
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEco-friendly reactions in PEG-400: a highly efficient and green approach for stereoselective access to multisubstituted 3,4-dihydro-2(1H)-quinazolines using 2-aminobenzylamines†
Nutan Sharma,
Pankaj Sharma and
Sunita Bhagat *
*
Organic Synthesis Research Laboratory, Department of Chemistry, A. R. S. D. College, University of Delhi, New Delhi-110021, India. E-mail: sunitabhagat28@gmail.com
First published on 26th February 2018
Abstract
An efficient and stereoselective synthesis of novel 3,4-dihydro-2(1H)-quinazolines has been developed through cyclization reactions of 2-aminobenzylamines with α-oxoketene dithioacetals using PEG-400 as an inexpensive, easy to handle, non-toxic and recyclable reaction medium. The developed protocol is operationally simple and tolerates various substrates having different functionalities. This protocol features several attributes such as excellent yields, no work up, green reaction conditions, and being environmentally benign. The attractive feature of this new strategy is that all the reported final compounds have been isolated as single (E)-stereoisomeric forms, which was confirmed by 1HNMR and X-ray crystallographic studies.
Introduction
Quinazolines are a class of fused heterocyclic ring systems containing multiple pharmacophores which are undoubtedly well established as biologically and pharmaceutically important compounds.1 Quinazoline-based heterocycles and their hydrated congeners have been reported to possess diverse biological and therapeutic properties such as inhibition of the epidermal growth factor (EGF) receptors of tyrosine kinase,2a and analgesic,2b–d anticancer,3 anti-inflammatory,4 antidiuretic,5 anticonvulsant6 and anti-Alzheimer7 activities. Commercialized drug compounds such as gefitinib (I), erlotinib (II), alfuzosin (III), and afloqualone (IV) are some notable examples of drugs containing quinazoline as a core nucleus (Fig. 1). Considering these applications, development of novel synthetic routes towards the formation of these pharmaceutically important scaffolds attracts the interest of synthetic organic chemists.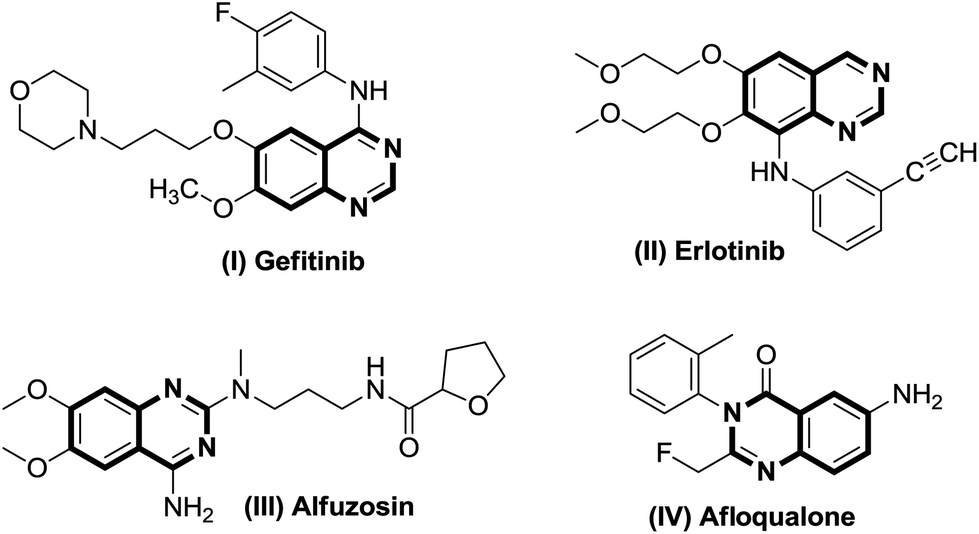 | ||
| Fig. 1 Some popular marketed drugs containing quinazoline as a core nucleus. | ||
Substituted quinazolines have been reported to be constructed by numerous methods involving several substrates.8–10 Among them, many of the synthetic methodologies have craftily employed 2-aminobenzylamines as substrates with aldehydes for the synthesis of quinazolines (Scheme 1, i–iv). These reactions have been carried out by using (i) [Cp*IrCl2]2 as a catalyst, four equivalents of styrene in xylene solvent under inert atmosphere of N2, 24 h, reflux conditions11a (Scheme 1, a); (ii) nanoclusters of bi-metallic Pt/Ir alloy as a catalyst, 5,5′,6,6′-tetrahydroxy-3,3,3′,3′-tetramethyl-1,1′-spiro-bisindane, (TTSBI) as a co-catalyst, K2CO3 as a base with CDCl3/H2O as solvent for a longer reaction duration11b (Scheme 1, b); (iii) a CuCl/DABCO/4-HO-TEMPO/O2 catalytic system in CH3CN at 80 °C (ref. 12a) (Scheme 1, c) and (iv) four equivalents of NaOCl as oxidant in MeOH solvent for longer reaction hours12b (Scheme 1, d). However, despite the synthetic utility of these protocols, the reported techniques suffer from drawbacks such as the use of expensive metal catalyst along with a co catalyst and requirement of a stoichiometric amount of toxic oxidant NaOCl for the transformation to take place. In such a scenario, development of a catalyst free and eco friendly methodology to deliver functionalized quinazolines will provide an intriguing and green alternative to the conventional approach.
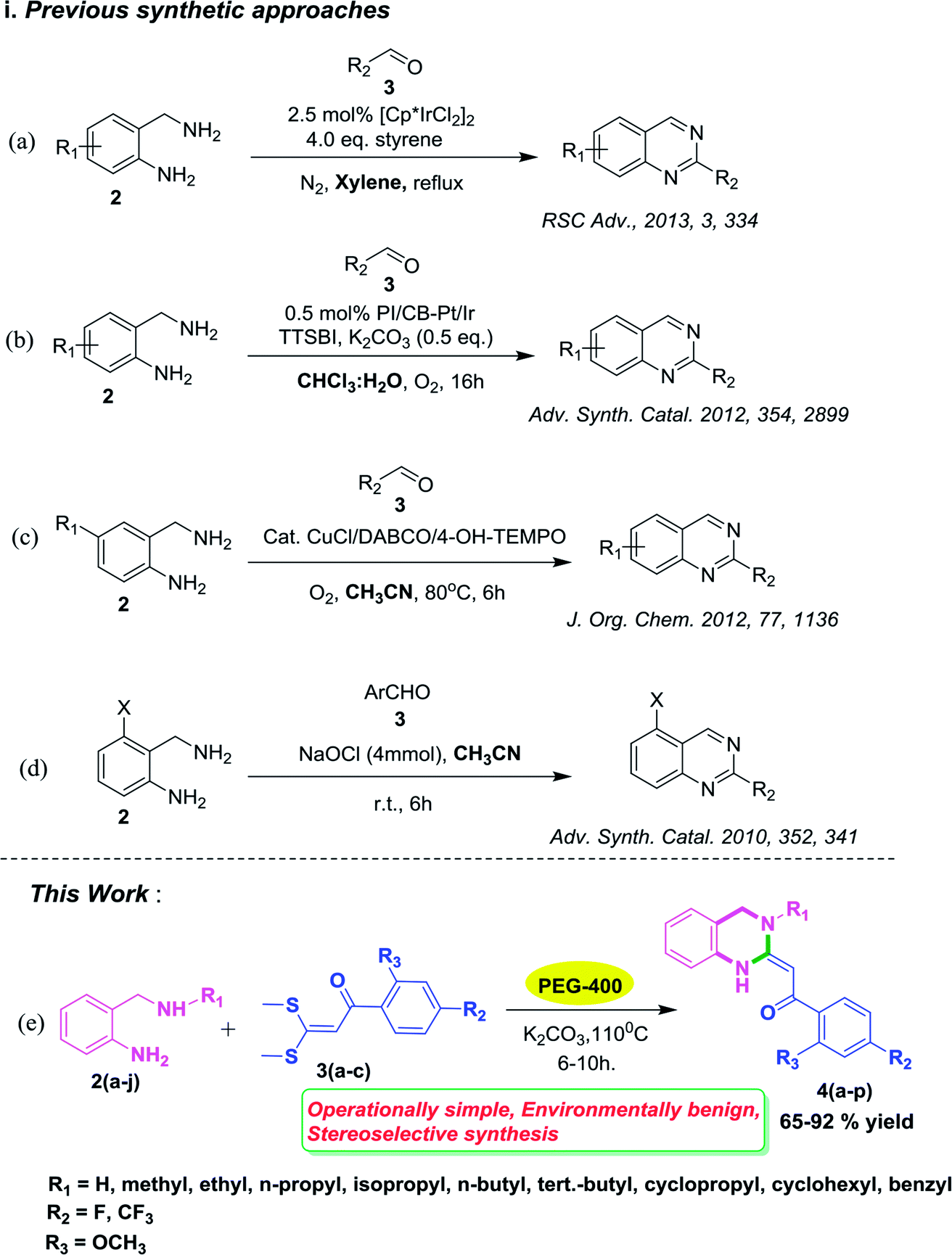 | ||
| Scheme 1 Previous synthetic approaches and our designed approach. | ||
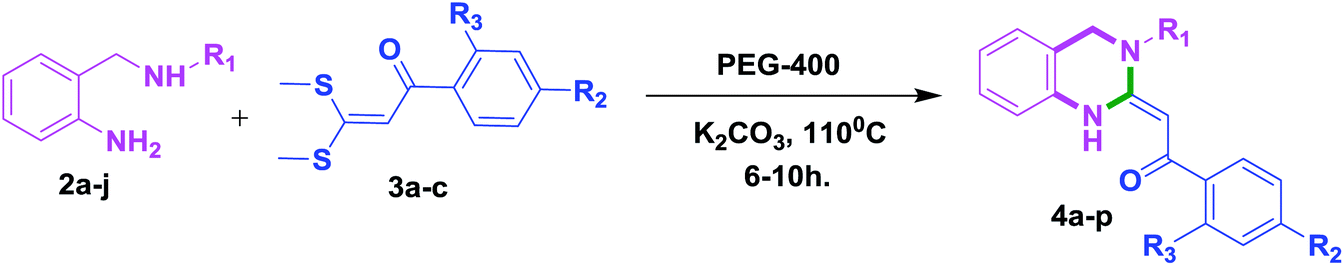
Over the years, there has been a growing public concern towards the adverse effects of toxic and volatile organic solvents and the search for alternate nonhazardous reaction media is gaining progress. Meanwhile, using water as a green solvent has been well documented13 but its practical use is limited due to the hydrophobic nature of organic compounds.14 On the contrary, PEGs have been explored as powerful, novel, eco-friendly reaction media for innumerable organic transformations15 owing to their non toxic, inexpensive, thermally stable, readily recyclable, and biodegradable nature.16 In continuation of our research studies aimed at developing environmentally benign and improved synthetic methodologies for organic reactions,17 we herein report for the first time an efficient and green approach for the stereoselective synthesis of 3,4-dihydro-2(1H)-quinazolines via substituted 2-amino benzylamines and α-oxoketene dithioacetals as building blocks using PEG-400 as an eco-friendly reaction medium (Scheme 1, e). To the best of our knowledge, there have been no reports on the synthesis of 3,4-dihydro-2(1H)-quinazolines using PEG-400 as a solvent under catalyst-free conditions.
Results and discussion
To check feasibility of this route, a model reaction between 2b and 3a was performed by varying different protic and aprotic solvents and various bases (Table 1). When the reaction was performed using EtOH as polar protic solvent and CH3CN as low boiling aprotic solvent under reflux conditions using K2CO3 as base, the desired product 4b was obtained in lower yields (Table 1, entries 1 and 2). Whereas under similar conditions, high boiling aprotic solvent such as toluene was not proven very effective and gave the desired product 4b in 50% yield after 12 h (Table 1, entry 3). High boiling polar aprotic solvents like DMF and DMSO afforded the desired product in higher yield taking less time (Table 1, entries 4 and 5). It should be noted that use of polar protic solvents like n-BuOH and t-BuOH increased the yield to 65% and 70% in the presence of K2CO3 as base in 10 h (Table 1, entries 6 and 7). Keeping in view the economical and environmental concerns, H2O was screened as the reaction medium but observations indicated trace amount of product formation due to solubility problem (Table 1, entry 8). In order to improve the efficiency of the reaction, we next performed our model reaction in PEG-400 at 110 °C using K2CO3 as base, and to our delight corresponding product 4b was obtained in a maximum yield of 90% within 8 h (Table 1, entry 9). Encouraged by the above result, a mixture of PEG-400 and H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), was used as reaction medium which resulted in the formation of product 4b in 81% yield (Table 1, entry 10). Moreover, change of combination of solvents from PEG-400 and H2O to PEG-400 and EtOH (1:1) also had a detrimental effect on the yield of the product (Table 1, entry 11). Further increase or decrease in the temperature of the reaction could not increase the yield of the desired product 4b (Table 1, entries 12 and 13). From Table 1, it is clear that PEG-400 unanimously dominates as effective solvent medium because it increases the solubility of reactants, which leads to larger interfacial area and lower mass transfer resistance.18 When base was changed from K2CO3 to Na2CO3 at 110 °C, 85% yield was observed in 8 h (Table 1, entry 14).
:1), was used as reaction medium which resulted in the formation of product 4b in 81% yield (Table 1, entry 10). Moreover, change of combination of solvents from PEG-400 and H2O to PEG-400 and EtOH (1:1) also had a detrimental effect on the yield of the product (Table 1, entry 11). Further increase or decrease in the temperature of the reaction could not increase the yield of the desired product 4b (Table 1, entries 12 and 13). From Table 1, it is clear that PEG-400 unanimously dominates as effective solvent medium because it increases the solubility of reactants, which leads to larger interfacial area and lower mass transfer resistance.18 When base was changed from K2CO3 to Na2CO3 at 110 °C, 85% yield was observed in 8 h (Table 1, entry 14).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | Temp (°C) | Time (h) | Yieldb (%) |
| a Reactions were performed using 1 equiv. of 3a, 1.2 equiv. of 2b, 3.0 equiv. of base.b Isolated yield. | |||||
| 1 | EtOH | K2CO3 | 80 | 15 | 40 |
| 2 | CH3CN | K2CO3 | 90 | 18 | 45 |
| 3 | Toluene | K2CO3 | 110 | 12 | 50 |
| 4 | DMF | K2CO3 | 120 | 10 | 60 |
| 5 | DMSO | K2CO3 | 120 | 10 | 62 |
| 6 | n-BuOH | K2CO3 | 120 | 10 | 65 |
| 7 | t-BuOH | K2CO3 | 85 | 10 | 70 |
| 8 | H2O | K2CO3 | 100 | 15 | Trace |
| 9 | PEG 400 | K2CO3 | 110 | 8 | 90 |
| 10 | PEG 400:H2O |
K2CO3 | 110 | 8 | 81 |
| 11 | PEG 400:EtOH |
K2CO3 | 110 | 10 | 72 |
| 12 | PEG 400 | K2CO3 | 120 | 8 | 90 |
| 13 | PEG 400 | K2CO3 | 90 | 8 | 80 |
| 14 | PEG 400 | Na2CO3 | 110 | 8 | 85 |
| 15 | PEG 400 | TEA | 110 | 8 | 65 |
| 16 | PEG 400 | DIPEA | 110 | 8 | 68 |
| 17 | PEG 400 | — | 110 | 8 | 20 |
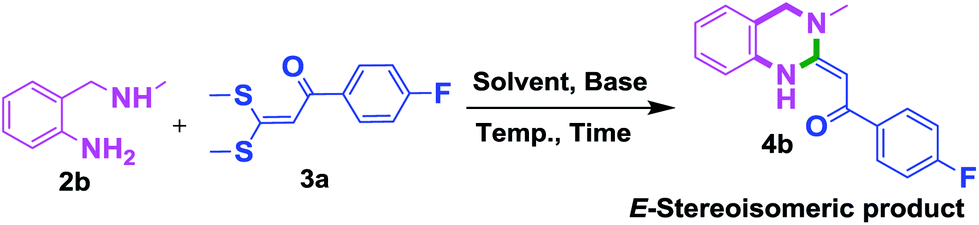
Screening of other organic bases such as TEA and DIPEA in same reaction conditions resulted in 65% and 68% yields respectively (Table 1, entries 15 and 16). However the yield of reaction was drastically reduced to 20% when the reaction was performed in the absence of base (Table 1, entry 17). These optimization studies led to the conclusion that PEG-400 with K2CO3 is the reaction medium of choice for this transformation. The reaction product was fully characterized by the IR, 1H NMR, 13C NMR & HRMS data. Compound 4b existed in one stereoisomeric form (E-form) as evident from its IR & 1HNMR data. IR spectra strongly indicated a hydrogen-bonded carbonyl stretching vibration, which was merged with bands around and below 1600 cm−1 (1594 cm−1 in this case). The signals for vinylic proton appeared as sharp singlets, indicating the purity of the geometrical isomers. 1HNMR spectrum showed a characteristic chelated –NH proton far downfield at δ 13.66, assigned to the amino group which participated in a strong hydrogen bond with oxygen of the carbonyl group in a six membered, planar chelate19 (Fig. 2). Moreover, 1HNMR analysis of crude reaction mixture of 4g (see ESI,† page 31) also indicated the formation of one stereoisomeric form of the product. Its spectral data analysis confirmed the structure.
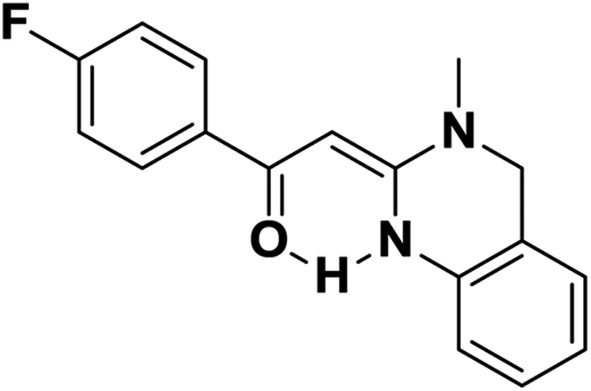 | ||
| Fig. 2 Stereoselectivity via formation of six membered planar chelate. | ||
In the 1H NMR spectrum, presence of peaks as sharp singlets at δ 5.43 (s, 1H) and δ 4.45 (s, 2H), were characterized for vinylic proton and methylene group –NCH2− in 4b. In the 13C NMR spectrum of 4b, characteristic peak at δ 75.9 and δ 50.7 was observed for vinylic carbon and methylene carbon. HRMS data of the compound showed M + 1 peak. Similarly all other compounds were also characterized by IR, 1H NMR, 13C NMR and HRMS studies. The structure of the product 4o was determined by single crystal X-ray diffraction (Fig. 3). The structures of the other products were concluded by analogy.
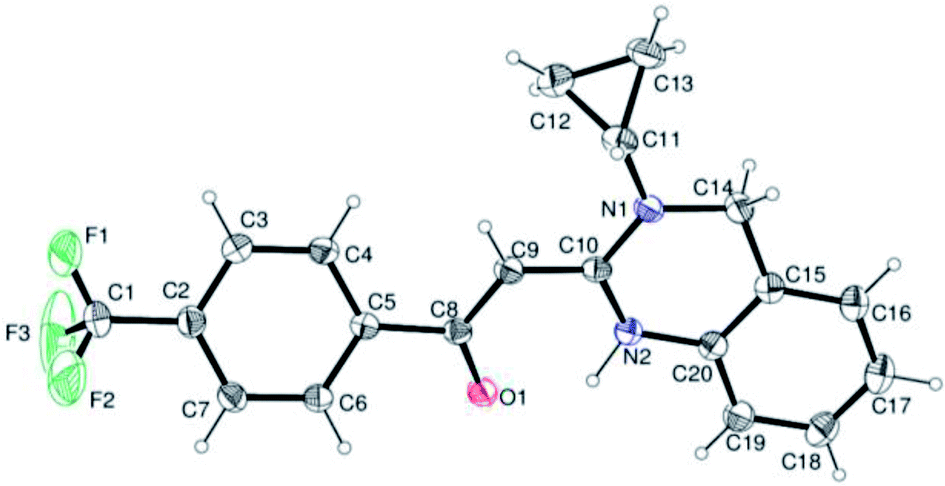 | ||
| Fig. 3 Ortep diagram of compound 4o. | ||
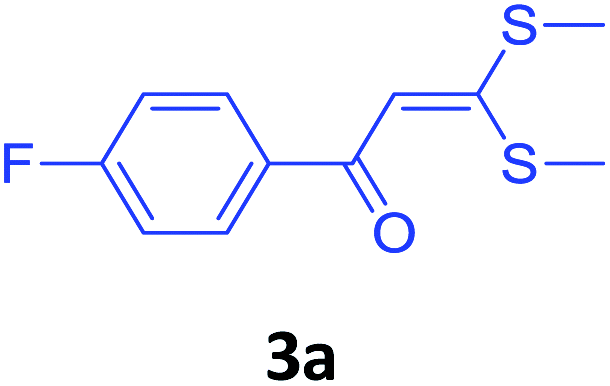
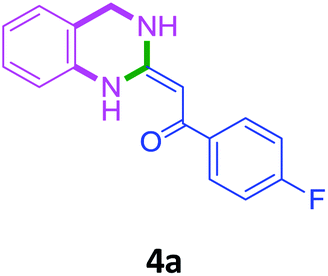
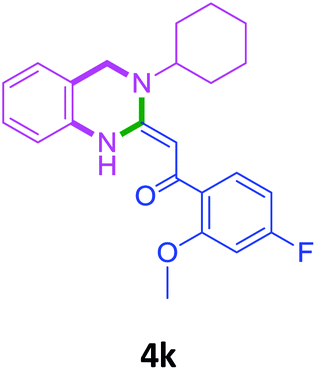
With these optimized conditions in hand, the scope and robustness of the protocol was explored by employing diversely substituted 2-amino benzylamines 2a–j with α-oxoketene dithioacetals 3a–c (Table 2). We observed that amines 2a–g having aliphatic substitution such as methyl, ethyl, n-propyl, isopropyl, n-butyl and tert-butyl at R1 afforded the desired stereoselective products 4a–g, 4l & 4n in good to excellent yields of 80–92% with α-oxoketene dithioacetals 3a–c (Table 2, entries 1–7, 12, 14). However, the same α-oxoketene dithioacetals 3a–c provided the products 4h–k, 4m & 4o–p in comparatively lower yields of 65–71% when amines 2h–j bearing aromatic and cyclic groups such as benzyl, cyclopropyl and cyclohexyl at R1 were used (Table 2, entries 8–11, 13, 15–16). This can be attributed to the lesser reactivity of aromatic and cyclic amines due to the involvement of lone pair of –NH2 in conjugation in the former and attachment of –NH2 group to a sp2 hybridized carbon atom in the later as compared to aliphatic amines.
|
||||
|---|---|---|---|---|
| Entry | Substrate (2) | Amine (3) | Product (4) | Yieldb (%) |
| a Unless otherwise specified, reactions were performed using 1 equiv. of 3a, 1.2 equiv. of 2b, 3.0 equiv. of base.b Isolated yield. | ||||
| 1 | 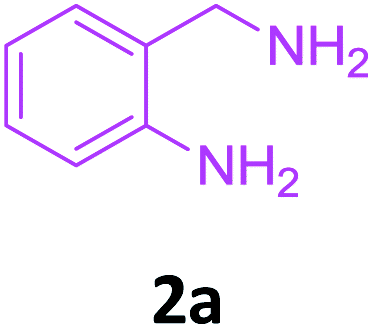 |
 |
 |
80 |
| 2 | 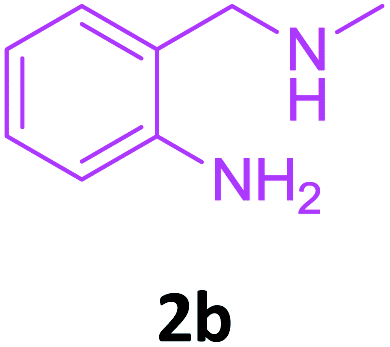 |
3a | 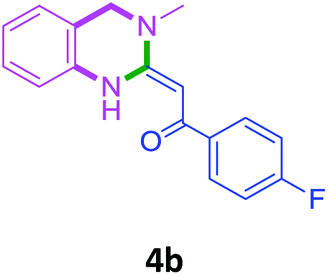 |
90 |
| 3 |  |
3a |  |
88 |
| 4 |  |
3a |  |
85 |
| 5 |  |
3a |  |
82 |
| 6 |  |
3a |  |
92 |
| 7 |  |
3a |  |
86 |
| 8 |  |
3a |  |
70 |
| 9 |  |
3a |  |
71 |
| 10 | 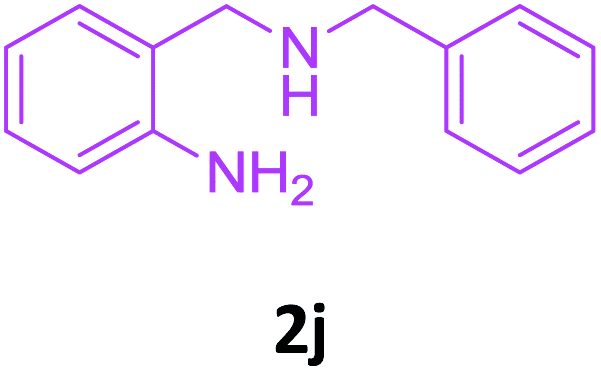 |
3a | 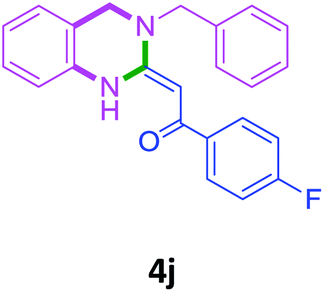 |
68 |
| 11 | 2i | 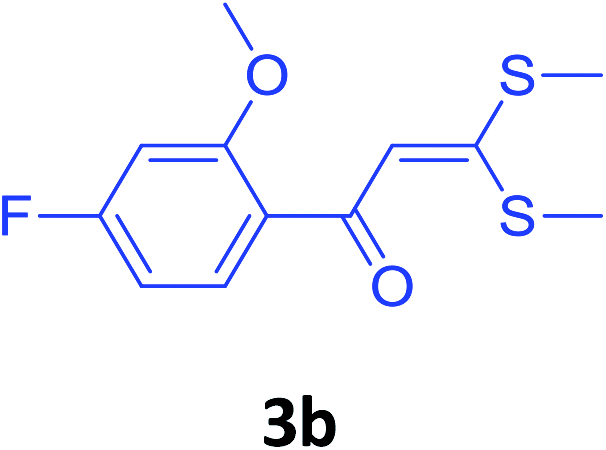 |
 |
65 |
| 12 | 2f | 3b | 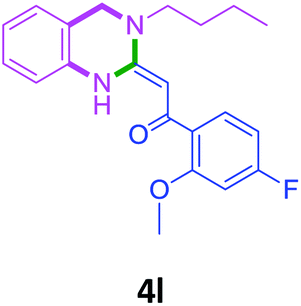 |
82 |
| 13 | 2h | 3b | 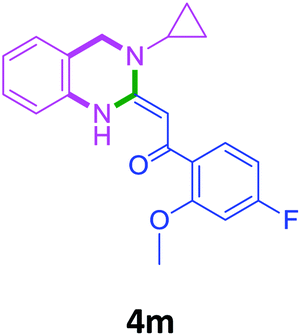 |
70 |
| 14 | 2f | 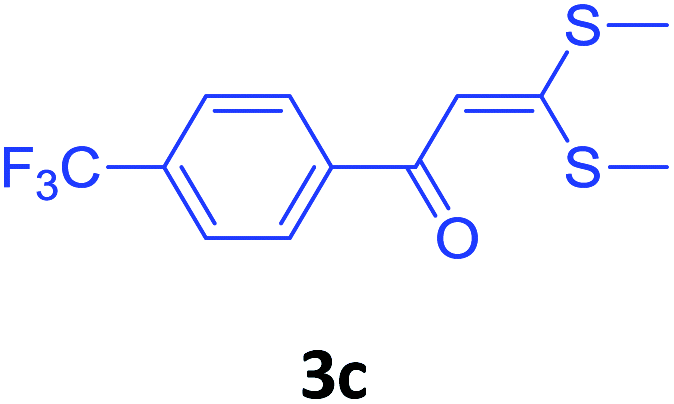 |
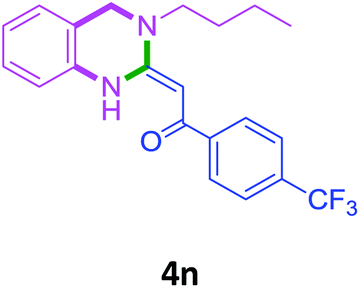 |
80 |
| 15 | 2h | 3c | 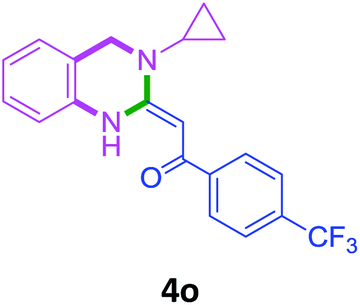 |
65 |
| 16 | 2i | 3c | 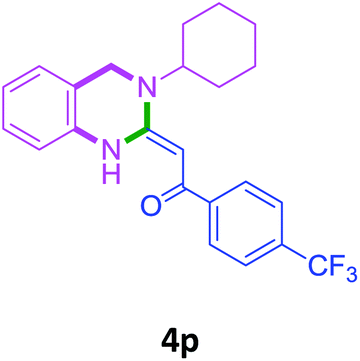 |
66 |
In order to justify sustainable chemistry issues, recovery and reusability of PEG-400 was studied using the reaction of 2g & 3a in the presence of PEG-400 (10 ml) at 110 °C for 6–10 h. After the completion of reaction, reaction mass was then cooled and solvent was evaporated under vacuum. Since PEG is immiscible with diethyl ether, it was washed (two times) with this solvent to remove any unwanted organic impurities and was used successfully in consecutive runs (the yield decreased from 86% to 72% after 5 runs, Table 3), although a weight loss of approximately 5% of PEG was observed from cycle to cycle due to mechanical loss.
| Runs | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Yield (%) | 86 | 84 | 80 | 76 | 72 |
A plausible mechanism for the 3,4-dihydro-2(1H)-quinazoline ring formation is depicted in Scheme 2. The mechanism first involves the substitution of the thiomethyl group of α-oxoketene dithioacetal 3 by that amino group of reactant 2 which is directly attached to aromatic ring, resulting in the formation of intermediate 4A at first.
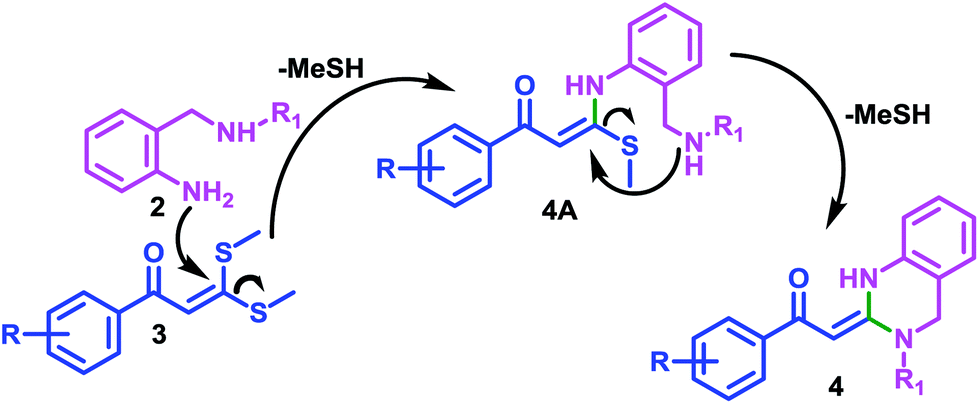 | ||
| Scheme 2 Plausible Mechanism. | ||
This can be assigned to the presence of characteristic chelated proton of –NH at very high chemical shift values (δ 14–15 ppm) in the 1HNMR data of compounds 4(a–p). After that, second thiomethyl group is replaced by second amino group of reactant 2 resulting in formation of cyclized product 4 with subsequent elimination of –MeSH.
Conclusions
In conclusion, we have succeeded in developing first novel, inexpensine, cleaner and green methodology for the synthesis of 3,4-dihydro-2(1H)-quinazolines. Moreover, avoid of work-up make this new strategy attractive, easy to execute and facile for the assembly of a wide variety of biologically relevant dihydroquinazolines. As the methodology is completely devoid of anhydrous solvents or any catalysts, this approach therefore exemplifies the reconciliation of operational simplicity and economic viability in an environmental friendly time and cost effective manner. It is noteworthy that α-oxoketene dithioacetals and substituted 2-amino benzylamines selectively afforded a single isomer. Further applicability of PEG-400 as solvent system to a wide range of substrates towards the development of catalyst free pathways for synthesizing diverse range of biologically important heterocycles is an ongoing goal of research in our laboratory.Experimental
General information and method
All the reactions were performed in an oven-dried Schlenk flask under nitrogen atmosphere. Column chromatography was performed using silica gel (mesh 100–200). TLC analysis was performed on commercially prepared 60 F254 silica gel plates. Visualization of spots on TLC plate was accomplished with UV light (254 nm) and staining over I2 chamber. IR spectra was recorded in CHCl3 on a Perkin Elmer Spectrum RX-1 FT-IR spectrophotometer. CDCl3 was used as NMR solvent for characterization of compounds. The 1H NMR spectra were recorded on a Jeol JNM-ECX400P at 400 MHz. The 13C NMR spectra on Jeol JNM-ECX400P at 100 MHz. Chemical shifts for carbons are reported in ppm from tetramethylsilane and are referenced to the carbon resonance of the solvent. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublet, br s = broad singlet), coupling constants in Hertz, and integration. HRMS (ESI) were recorded with Q-TOF electrospray mass spectrometer. All purchased chemicals were used as received. All melting points are uncorrected.General procedure for synthesis of 2-N-aryl/alkyl substituted anilines (2a–j)
Compounds 2a–j were synthesized by reductive amination20 of 2-nitro benzaldehyde with various aliphatic, aromatic and cyclic amines followed by reduction21 of nitro group using 10% Pd–C/H2 in EtOH at 50 Psi for 2–3 h. Formation of compounds 2a–j was confirmed by 1H NMR, 13C NMR and HRMS data.Analytical data of 2-amino benzylamines
General procedure for the synthesis 3,4-dihydro-1H-quinazolin-2-ylidenes (4a–p)
In an oven-dried two-neck RBF, α-oxoketene dithioacetal 3a–c (1 equiv.), 1.2 equiv. of 2-substituted aniline 2a–j, 3.0 equiv. of K2CO3 was added in 10 mL of PEG-400. Reaction mass was heated at 110 °C for 6–10 h. Progress of reaction was monitored by TLC. The reaction mixture was then cooled and solvent was evaporated under vacuum. Crude was directly purified by column chromatography using silica gel (100:200 mesh) in 5–10% ethyl acetate/hexane as eluent to afford the corresponding product. Since PEG is immiscible with diethyl ether, it was washed (two times) with this solvent to remove any unwanted organic impurities and was used successfully in consecutive runs. Although a weight loss of approximately 5% of PEG was observed from cycle to cycle due to mechanical loss.
Analytical data of 3,4-dihydro-1H-quinazolin-2-ylidenes
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 165.8, 164.2, 135.7, 132.8, 129.5, 129.0, 124.5, 123.8, 119.3, 115.3, 115.1, 79.1 (vinylic CH), 45.7 (–NHCH2); HRMS (ESI) (M + H)+ calcd for C16H13FN2O: 269.1090, found 269.1085.O), 165.2, 162.7, 157.4, 137.5, 134.2, 128.6, 125.4, 122.8, 117.9, 115.3, 115.0, 114.8, 75.9 (vinylic CH), 50.7 (–NCH2), 37.5 (–CH3); HRMS (ESI) (M + H)+ calcd for C17H15FN2O: 283.1246, found 283.1245.O), 165.1, 162.6, 156.7, 137.7, 134.3, 128.6, 128.5, 125.4, 122.8, 117.9, 115.2, 114.9, 114.7, 75.7 (vinylic CH), 48.1 (–NCH2), 45.0 (–CH2CH3), 11.2 (–CH3); HRMS (ESI) (M + H)+ calcd for C18H17FN2O: 297.1403, found 297.1396.O), 165.1, 162.6, 157.1, 137.7, 134.4, 128.6, 125.3, 122.8, 117.9, 115.2, 115.0, 114.7, 75.9 (vinylic CH), 52.1 (–CH2CH2CH3), 49.0 (–NCH2), 19.8 (–CH2CH3), 11.3 (–CH3); HRMS (ESI) (M + H)+ calcd for C19H19FN2O: 311.1559, found 311.1553.O), 166.9, 164.4, 156.5, 135.3, 133.1, 131.7, 131.4, 128.9, 126.8, 126.5, 117.6, 116.1, 115.9, 115.7, 79.0 (vinylic CH), 52.3 (–NCH), 41.5 (–NCH2), 18.6 –CH3); HRMS (ESI) (M + H)+ calcd for C19H19F2O: 311.1559, found 311.1553; ESI-MS (m/z).O), 165.1, 162.6, 157.0, 137.7, 134.5, 128.6, 125.3, 122.8, 117.9, 115.2, 115.0, 114.7, 75.8 (vinylic CH), 50.3 (–CH2CH2CH2CH3), 48.9 (–NCH2), 28.4 (–CH2CH2CH3), 20.1 (–CH2CH3), 13.8 (–CH3); HRMS (ESI) (M + H)+ calcd for C20H21FN2O: 325.1716, found 325.1724.O), 159.0, 137.8, 135.4, 128.6, 124.6, 122.9, 121.6, 115.1, 114.8, 82.4 (vinylic CH), 57.1 (–NCH2), 43.9 (–C(CH3)3), 29.7 (–CH3); HRMS (ESI) (M + H)+ calcd for C20H21FN2O: 325.1716, found 325.1716.O), 165.2, 162.7, 159.2, 137.4, 134.1, 128.7, 125.3, 122.8, 119.2, 115.1, 115.0, 114.8, 77.9 (vinylic CH), 48.7 (–NCH2), 30.5 (–NCH), 9.0 (cyclopropyl CH2); HRMS (ESI) (M + H)+ calcd for C19H17FN2O: 309.1403, found 309.1405.O), 165.1, 162.6, 157.8, 137.9, 134.9, 128.6, 125.2, 122.8, 118.7, 115.0, 114.8, 75.9 (vinylic CH), 56.5 (–NCH), 41.7 (–NCH2), 29.5 (–NCHCH2), 25.7 (–NCHCH2CH2CH2), 25.4 (–NCHCH2CH2CH2); HRMS (ESI) (M + H)+ calcd for C22H23FN2O: 351.1872, found 351.1892.O), 165.2, 162.7, 157.5, 137.4, 134.9, 134.2, 130.3, 129.0, 128.6, 127.9, 126.8, 125.4, 123.0, 117.9, 115.4, 115.0, 114.7, 76.3 (vinylic CH), 48.7 (–NCH2), 44.5 (–NCH2,ring); HRMS (ESI) (M + H)+ calcd for C23H19FN2O: 359.1559, found 359.1583.O), 165.1, 162.7, 158.2, 157.5, 134.9, 131.8, 128.5, 128.1, 125.2, 122.6, 118.6, 115.1, 107.0, 106.7, 99.4, 99.1, 81.0 (vinylic CH), 56.4 (–NCH), 55.8 (Ar–OCH3), 41.6 (–NCH2), 29.5 (–NCHCH2), 25.7 (–NCHCH2CH2CH2), 25.4 (–NCHCH2CH2CH2); HRMS (ESI) (M + H)+ calcd for C21H23FN2O2: 381.1978, found 381.1975.O), 158.1, 156.6, 153.6, 134.5, 131.0, 128.6, 125.2, 122.6, 115.3, 107.0, 106.7, 99.4, 99.1, 80.9 (vinylic CH), 55.8 (Ar–OCH3), 50.4 (–NCH2,ring), 29.6 (–CH2CH2CH2CH3), 20.1 (–CH2CH3), 13.8 (–CH3); HRMS (ESI) (M + H)+ calcd for C21H23FN2O2: 355.1822, found 355.1820.O), 165.2, 162.8, 158.8, 158.3, 134.3, 131.1, 128.5, 127.8, 125.3, 122.6, 119.2, 115.1, 106.9, 99.4, 83.0 (vinylic CH), 55.7 (Ar–OCH3), 48.7 (–NHCH2), 30.5 (–NCH), 8.8 (cyclopropyl CH2); HRMS (ESI) (M + H)+ calcd for C20H19FN2O2: 339.1509, found 339.1498.O), 157.2, 145.1, 134.2, 128.8, 126.8, 125.4, 125.1, 123.1, 117.8, 115.4 (vinylic CH), 50.4 (–NCH2), 49.0 (–CH2CH2CH2CH3), 28.5 (CH2CH2CH3), 20.1 (–CH2CH3), 13.8 (CH3); HRMS (ESI) (M + H)+ calcd for C21H21F3N2O: 375.1684, found 375.1680.O), 159.4, 144.6, 133.9, 128.7, 126.9, 125.4, 125.1, 123.1, 119.1, 115.3, 78.7 (vinylic CH), 48.7 (–NCH2), 30.6 (–NCH), 9.0 (cyclopropyl CH2); HRMS (ESI) (M + H)+ calcd for C20H17F3N2O: 359.1371, found 359.1394.O), 157.9, 145.1, 134.6, 131.5, 131.1, 128.7, 126.8, 125.3, 125.1, 123.1, 118.5, 115.1, 76.9 (vinylic CH), 56.6 (–NCH), 41.8 (–NCH2), 29.5 (–NCHCH2), 25.7 (–NCHCH2CH2CH2), 25.3 (–NCHCH2CH2CH2); HRMS (ESI) (M + H)+ calcd for C23H23F3N2O: 401.1840, found 401.1832.
O), 165.8, 164.2, 135.7, 132.8, 129.5, 129.0, 124.5, 123.8, 119.3, 115.3, 115.1, 79.1 (vinylic CH), 45.7 (–NHCH2); HRMS (ESI) (M + H)+ calcd for C16H13FN2O: 269.1090, found 269.1085.O), 165.2, 162.7, 157.4, 137.5, 134.2, 128.6, 125.4, 122.8, 117.9, 115.3, 115.0, 114.8, 75.9 (vinylic CH), 50.7 (–NCH2), 37.5 (–CH3); HRMS (ESI) (M + H)+ calcd for C17H15FN2O: 283.1246, found 283.1245.O), 165.1, 162.6, 156.7, 137.7, 134.3, 128.6, 128.5, 125.4, 122.8, 117.9, 115.2, 114.9, 114.7, 75.7 (vinylic CH), 48.1 (–NCH2), 45.0 (–CH2CH3), 11.2 (–CH3); HRMS (ESI) (M + H)+ calcd for C18H17FN2O: 297.1403, found 297.1396.O), 165.1, 162.6, 157.1, 137.7, 134.4, 128.6, 125.3, 122.8, 117.9, 115.2, 115.0, 114.7, 75.9 (vinylic CH), 52.1 (–CH2CH2CH3), 49.0 (–NCH2), 19.8 (–CH2CH3), 11.3 (–CH3); HRMS (ESI) (M + H)+ calcd for C19H19FN2O: 311.1559, found 311.1553.O), 166.9, 164.4, 156.5, 135.3, 133.1, 131.7, 131.4, 128.9, 126.8, 126.5, 117.6, 116.1, 115.9, 115.7, 79.0 (vinylic CH), 52.3 (–NCH), 41.5 (–NCH2), 18.6 –CH3); HRMS (ESI) (M + H)+ calcd for C19H19F2O: 311.1559, found 311.1553; ESI-MS (m/z).O), 165.1, 162.6, 157.0, 137.7, 134.5, 128.6, 125.3, 122.8, 117.9, 115.2, 115.0, 114.7, 75.8 (vinylic CH), 50.3 (–CH2CH2CH2CH3), 48.9 (–NCH2), 28.4 (–CH2CH2CH3), 20.1 (–CH2CH3), 13.8 (–CH3); HRMS (ESI) (M + H)+ calcd for C20H21FN2O: 325.1716, found 325.1724.O), 159.0, 137.8, 135.4, 128.6, 124.6, 122.9, 121.6, 115.1, 114.8, 82.4 (vinylic CH), 57.1 (–NCH2), 43.9 (–C(CH3)3), 29.7 (–CH3); HRMS (ESI) (M + H)+ calcd for C20H21FN2O: 325.1716, found 325.1716.O), 165.2, 162.7, 159.2, 137.4, 134.1, 128.7, 125.3, 122.8, 119.2, 115.1, 115.0, 114.8, 77.9 (vinylic CH), 48.7 (–NCH2), 30.5 (–NCH), 9.0 (cyclopropyl CH2); HRMS (ESI) (M + H)+ calcd for C19H17FN2O: 309.1403, found 309.1405.O), 165.1, 162.6, 157.8, 137.9, 134.9, 128.6, 125.2, 122.8, 118.7, 115.0, 114.8, 75.9 (vinylic CH), 56.5 (–NCH), 41.7 (–NCH2), 29.5 (–NCHCH2), 25.7 (–NCHCH2CH2CH2), 25.4 (–NCHCH2CH2CH2); HRMS (ESI) (M + H)+ calcd for C22H23FN2O: 351.1872, found 351.1892.O), 165.2, 162.7, 157.5, 137.4, 134.9, 134.2, 130.3, 129.0, 128.6, 127.9, 126.8, 125.4, 123.0, 117.9, 115.4, 115.0, 114.7, 76.3 (vinylic CH), 48.7 (–NCH2), 44.5 (–NCH2,ring); HRMS (ESI) (M + H)+ calcd for C23H19FN2O: 359.1559, found 359.1583.O), 165.1, 162.7, 158.2, 157.5, 134.9, 131.8, 128.5, 128.1, 125.2, 122.6, 118.6, 115.1, 107.0, 106.7, 99.4, 99.1, 81.0 (vinylic CH), 56.4 (–NCH), 55.8 (Ar–OCH3), 41.6 (–NCH2), 29.5 (–NCHCH2), 25.7 (–NCHCH2CH2CH2), 25.4 (–NCHCH2CH2CH2); HRMS (ESI) (M + H)+ calcd for C21H23FN2O2: 381.1978, found 381.1975.O), 158.1, 156.6, 153.6, 134.5, 131.0, 128.6, 125.2, 122.6, 115.3, 107.0, 106.7, 99.4, 99.1, 80.9 (vinylic CH), 55.8 (Ar–OCH3), 50.4 (–NCH2,ring), 29.6 (–CH2CH2CH2CH3), 20.1 (–CH2CH3), 13.8 (–CH3); HRMS (ESI) (M + H)+ calcd for C21H23FN2O2: 355.1822, found 355.1820.O), 165.2, 162.8, 158.8, 158.3, 134.3, 131.1, 128.5, 127.8, 125.3, 122.6, 119.2, 115.1, 106.9, 99.4, 83.0 (vinylic CH), 55.7 (Ar–OCH3), 48.7 (–NHCH2), 30.5 (–NCH), 8.8 (cyclopropyl CH2); HRMS (ESI) (M + H)+ calcd for C20H19FN2O2: 339.1509, found 339.1498.O), 157.2, 145.1, 134.2, 128.8, 126.8, 125.4, 125.1, 123.1, 117.8, 115.4 (vinylic CH), 50.4 (–NCH2), 49.0 (–CH2CH2CH2CH3), 28.5 (CH2CH2CH3), 20.1 (–CH2CH3), 13.8 (CH3); HRMS (ESI) (M + H)+ calcd for C21H21F3N2O: 375.1684, found 375.1680.O), 159.4, 144.6, 133.9, 128.7, 126.9, 125.4, 125.1, 123.1, 119.1, 115.3, 78.7 (vinylic CH), 48.7 (–NCH2), 30.6 (–NCH), 9.0 (cyclopropyl CH2); HRMS (ESI) (M + H)+ calcd for C20H17F3N2O: 359.1371, found 359.1394.O), 157.9, 145.1, 134.6, 131.5, 131.1, 128.7, 126.8, 125.3, 125.1, 123.1, 118.5, 115.1, 76.9 (vinylic CH), 56.6 (–NCH), 41.8 (–NCH2), 29.5 (–NCHCH2), 25.7 (–NCHCH2CH2CH2), 25.3 (–NCHCH2CH2CH2); HRMS (ESI) (M + H)+ calcd for C23H23F3N2O: 401.1840, found 401.1832.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to SERB, Department of Science & Technology, INDIA for providing financial support and USIC, University of Delhi for providing instrumentation facilities.Notes and references
- (a) H. A. Burris III, Oncologist, 2004, 9, 10 CrossRef; (b) T. P. Selvam and P. V. Kumar, Res. Pharm., 2011, 1, 1 Search PubMed; (c) S. L. Schreiber, Nat. Chem. Biol., 2005, 1, 64 Search PubMed; (d) I. Collins, P. Workman, Nat. Chem. Biol., 2006, 2, 689 Search PubMed; (e) A. Foster, H. A. Coffrey, M. J. Morin and F. Rastinejad, Science, 1999, 286, 2507 CrossRef PubMed.
- (a) P. A. Ple, T. P. Green, L. F. Hennequin, J. Curwen, M. Fennell, J. Allen, C. Lambertvan der Brempt and G. Costello, J. Med. Chem., 2004, 47, 871 CrossRef CAS PubMed; (b) A. M. Alafeefy, A. A. Kadi, O. A. Al-Deeb, K. E. H. El-Tahir and N. A. Al-Jaber, Eur. J. Med. Chem., 2010, 45, 4947 CrossRef CAS PubMed; (c) V. Alagarsamy, V. Muthukumar, N. Pavalarani, P. Vasanthanathan and R. Revathi, Biol. Pharm. Bull., 2003, 26, 557 CrossRef CAS PubMed; (d) K. M. Amin, M. M. Kamel, M. M. Anwar, M. Khedr and Y. M. Syam, Eur. J. Med. Chem., 2010, 45, 2117 CrossRef CAS PubMed.
- (a) A. J. Barker, E.P. Patent, 0635498, 2001; (b) K. Scherlach, H. W. Nuetzmann, V. Schroeckh, H. M. Dahse, A. A. Brakhage and C. Hertweck, Angew. Chem., Int. Ed., 2011, 50, 9843 CrossRef CAS PubMed; (c) P. Thanigaimalai, V. K. Sharma, K. C. Lee, C. Y. Yun, Y. Kim and S. H. Jung, Bioorg. Med. Chem. Lett., 2010, 20, 4771 CrossRef CAS PubMed; (d) L. A. Doyle and D. D. Ross, Oncogene, 2003, 22, 7340 CrossRef PubMed.
- (a) P. M. Chandrika, T. Yakaiah, A. R. Ram Rao, B. Narsaiah, N. C. Reddy, V. Sridhar and J. V. Rao, Eur. J. Med. Chem., 2008, 43, 846 CrossRef CAS PubMed; (b) A. Baba, N. Kawamura, H. Makino, Y. Ohta, S. Taketomi and S. Sohda, J. Med. Chem., 1996, 39, 5176 CrossRef CAS PubMed.
- J. Rosenberg, F. Gustafsson, S. Galatius and P. R. Hilderbrandt, Cardiovasc. Drugs Ther., 2005, 19, 301 CrossRef CAS PubMed.
- V. G. Ugale and S. B. Bari, Eur. J. Med. Chem., 2014, 80, 447 CrossRef CAS PubMed.
- (a) T. Mohamed, M. K. Mann and P. P. N. Rao, RSC Adv., 2017, 7, 22360 RSC; (b) R. Subramaniam, G. Rao and S. P. P. Nagesh, Pharm. Sin., 2011, 2, 146 CAS.
- (a) F. Portela-Cubillo, J. S. Scott and J. C. Walton, Chem. Commun., 2008, 2935 RSC; (b) F. Portela-Cubillo, J. S. Scott and J. C. Walton, J. Org. Chem., 2009, 74, 4934 CrossRef CAS PubMed; (c) Y. P. Patil, P. J. Tambade, K. D. Parghi, R. V. Jayaram and B. M. Bhanage, Catal. Lett., 2009, 133, 201 CrossRef CAS; (d) T. Mizuno, M. Mihara, T. Nakai, T. Iwai and T. Ito, Synthesis, 2007, 2524 CrossRef CAS; (e) R. K. Saunthwal, M. Patel, A. K. Danodia and A. K. Verma, Org. Biomol. Chem., 2015, 13, 1521 RSC.
- (a) Y. Lv, Y. Li, T. Xiong, W. Pu, H. Zhang, K. Sun, Q. Liu and Q. Zhang, Chem. Commun., 2013, 49, 6439 RSC; (b) C. Huang, Y. Fu, H. Fu, Y. Jiang and Y. Zhao, Chem. Commun., 2008, 6333 RSC; (c) R. Rohlmann, T. Stopka, H. Richter and O. G. Mancheño, J. Org. Chem., 2013, 78, 6050 CrossRef CAS PubMed.
- (a) C. Wang, S. Li, H. Liu, Y. Jiang and H. Fu, J. Org. Chem., 2010, 75, 7936 CrossRef CAS PubMed; (b) J. Zhang, D. Zhu, C. Yu, C. Wan and Z. Wang, Org. Lett., 2010, 12, 2841 CrossRef CAS PubMed; (c) L. Jiarong, C. Xian, S. Daxin, M. Shuling, L. Qing, Z. Qi and T. Jianhong, Org. Lett., 2009, 11, 1193 CrossRef PubMed; (d) Z. G. Li, H. Huang, H. B. Sun, H. L. Jiang and H. Liu, J. Comb. Chem., 2008, 10, 484 CrossRef CAS PubMed; (e) C. C. Malakar, A. Baskakova, J. Conrad and U. Beifuss, Chem.–Eur. J., 2012, 18, 8882 CrossRef CAS PubMed.
- (a) J. Fang, J. Zhou and Z. Fang, RSC Adv., 2013, 3, 334 RSC; (b) H. Yuan, W.-J. Yoo, H. Miyamura and S. Kobayashi, Adv. Synth. Catal., 2012, 354, 2899 CrossRef CAS.
- (a) B. Han, X.-L. Yang, C. Wang, Y.-W. Bai, T.-C. Pan, X. Chen and W. Yu, J. Org. Chem., 2012, 77, 1136 CrossRef CAS PubMed; (b) C. U. Maheshwari, G. S. Kumar, M. Venkateshwar, R. A. Kumar, M. L. Kantam and K. R. Reddy, Adv. Synth. Catal., 2010, 352, 341 CrossRef.
- (a) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725 CrossRef CAS PubMed; (b) S. Chitra, N. Paul, S. Muthusbramanian and P. Manisankar, Green Chem., 2011, 13, 2777 RSC; (c) C.-J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS PubMed; (d) U. M. Lindström, Chem. Rev., 2002, 102, 2751 CrossRef; (e) B. Das, P. Jangili, J. Kashanna and R. A. Kumar, Synthesis, 2011, 3267 CrossRef CAS.
- R. Breslow, Acc. Chem. Res., 1991, 24, 159 CrossRef CAS.
- (a) N. R. Modugu and P. Kumar Pittala, New J. Chem., 2017, 41, 14062 RSC; (b) H. Zeng, Q. Tian and H. Shao, Green Chem. Lett. Rev., 2011, 4, 281 CrossRef CAS; (c) R. Kumar, P. Chaudhary, S. Nimesh and R. Chandra, Green Chem., 2006, 8, 356 RSC; (d) S. Chandrasekhar, C. Narsihmulu, S. S. Sultana and N. R. K. Reddy, Org. Lett., 2002, 4, 4399 CrossRef CAS PubMed; (e) R. Ballini, L. Barboni and A. Palmieri, Green Chem., 2008, 10, 1004 RSC; (f) A. Kamal, D. R. Reddy and S. Rajendar, Tetrahedron Lett., 2005, 46, 7951 CrossRef CAS; (g) S. Chandrasekhar, C. Narsihmulu, G. Chandrashekar and T. Shyamsunder, Tetrahedron Lett., 2004, 45, 2421 CrossRef CAS.
- (a) L. C. C. Vieira, M. W. Paixão and A. G. Corrêa, Tetrahedron Lett., 2012, 53, 2715 CrossRef CAS; (b) K. Knop, R. Hoogenboom, D. Fisher and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288 CrossRef CAS PubMed.
- (a) N. Sharma, T. S. Chundawat, S. C. Mohapatra and S. Bhagat, RSC Adv., 2013, 3, 16336 RSC; (b) P. Kumari, N. Sharma, A. Kumar and S. C. Mahopatra, Synlett, 2017 Search PubMed.
- G. Karlstrom and O. Engkvist, in Theory of Poly(ethylene glycol) in Solution, American Chemical Society, 1997, vol. 680, pp. 16–30 Search PubMed.
- O. M. Singh, H. Junjappa and H. Ila, J. Chem. Soc., Perkin Trans. 1, 1997, 1, 3561 RSC.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849 CrossRef CAS PubMed.
- R. H. Bahekar, M. R. Jain, A. Goel, D. N. Patel, V. M. Prajapati, A. A. Gupta, P. A. Jadav and P. R. Patel, Bioorg. Med. Chem., 2007, 15, 3248 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Datas and spectral copies of 1H, 13C NMR for target compounds. X-ray crystallographic data of compound 4o with CCDC No. 1055453. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra13487h |
| This journal is © The Royal Society of Chemistry 2018 |