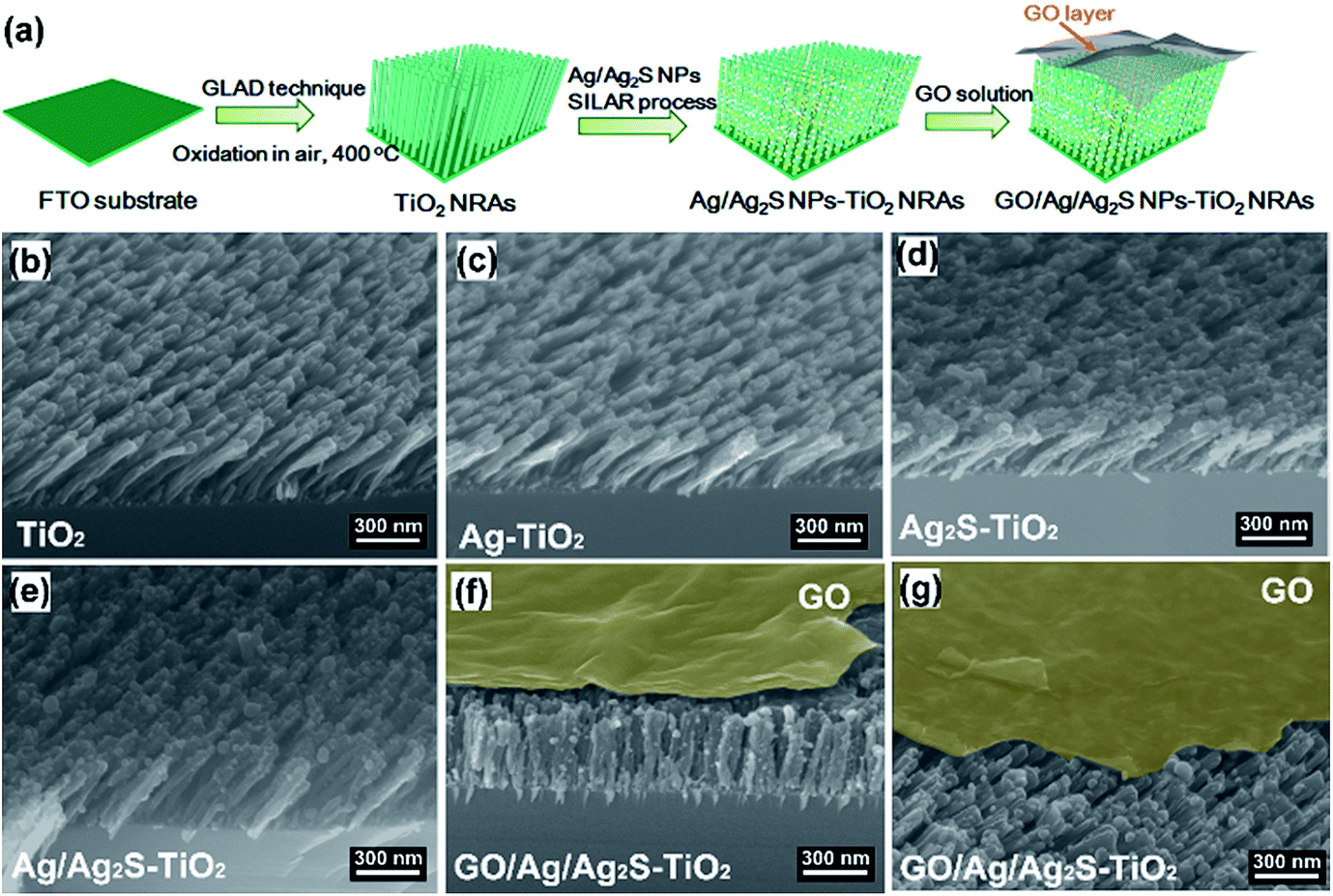 | ||
| Fig. 1 (a) Schematic illustration of the preparation of GO/Ag/Ag2S–TiO2 nanorod arrays (NRAs); scanning electron microscope (SEM) images of the different samples: (b) TiO2 NRAs; (c) Ag–TiO2 NRAs; (d) Ag2S–TiO2 NRAs; (e) Ag/Ag2S–TiO2 NRAs; (f) GO/Ag/Ag2S–TiO2 NRAs; (g) cross-section of sample GO/Ag/Ag2S–TiO2 NRAs. | ||
Materials characterization
The morphology and nanostructure of the fabricated samples were examined by field-emission scanning electron microscope (FE-SEM, JEOL-7001F), high-resolution transmission electron microscope (HRTEM, JEOL-2011) and Raman spectroscopy (LABRAM HR800, excitation wavelength of 633 nm), respectively. A Rigaku 2500 X-ray diffractometer was used to investigate the crystallographic characteristics of samples. The optical properties of samples were examined by a UV-vis spectrometer (Perkin Elmer Lambda 35) in a wavelength range from 400 nm to 900 nm at room temperature. The photoluminescence (PL) spectra were recorded by using Raman spectrometer (LabRAM ARMIS) with a 325 nm excitation. X-ray photoemission spectra were recorded by using ESCALAB 250Xi (Thermo Fisher). Monochromatized Al Kα was employed as anode. Survey spectra of all the samples were recorded from 0 eV to 1350 eV (binding energy) with each step at 1.0 eV, while the spectra for every specific element were collected with 0.05 eV/step. Mass Spectrometer (MS) analysis was performed with equipment of Thermo Scientific Q Exactive mass spectrometer.Photocatalytic degradation of crystal violet
The photocatalytic activity of different samples was evaluated by the photodegradation of crystal violet (CV) under visible light at ambient temperature. The sample on quartz substrate (15 mm × 15 mm) was placed in a quartz cell containing 5 mL of CV (5 μM) solution. Prior to light irradiation, the photocatalyst was immersed in a CV solution in dark for 30 min to reach an adsorption/desorption equilibrium, and Xe lamp with an ultraviolet filter was turned on for different time spans. After that, the concentration of CV was monitored using UV-vis spectroscopy at 584 nm. The degradation efficiency was calculated by (1 − C/Co) × 100%.Electrochemical measurements
The steady sate current density and electrochemical impedance spectroscopy (EIS) measurements were carried out in a three-electrode-cell controlled by an electrochemistry workstation (CHI 660D, Chenhua instrument). The nanostructured films were used as a working electrode with an exposed area of 1.5 cm2 in 0.1 M KOH solution (pH = 13.0). An Ag/AgCl electrode (saturated KCl) and Pt sheet were used as the reference and counter electrodes, respectively. In this paper, all the reported potentials refer to the Ag/AgCl electrode. Before the electrochemical measurements, cyclic voltammetry tests were scanned for several cycles to confirm the stability of the samples. EIS spectra were recorded from 0.1 Hz to 105 Hz at open circle potentials for all the samples under visible light irradiation (>420 nm). Photocurrent densities with bias in the range of −0.8–0.0 V were measured with and without the visible light.Results and discussions
Characterization of photocatalysts
Fig. 1(b–g) show the scanning electron microscope (SEM) images of all the prepared samples. It can be seen that these TiO2 NRAs regularly aligned on Si substrate having diameters of ∼50 nm and lengths of ∼200 nm (Fig. 1(b)). Ag and Ag2S NPs distributes randomly on TiO2 NRAs surface (Fig. 1(c–e)). GO of the sample GO/Ag/Ag2S–TiO2 NRAs, spreads smoothly like a blanket and covers Ag/Ag2S–TiO2 NRAs, which shows the connection of every single NRs to each other and with also some Ag and Ag2S NPs (Fig. 1(f), (g)).Moreover, transmission electron microscopy (TEM) images presented in Fig. 2 indicate Ag and Ag2S NPs were separately dispersed on the surface of TiO2. Their average sizes were about ∼4 nm, having a regular elliptical shape. According to the measurement of lattice fringes, d = 0.233 nm, 0.244 nm, 0.345 nm and 0.325 nm match well with the crystallographic planes of Ag (111), Ag2S (200), anatase (101) and rutile (110), respectively. The formation of metal–semiconductor nanojunctions, including Ag–TiO2 NRAs, could be favorable for interfacial charge transfer among the three components, enhancing photocatalytic activities of the composites. In addition, the existence of anatase–rutile heterojunction in the NRAs could help the rutile particles to efficiently collect photon-induced electrons from the anatase particles to reduce the carrier recombination.34
 | ||
| Fig. 2 Transmission electron microscopy (TEM) images and high-resolution TEM (HRTEM) images of (a, b) Ag/Ag2S–TiO2 NRAs; (c, d) GO/Ag/Ag2S–TiO2 NRAs. | ||
The Ti, Ag and S energy dispersive X-ray spectroscopy (EDX) element mapping of GO/Ag/Ag2S–TiO2 NRAs are shown in Fig. 3(a–d). The existence of Ag, S, Ti elements can be confirmed. Moreover, it can be concluded that Ag2S NPs adhere on the surface of TiO2 nanorods. X-ray diffraction (XRD) patterns of different samples are shown in Fig. 3(e). The spectra exhibited diffraction peaks at 25.5° and 27.6° corresponding to the (101) crystal planes of the anatase phase TiO2 (JCPDS no. 21-1272) and (110) crystal planes of the rutile phase TiO2 (JPCDS no. 21-1276). Beside this, the diffraction peak at 38.2° of Ag (111) (JCPDS no. 04-0783) can be found in the XRD patterns of Ag–TiO2 NRAs, Ag/Ag2S–TiO2 NRAs, and GO/Ag/Ag2S–TiO2 NRAs. However, characteristic peaks of Ag2S are very weak in all the samples. The intensive peak at 10.3° can be assigned to the (001) diffraction peak of graphene oxide. The full XRD spectrum of GO/Ag/Ag2S–TiO2 NRAs is shown in Fig. S1 as ESI.†
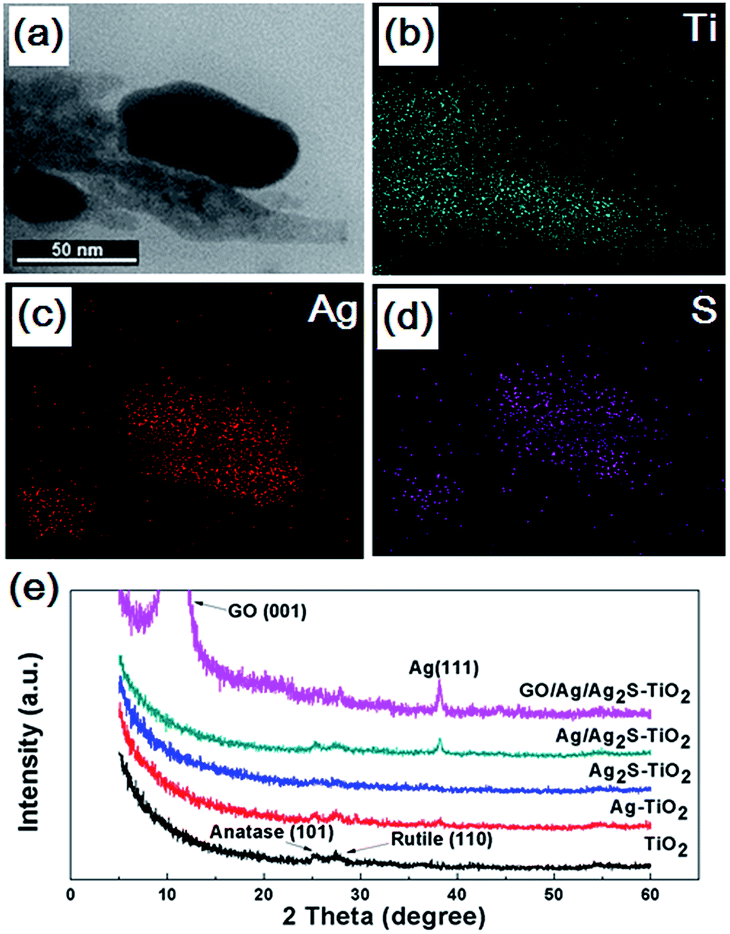 | ||
| Fig. 3 Morphology (a) and energy dispersive X-ray spectroscopy (EDX) elemental mapping of Ag/Ag2S–TiO2 NRAs sample: (b) Ti; (c) Ag; (d) S; (e) X-ray diffraction (XRD) patterns of the TiO2 NRAs, Ag–TiO2 NRAs, Ag2S–TiO2 NRAs, Ag/Ag2S–TiO2 NRAs and GO/Ag/Ag2S–TiO2 NRAs. | ||
Fig. 4(a–e) are the O 1s, S 2p, Ag 3d, C 1s, Ti 2p X-ray photoelectron spectroscopy (XPS) fine scan spectra of GO/Ag/Ag2S–TiO2 NRAs. XPS surveys of all samples are shown in Fig. 4(g). The O 1s spectrum can be fitted by two peaks: main peak located at the binding energy 530.0 eV which can be attributed to the lattice ‘O’; another one located at 531.3 eV referring to the hydroxyls or water adsorbed on the surface of the nanostructure. S 2p spectrum includes doublets containing S 2p3/2 and S 2p1/2 locating at 161.2 and 162.5 eV respectively, which can be assigned to S2−.35 Ag 3d spectra can be deconvoluted into two sets of doublet (Ag 3d5/2 and Ag 3d3/2 with spin orbit splitting of 6.1 eV (ref. 36)): Ag 3d5/2 peaks at 368.3 eV for metallic Ag and 368.1 eV for Ag+.37 C 1s spectra consist of the peaks at 284.9 eV, 286.3 eV and 288.9 eV, which are contributed by C–C; C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds from deposited GO.38 Ti 2p spectrum including doublets of Ti 2p3/2 and Ti 2p1/2 at binding energies 458.8 and 464.4 eV respectively confirm the presence of Ti4+ cations in TiO2.
O bonds from deposited GO.38 Ti 2p spectrum including doublets of Ti 2p3/2 and Ti 2p1/2 at binding energies 458.8 and 464.4 eV respectively confirm the presence of Ti4+ cations in TiO2.
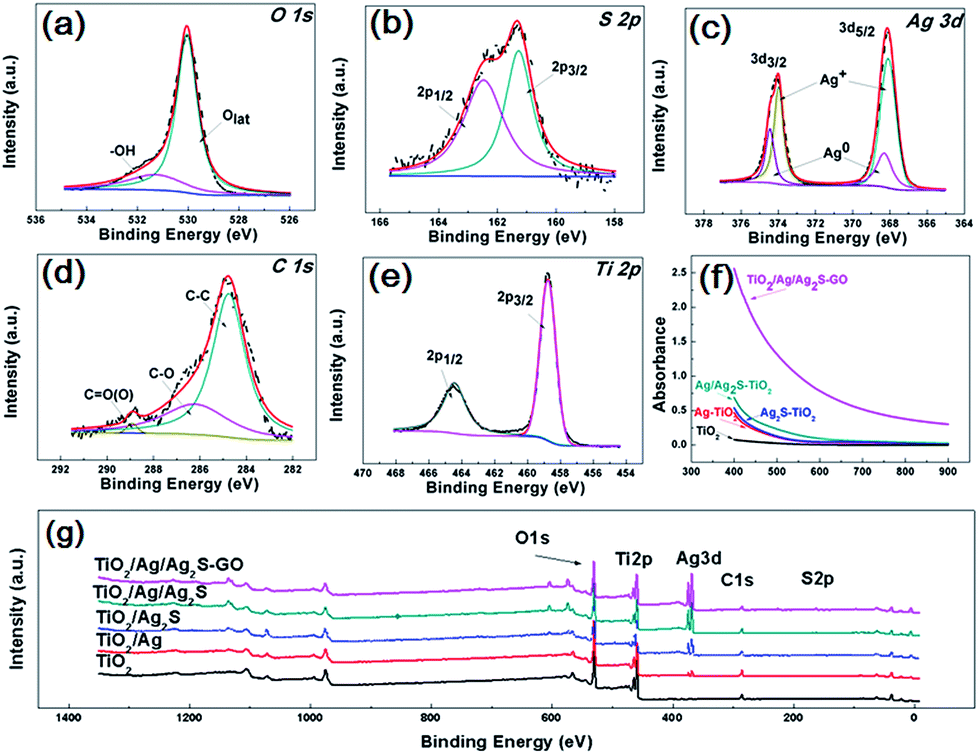 | ||
| Fig. 4 X-ray photoelectron spectroscopy (XPS) spectra of all samples: (a) O 1s; (b) S 2p; (c) Ag 3d; (d) C 1s; (e) Ti 2p; (g) survey of GO/Ag/Ag2S–TiO2 NRAs; (f) diffusion reflectance UV-vis spectra of TiO2 NRAs, Ag–TiO2 NRAs, Ag2S–TiO2 NRAs, Ag/Ag2S–TiO2 NRAs and GO/Ag/Ag2S–TiO2 NRAs. | ||
Diffusion reflectance UV-vis spectra of all typical resultants are shown in Fig. 4(f), which are fitted with Kubelka–Munk function. All the samples exhibit the absorption band around 400–900 nm. The spectra of Ag–TiO2 NRAs, Ag2S–TiO2 NRAs and Ag/Ag2S–TiO2 NRAs demonstrate obvious enhancement on light absorption in the visible region compared with TiO2 NRAs, implying the addition of Ag and Ag2S can increase the adsorption efficiency for TiO2. However, the surface plasmon resonance (SPR) absorption band is hardly observed in the spectrum of Ag–TiO2 NRAs because of the low imaginary part of the dielectric function of Ag. It can be clearly seen that the absorbance of GO/Ag/Ag2S–TiO2 NRAs is remarkably enhanced, which further confirms the contribution of GO for visible light utilization.
Photodegradation of CV
To evaluate the effect of composite decoration on the photocatalytic activity of TiO2, the photodegradation of CV was carried out under visible light irradiation. For comparison, we also tested Ag/Ag2S–TiO2 NRAs, Ag–TiO2 NRAs, Ag2S–TiO2 NRAs and TiO2 NRAs. All these samples combined with GO were also tested. As shown in Table 1, after 120 min illumination under visible lights, neither TiO2 NRAs nor GO-TiO2 NRAs exhibited any activity for the CV degradation due to nonabsorbance of TiO2 in visible light range, while 16.32%, 13.51% and 28.92% of CV were degraded by Ag–TiO2 NRAs, Ag2S–TiO2 and Ag/Ag2S–TiO2 NRAs. These results indicated that both Ag and Ag2S NPs could be excited by visible light. Ag NPs transform the irradiation photons energy into localized SPR oscillations and hot electrons move quickly to other part of nanostructured electrode.39–42 Higher activity of Ag/Ag2S–TiO2 NRAs could be attributed to the photoexcited Ag2S semiconductor and plasmon-induced Ag NPs. Moreover, further enhancement of photocatalytic activities was observed after the introduction of GO. No matter what kind of combinations, after covering with GO, the photocatalytic degradation efficiencies were improved by 200–300%. With the combination of GO/Ag/Ag2S–TiO2 NRAs, the degradation efficiency of CV reached maximum to 80.01%. Therefore, GO played an important role in the enhanced activity of Ag/Ag2S–TiO2 NRAs.| Sample | TiO2 | GO–TiO2 | Ag–TiO2 | GO/Ag–TiO2 |
| Degradation efficiency (%) | 0 | 0 | 16.32 | 47.00 |
| Sample | Ag2S–TiO2 | GO/Ag2S–TiO2 | Ag/Ag2S–TiO2 | GO/Ag/Ag2S–TiO2 |
| Degradation efficiency (%) | 13.51 | 31.77 | 28.92 | 80.01 |
Fig. 5 (a and b) show the comparison of the photocurrent density of different samples with light on/off under simulated solar irradiation. The photocurrent of pure TiO2 was zero, while the photocurrent density of Ag–TiO2 NRAs and Ag2S–TiO2 NRAs increased. And Ag/Ag2S–TiO2 NRAs raised to about 1.92 mA cm−2 at 0 V. After composited with GO, the photocurrent of GO/Ag/Ag2S–TiO2 NRAs was remarkably improved 10 times to 6.77 mA cm−2 at 0 V, which indicated that higher efficient separation of photogenerated carriers occurred. At the same condition, dark current density of GO/Ag/Ag2S–TiO2 NRAs (4.64 mA cm−2 at 0 V) was only 68% of that under visible light. Thus reasonable amount of GO in nanostructure could act as a sinker for photoinduced charge carriers, promoting charge separation to enhance the overall photocatalytic efficiency in contact with TiO2. The promotion could be contributed to the addition of p-type semiconductor of GO which has hollow sites.14 In the GO/Ag/Ag2S–TiO2 NRAs sample, Ag2S and TiO2 are both n-type,43 the introduction of GO will compose p–n junction in the sample, then the junction could enhance the current transferring through the electron and hole sites among the composites of the sample. Ning et al. have reported a core–shell heterostructure TiO2/rGO/NiFe-LDH NRAs which own an enhanced photoconversion efficiency (0.58% at 0.13 V vs. SCE) and photocurrent density (1.74 mA cm−2 at 0.6 V vs. SCE).44 For comparison, we plot current value at same voltage as red star on our PEC figures. Moreover, in site doped Au/TiO2 nanotube photoanode made by Wu.45 and GO-coated TiO2 nanoparticles fabricated by Kim et al.46 were all tested by photocurrent. The relative datas were organized in Table 2. Compared with recent results, our material's performance on separation of electrons and holes make a quite big progress.
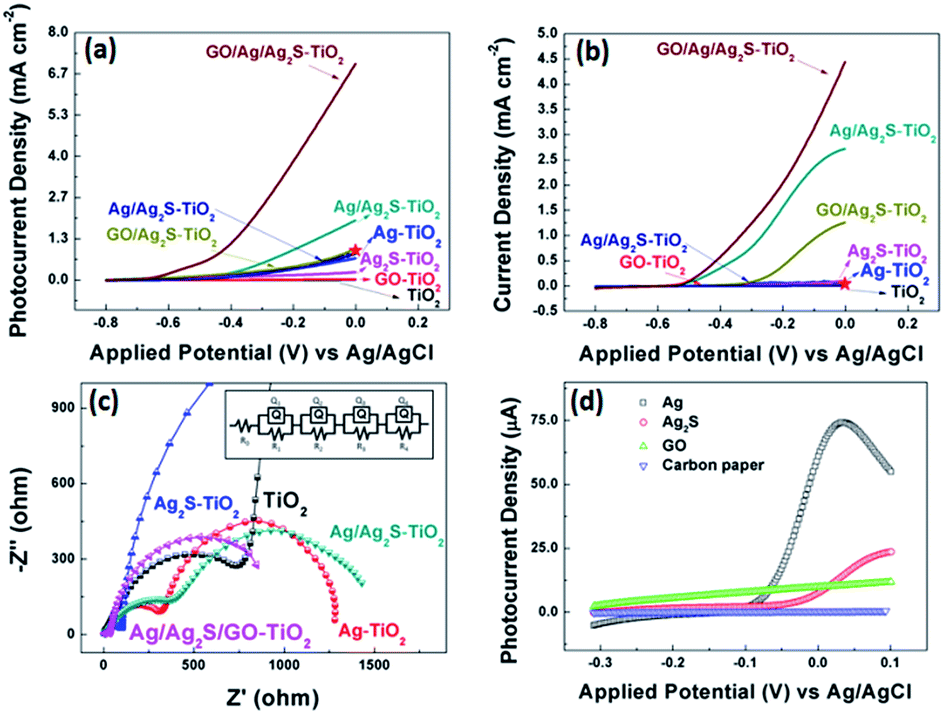 | ||
| Fig. 5 (a) j–V characteristics of all samples under visible light illuminations; (b) j–V characteristics of all samples without light illuminations. The red stars in (a, b) indicate the current density of TiO2/rGO/NiFe-LDH NRAs reported by Ning et al. under same voltage condition;36 (c) partial enlarged detail Nyquist plots under visible light at open circuit voltage of all decorated sample and the equivalent circuit for samples; (d) j–V characteristics for various combination of Ag, Ag2S and GO decoration on carbon paper under visible light illuminations. | ||
| Group/system | Photocurrent (mA cm−2) | Voltage (V) | Light resource | Electrolyte |
|---|---|---|---|---|
| Ning et al. TiO2/rGO/NiFe-LDH nanorods | 1.74 | 0.6 | 150 W Xe lamp | 0.5 M Na2SO4 solution |
| Wu et al. Au/TiO2 nanotubes | 0.50 | 0 | 300 W Xe lamp | 0.1 M Na2SO4 solution |
| Kim et al. GO-coated TiO2 nanoparticles | 1.25 | 0 | 300 W Xe arc lamp | 0.1 M HClO4 solution |
| This work | 6.98 | 0 | 300 W Xe lamp | 0.1 M KOH solution |
EIS measurements for all samples were also conducted under visible light irradiation, as shown in Fig. 5(c) as the enlarged image of full spectra (see Fig. S2†). This method can characterize the charge-carrier migration. An equivalent circuit was used to fit the Nyquist plots as shown in the inset of Fig. 5(c) and simulated data were shown in Table S2.† All the hybrid nanostructured samples showed depressed semicircles at high frequencies compared with pure TiO2 counterparts. The reduced semicircles indicate diminished resistance of working electrodes, suggesting a decrease in the solid state interface layer resistance and the charge transfer resistance across the solid–liquid junction on the surface by forming hybrid structures of decorated TiO2 NRAs with GO.47 The R0 are the resistance of FTO substrate. Q1 and R1 represent the double layer on the reference electrode. The obtained Nyquist spectra show two arcs, the first arc (R2), at higher frequencies, is related to the interfacial resistance between TiO2 nanorod array and electrolyte. And the arcs at the intermediate frequencies represent the resistance (R3) of holes transferred to the electrolyte through surface states (such as Ag or Ag2S NPs) and the capacitance of surface states. For Ag/Ag2S–TiO2 NRAs, Ag NPs was deposited after Ag2S decoration.
However, based on HRTEM analysis, the Ag and Ag2S are separately attached on the TiO2 arrays surface. So, an extra resistance (R4) was added and can be described as electron or hole transfer between NPs of Ag and Ag2S. When layer of GO was coated, the R4 can also represents the transfer between the decorated TiO2 and GO. All simulated data are listed in Table S2.†48 Based on the results, it can be seen that the additions of Ag or/and Ag2S can decrease the R2 and R3, implying the reduced transferring resistance between the deposition substrate and TiO2. The extra introduction of GO can decrease the hole transfer resistance (R3) dramatically. By comparing the EIS data, GO/Ag/Ag2S–TiO2 NRAs are superior to other samples with smaller semicircles, suggesting a rapid transport of charge carriers and an effective charge separation, which is in agreement with the PEC analysis.
In the photocurrent measurements, the obtained currents tested under visible light include photocurrent for oxygen evolution reaction (OER), and electrochemical current caused by applied potential in OER process. So, we use current with light minus the one without light in order to investigate more specific on photo-catalysis progress. Then, the largest current value obtained on GO/Ag/Ag2S–TiO2 NRAs was used as reference. The currents of all the other samples were divided by the reference to calculate the relative values, which are listed in Table 3. According to data, it can be easily obtained that simply composing between Ag and Ag2S particles doesn't improve the photocatalytic performance so much. Only after the addition of GO layer, the activities of all system enhance remarkably, while the GO/Ag/Ag2S–TiO2 NRAs has maximum increment up to 7.7 times. GO layer could both enhanced transmission of charges and protect NPs from corrosion. What's more, to confirm the reaction mechanism and active reaction sites among all the possible composites, we deposited the single composite on carbon paper rather than TiO2 NRAs. Then the photocurrents of the prepared samples were recorded under the same test condition which are shown in Fig. 5(d). Basing on the tests, the onset potentials of Ag is most negative among all the prepared samples, implying that Ag has lowest OER overpotential among the four species.
| Sample | TiO2 | GO-TiO2 | Ag–TiO2 | GO/Ag–TiO2 |
| Percentage (%) | 0 | 0.22 | 12.89 | 2.11 |
| Sample | Ag2S–TiO2 | GO/Ag2S–TiO2 | Ag/Ag2S–TiO2 | GO/Ag/Ag2S–TiO2 |
| Percentage (%) | 7.59 | 16.11 | 12.97 | 100.00 |
The evolution of the CV solution's absorption spectra for GO/Ag/Ag2S–TiO2 NRAs are recorded during the whole reaction process every 30 min time point. From Fig. 6(a), the intensity of peak gradually declines with longer irradiation time which indicates reduced concentrate of dye concentrate. To analyse the products after reaction, mass spectrometer (MS) for CV in aqueous solution before and after prolonged irradiation (120 min) are also studied, shown in Fig. S3.† As standard CV molecular (M = 407.99) concerned, peak at m/z = 372.23 indicate CV molecular which equals to formula weight minus chloridion. When we infer product after reaction from m/z = 152.12, it is exactly coincident to 1/3 part of whole molecular that three C–C bonds of C atom in central broken up. The result means there is a single product after degradation reaction.
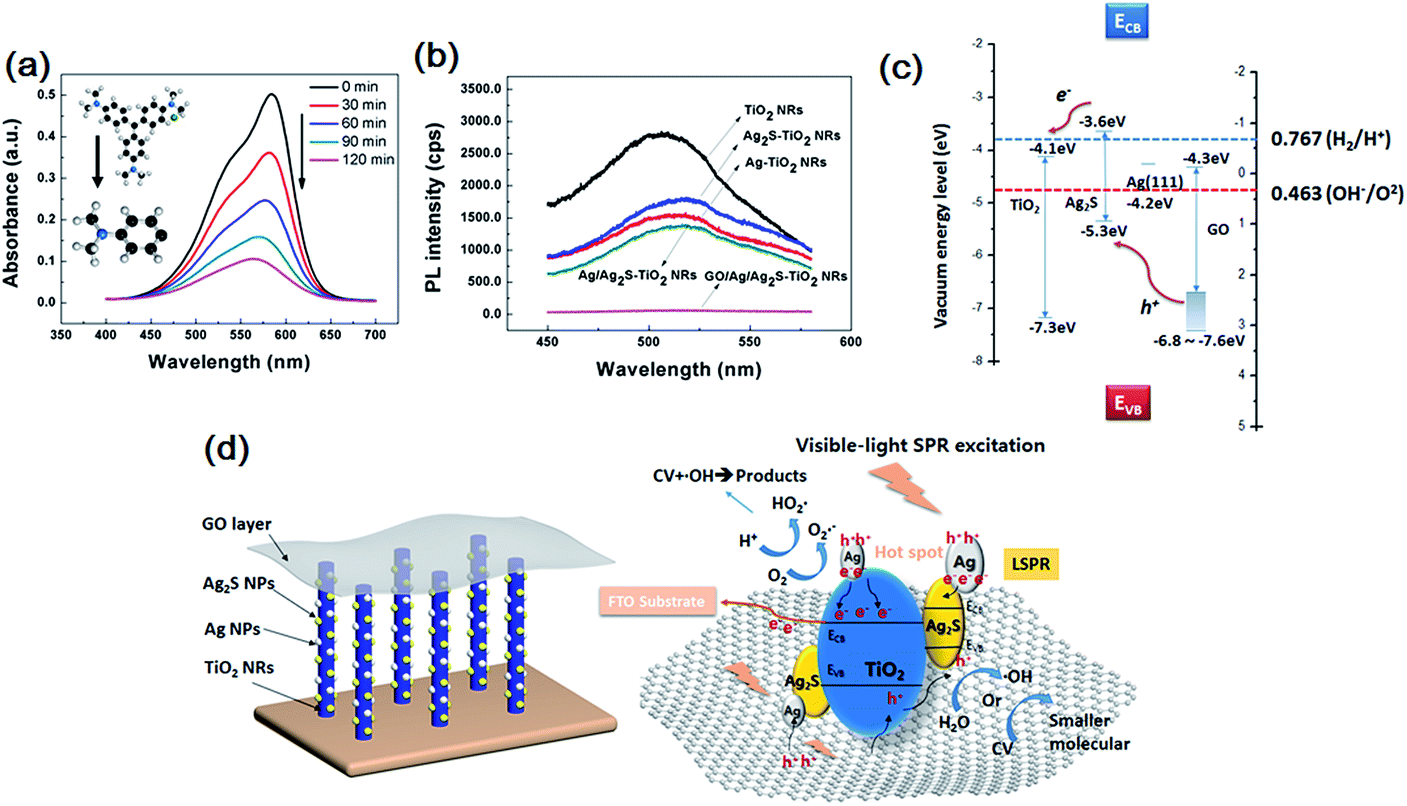 | ||
| Fig. 6 (a) UV-visible absorption spectra recorded during the catalytic degradation of CV over GO/Ag/Ag2S–TiO2 NRAs; (b) photoluminescence (PL) spectrum of single TiO2 NRAs, Ag–TiO2 NRAs, Ag2S–TiO2 NRAs, Ag/Ag2S–TiO2 NRAs and GO/Ag/Ag2S–TiO2 NRAs; (c) band edge positions of TiO2, Ag2S, Ag and GO; (d) photocatalytic process for GO/Ag/Ag2S–TiO2 NRAs under visible light. | ||
Photoluminescence (PL) technique is an effective way to study the efficiency of the charge carrier trapping, migration and transfer, as PL signals result from the recombination of photo-induced carriers. Fig. 6(b) presents the PL spectra of all test samples. The peaks at ∼515 nm can be attributed to the self-trapped excitations and the oxygen vacancies (Vo) in TiO2.49,50 Due to the metallic particle decoration on TiO2, the PL peaks of TiO2 become weaker. The PL intensity of GO/Ag/Ag2S–TiO2 NRAs is much lower than that of other nanostructures, implying a lower recombination rate of photo-induced electron–hole pairs, and thus a better photocatalytic performance. While PL intensity of Ag–TiO2 NRAs, Ag2S–TiO2 NRAs and Ag/Ag2S–TiO2 NRAs are between TiO2 NRAs and GO/Ag/Ag2S–TiO2 NRAs which is corresponding with dye degradation results.
Photocatalytic mechanism
Basing on above results, the photocatalytic mechanism of GO/Ag/Ag2S–TiO2 NRAs can be proposed as below. The valence band-edges of materials are shown in Fig. 6(c). In the composite, Ag NPs, Ag2S NPs (always n-type) and GO sheets (typical p-type) are active to visible light. The higher activity of GO/Ag/Ag2S–TiO2 NRs could be contributed to the photoexcited.Ag2S semiconductor, plasmon-induced Ag NPs, effective electron–hole separation of GO and clever combination of all the composites. Under visible light irradiation, Ag nanoparticles are photoexcited owing to their SPR effect, and then charge separation is accomplished, where the electrons transfer from Ag nanoparticles to the TiO2 CB, leading to the generation of holes in Ag nanoparticles for oxidation of OH−. Meanwhile, Ag2S NPs and GO are also excited to form electrons and holes.51 Considering that the CB of Ag2S is more positive than that of TiO2,52,53 electrons generated from Ag2S also tend to transfer into the CB of TiO2. This facilitates the separation of electrons and holes. Besides, covering of GO layer remarkably enhances full-wavelength absorption from UV-vis spectra reported in Fig. 4(f), which accomplishes high-usage of visible light. Since work electrode surface is positively charged, excited electrons from GO are easier to move into TiO2. Then, vast holes tend to move directly into Ag NPs which consume holes quickly because of its lowest OER overpotential among the four species in the composite, resulting acceleration of the whole reaction. Thus Ag NPs is probably active OER reaction sites for the whole system. Ag nanoparticles also act as the charge transmission bridges, which can efficiently promote the separation of electron–hole pairs. Also, both Ag and Ag2S combination with TiO2 could narrow down its original band gap, it may enhance the adsorption efficiency of the composite.
Then electrons can either reduce the dye or can react with electron acceptors (O2 absorbed on the surface of Ti3+ or dissolved in water) to create superoxide radicals (O2˙−). Meanwhile, the resultant electron-deficient Ag particles can oxidize the organic molecule or react with OH− to form hydroxyl radicals, OH˙. The process is demonstrated in Fig. 6(d). The co-decoration of Ag/Ag2S NPs and GO not only extends TiO2 to visible light region, but also increases the efficiency of charge separation and improves its photocatalytic efficiency.
Conclusions
In summary, we obtained the procedure for preparation of the TiO2 NRAs on various substrates including FTO, Si and et al., and fabricated successfully GO/Ag/Ag2S–TiO2 NRAs by using SILAR technique and impregnation method. By comparing with TiO2 NRAs, it exhibits remarkable improvement of photocatalytic activity for dye degradation and PEC performance. The enhanced photocatalytic activities through decoration of the NRAs are attributed to the synergy of matched band of Ag2S NPs, SPR effect of Ag NPs and rapid transport of charge carriers that improves charge separation of system. Our studies demonstrate that we can utilize photon in a wider range of solar spectrum to generate charge carriers for photocatalytic reactions through rational design of composite nanostructures. The combination of GO with traditional photocatalyst also provides a more effective way to harvest solar energy for decomposition reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Chinese Ministry of Science and Technology (grant No. 2016YFE0104000) and the National Natural Science Foundation of China (grant No. 51372135).References
- A. L. Linsebigler, G. Q. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- A. Wold, Chem. Mater., 1993, 5, 280–283 CrossRef CAS.
- H. Zhu, N. Goswami, Q. Yao, T. Chen, Y. Liu, Q. Xu, D. Chen, J. Lu and J. Xie, J Mater Chem A, 2018, 6, 1102–1108 CAS.
- Y. Ye, T. Chen, J. Zhen, C. Xu, J. Zhang and H. Li, Nanoscale, 2018 10.1039/C7NR07383F.
- C. Chen, X. Li, W. Ma, J. Zhao, H. Hidaka and N. Serpone, J. Phys. Chem. B, 2002, 106, 318–324 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- G. Wu, T. Nishikawa, B. Ohtani and A. Chen, Chem. Mater., 2007, 19, 4530–4537 CrossRef CAS.
- Q. Sun and Y. Xu, J. Phys. Chem. C, 2009, 113, 12387–12394 CAS.
- W. Zhao, Y. Sun and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 12566–12567 CrossRef CAS PubMed.
- G. Liu, L. Wang, H. G. Yang, H.-M. Cheng and G. Q. Lu, J. Mater. Chem., 2010, 20, 831–843 RSC.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- H. Wei, Y. Wu, N. Lun and F. Zhao, J. Mater. Sci., 2004, 39, 1305–1308 CrossRef CAS.
- X.-B. Xiang, Y. Yu, W. Wen and J.-M. Wu, New J. Chem., 2018, 42, 265–271 RSC.
- Y. Hu, X. Song, S. Jiang and C. Wei, Chem. Eng. J., 2015, 274, 102–112 CrossRef CAS.
- S. Shuang, R. Lv, Z. Xie and Z. Zhang, Sci. Rep., 2016, 6, 26670 CrossRef CAS PubMed.
- W. Wan, R. Zhang, M. Ma and Y. Zhou, J Mater Chem A, 2018, 6, 754–775 CAS.
- B. Wang, J.-T. Cao, Y.-X. Dong, F.-R. Liu, X.-L. Fu, S.-W. Ren, S.-H. Ma and Y.-M. Liu, Chem. Commun., 2018, 54, 806–809 RSC.
- L. Jin, X. Zhao, J. Xu, Y. Luo, D. Chen and G. Chen, RSC Adv., 2018, 8, 2065–2071 RSC.
- K. Yuan, Q. Cao, H.-L. Lu, M. Zhong, X. Zheng, H.-Y. Chen, T. Wang, J.-J. Delaunay, W. Luo, L. Zhang, Y.-Y. Wang, Y. Deng, S.-J. Ding and D. W. Zhang, J Mater Chem A, 2017, 5, 14697–14706 CAS.
- J. Zheng, L. Calvillo, G. A. Rizzi and G. Granozzi, ChemPlusChem, 2016, 81, 391–398 CrossRef CAS.
- C.-J. Chang, K.-W. Chu, M.-H. Hsu and C.-Y. Chen, Int. J. Hydrogen Energy, 2015, 40, 14498–14506 CrossRef CAS.
- J. C. Yu, J. Yu, W. Ho and L. Zhang, Chem. Commun., 2001, 1942–1943, 10.1039/b105471f.
- C. Chen, W. Cai, M. Long, B. Zhou, Y. Wu, D. Wu and Y. Feng, ACS Nano, 2010, 4, 6425–6432 CrossRef CAS PubMed.
- A. Mathkar, D. Tozier, P. Cox, P. Ong, C. Galande, K. Balakrishnan, A. Leela Mohana Reddy and P. M. Ajayan, J. Phys. Chem. Lett., 2012, 3, 986–991 CrossRef CAS PubMed.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380–386 CrossRef CAS PubMed.
- C. Zhu, S. Guo, P. Wang, L. Xing, Y. Fang, Y. Zhai and S. Dong, Chem. Commun., 2010, 46, 7148–7150 RSC.
- L. Tong, F. Qiu, T. Zeng, J. Long, J. Yang, R. Wang, J. Zhang, C. Wang, T. Sun and Y. Yang, RSC Adv., 2017, 7, 47999–48018 RSC.
- S. Bagheri, A. TermehYousefi and T.-O. Do, Catal. Sci. Technol., 2017, 7, 4548–4569 CAS.
- O. Akhavan and E. Ghaderi, J. Phys. Chem. C, 2009, 113, 20214–20220 CAS.
- X.-Y. Zhang, H.-P. Li, X.-L. Cui and Y. Lin, J. Mater. Chem., 2010, 20, 2801–2806 RSC.
- Y.-B. Tang, C.-S. Lee, J. Xu, Z.-T. Liu, Z.-H. Chen, Z. He, Y.-L. Cao, G. Yuan, H. Song, L. Chen, L. Luo, H.-M. Cheng, W.-J. Zhang, I. Bello and S.-T. Lee, ACS Nano, 2010, 4, 3482–3488 CrossRef CAS PubMed.
- X. Cui, S. Yang, X. Yan, J. Leng, S. Shuang, P. M. Ajayan and Z. Zhang, Adv. Funct. Mater., 2016, 26, 5708–5717 CrossRef CAS.
- Z. Xie, X. X. Liu, W. P. Wang, X. J. Wang, C. Liu, Q. Xie, Z. C. Li and Z. J. Zhang, Nano Energy, 2015, 11, 400–408 CrossRef CAS.
- Y. Wang, R. Shi, J. Lin and Y. Zhu, Appl. Catal., B, 2010, 100, 179–183 CrossRef CAS.
- S. A. Pawar, D. S. Patil, J. H. Kim, P. S. Patil and J. C. Shin, Opt. Mater., 2017, 66, 644–650 CrossRef CAS.
- J. Zheng, L. Calvillo, G. A. Rizzi and G. Granozzi, ChemPlusChem, 2016, 81, 391–398 CrossRef CAS.
- N. D. Feng, Q. Wang, A. M. Zheng, Z. F. Zhang, J. Fan, S. B. Liu, J. P. Amoureux and F. Deng, J. Am. Chem. Soc., 2013, 135, 1607–1616 CrossRef CAS PubMed.
- P. Stathi, D. Gournis, Y. Deligiannakis and P. Rudolf, Langmuir, 2015, 31, 10508–10516 CrossRef CAS PubMed.
- D. Yang, Y. Y. Sun, Z. W. Tong, Y. Tian, Y. B. Li and Z. Y. Jiang, J. Phys. Chem. C, 2015, 119, 5827–5835 CAS.
- R. F. Dong, B. Z. Tian, C. Y. Zeng, T. Y. Li, T. T. Wang and J. L. Zhang, J. Phys. Chem. C, 2013, 117, 213–220 CAS.
- Q. Zhu, W. S. Wang, L. Lin, G. Q. Gao, H. L. Guo, H. Du and A. W. Xu, J. Phys. Chem. C, 2013, 117, 5894–5900 CAS.
- S. A. Ansari, M. M. Khan, M. O. Ansari, J. Lee and M. H. Cho, J. Phys. Chem. C, 2013, 117, 27023–27030 CAS.
- X. Yu, J. Liu, A. Genç, M. Ibáñez, Z. Luo, A. Shavel, J. Arbiol, G. Zhang, Y. Zhang and A. Cabot, Langmuir, 2015, 31, 10555–10561 CrossRef CAS PubMed.
- F. Y. Ning, M. F. Shao, S. M. Xu, Y. Fu, R. K. Zhang, M. Wei, D. G. Evans and X. Duan, Energy Environ. Sci., 2016, 9, 2633–2643 CAS.
- Z. Y. Wu, J. Wang, Z. Y. Zhou and G. H. Zhao, J Mater Chem A, 2017, 5, 12407–12415 CAS.
- H.-i. Kim, G.-h. Moon, D. Monllor-Satoca, Y. Park and W. Choi, J. Phys. Chem. C, 2012, 116, 1535–1543 CAS.
- B.-L. He, B. Dong and H.-L. Li, Electrochem. Commun., 2007, 9, 425–430 CrossRef CAS.
- E. Baran and B. Yazici, Thin Solid Films, 2017, 627, 82–93 CrossRef CAS.
- S. Khanchandani, P. K. Srivastava, S. Kumar, S. Ghosh and A. K. Ganguli, Inorg. Chem., 2014, 53, 8902–8912 CrossRef CAS PubMed.
- Z. Shan, D. Clayton, S. Pan, P. S. Archana and A. Gupta, J. Phys. Chem. B, 2014, 118, 14037–14046 CrossRef CAS PubMed.
- A. Mathkar, D. Tozier, P. Cox, P. J. Ong, C. Galande, K. Balakrishnan, A. L. M. Reddy and P. M. Ajayan, J. Phys. Chem. Lett., 2012, 3, 986–991 CrossRef CAS PubMed.
- H. R. Pant, B. Pant, J. K. Han, A. Amarjargal, H. P. Chan, L. D. Tijing, E. K. Kim and C. S. Kim, Ceram. Int., 2013, 39, 5083–5091 CrossRef.
- S. Pal, Y. K. Tak and J. M. Song, Appl. Environ. Microbiol., 2007, 73, 1712–1720 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13501g |
| This journal is © The Royal Society of Chemistry 2018 |