
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMiR-19b alleviates MPP+-induced neuronal cytotoxicity via targeting the HAPLN4/MAPK pathway in SH-SY5Y cells
Wei Liu†
a,
Lijiao Geng† *b and
Yong Chenb
*b and
Yong Chenb
aDepartment of Neurology, Huaihe Hospital of Henan University, Kaifeng, 475000, China
bDepartment of Rehabilitation Medicine, Huaihe Hospital of Henan University, No. 357 Ximen Street, Kaifeng, 475000, China. E-mail: angelcindtg@yahoo.com; Tel: +86-0371-23906882
First published on 16th March 2018
Abstract
Background: miR-19b has been reported to be involved in nervous system disease including Parkinson's disease (PD). However its molecular basis has not been exhaustively elucidated. Materials and Methods: SH-SY5Y cells were treated with 1-methyl-4-phenylpyridinium (MPP+) to construct PD model in vitro. RT-qPCR was performed to detect the expression of miR-19b and proteoglycan link protein 4 (HAPLN4) mRNA. Western blot analysis was used to measure the level of HAPLN4 and mitogen activated protein kinase (MAPK)-related protein. Cell viability and apoptosis were determined by MTT and flow cytometry. Commercial ELISA kits were applied to quantify caspase-3 activity, lactate dehydrogenase (LDH), reactive oxygen species (ROS), superoxide dismutase (SOD), tumor necrosis factor-α (TNF-α) and interleukin-1 beta (IL-1β). Dual-luciferase reporter assay was applied to assess the relationship between miR-19b and HAPLN4. Results: miR-19b was downregulated in MPP+-induced SH-SY5Y cells. miR-19b overexpression reversed MPP+-induced suppression of cell viability and promotion of cell apoptosis in SH-SY5Y cells. Moreover, miR-19b alleviated MPP+-induced cytotoxicity of SH-SY5Y cells, embodied by the decrease of LDH release, caspase-3 activity, ROS expression, TNF-α and IL-1β secretion, as well as the increase of SOD level. HAPLN4 was identified as a direct target of miR-19b and miR-19b repressed HAPLN4 expression in a post-transcriptional manner. In addition, miR-19b-mediated anti-apoptosis effect was abated following HAPLN4 expression restoration in MPP+-induced SH-SY5Y cells. Furthermore, MAPK signaling participated in miR-19b/HAPLN4-mediated regulation in MPP+-treated SH-SY5Y cells. Conclusion: the neuroprotective effect of miR-19b might be mediated by HAPLN4/MAPK pathway in SH-SY5Y cells.
1. Introduction
Parkinson's disease (PD) is an age-related neurodegenerative disease, characterized by a progressive loss of dopaminergic neurons and iron deposition in the substantia nigra.1 The symptoms of PD include movement disorders such as postural abnormalities, resting tremors, rigidity, and akinesia, and all of which develop as a result of the loss of 50–70% of dopaminergic neurons.2 Increased iron deposition was reported to cause oxidative stress and PD development.3 Moreover, accumulating evidence shows that occurrence of neuroinflammation and activation of apoptotic cascade contribute to the pathogenesis and progression of PD.4 Although many disease-modifying therapies of PD are discovered,5 it is still necessary to explore novel underlying molecular mechanism of PD for making further prevention and treatment.MicroRNAs (miRNAs), a class of small non-coding ribonucleic acids with 18–23 nucleotides (nt) in length, have been pointed to play vital roles in multiple physiological and pathological processes.6 miRNAs, widely expressed in brain tissues, are involved in the development and progression of many neurodegeneration diseases through regulating a series of biological events and cellular functions, including PD.7 For example, Zhou et al.8 demonstrated that miR-7 suppressed Nod-like receptor protein 3 (NLRP3) inflammasome activation and protected dopaminergic (DA) neurons against degeneration in ATP-induced BV2 cells and PD model mice. MiR-22 upregulation enhanced proliferation and survival of 6-hydroxydopamine-induced PC12 cells through targeting transient receptor potential melastatin 7 (TRPM7), indicating the neuroprotective and reversal effects of miR-22 in PD.9 MiR-155 was reported to regulate alpha-synuclein-induced inflammatory responses in models of PD, implicating miR-155 as a potential therapeutic target in PD treatment.10 MiR-19b, a key member of miR-17-92 cluster, is critical for brain development and function via modulating cell proliferation and apoptosis process.11 It was recently documented that dysregulation of miR-19 was implicated in nervous system disease including PD, Alzheimer's disease (AD), neurofibromatosis, spinocerebellar ataxia type 1 and some other neurodegenerative diseases.12 Gui et al.13 found that miR-19b-3p was significantly reduced in the brain of PD patients. Moreover, miR-19 inhibitor promoted apoptosis of aluminum (Al)-induced SH-SY5Y cells, and prevention of miR-19 downregulation by treatment with folic acid protected neuronal cells against Al-induced apoptosis.14 To date, however, the detailed function and molecular mechanism of miR-19b in PD have not yet been well investigated.
The present study aimed to explore the possible role and mechanism of miR-19b in 1-methyl-4-phenylpyridinium (MPP+)-induced SH-SY5Y cells. We demonstrated that miR-19b was downregulated in MPP+-induced SH-SY5Y cells. Moreover, miR-19b overexpression alleviated MPP+-induced cytotoxicity of SH-SY5Y cells. Furthermore, the neuroprotective effect of miR-19b might be mediated by Homo sapiens hyaluronan and proteoglycan link protein 4 (HAPLN4)/mitogen activated protein kinase (MAPK) pathway in SH-SY5Y cells.
2. Materials and methods
2.1. Cell culture and treatment
The SH-SY5Y neuroblastoma cell line was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Then, cells were maintained in Dulbecco's modified Eagle's medium (11995065, DMEM, Thermo Fisher Scientific, Waltham, MD, USA), supplemented with 10% fetal bovine serum (10099141, FBS, Thermo Fisher Scientific) and 1% penicillin/streptomycin (10378016, Thermo Fisher Scientific), in a humid incubator with 5% CO2 at 37 °C.To construct PD model in vitro, SH-SY5Y cells were exposed to different concentrations (0, 1, 2, and 4 mM) of MPP+ (ab144783, Abcam, Cambridge, UK) for 24 h or 2 mM of MPP+ for different treatment times (0, 6, 12, and 24 h), respectively. For flow cytometry, lactate dehydrogenase (LDH) release, caspase 3 activity, superoxide dismutase (SOD)/reactive oxygen species (ROS)/tumor necrosis factor-α (TNF-α)/interleukin-1 beta (IL-1β) levels, and protein levels assay, transfected or untransfected SH-SY5Y cells were treated with 2 mM MPP+ for 12 h.
2.2. Cell transfection
All miRNA mimics (miR-NC, miR-19b mimics), miRNA inhibitors (anti-miR-NC, anti-miR-19b), and siRNAs (si-NC, si-HAPLN4) were purchased from Qiagen (Hilden, Germany). The HAPLN4-overexpression vector (pcDNA-HAPLN4) and pcDNA3.1 empty vectors were purchased from Thermo Fisher Scientific (V87020). Gain-of-function experiments were performed with SH-SY5Y cells transfected with miR-19b mimics or pcDNA-HAPLN4, and loss-of-function experiments were performed with SH-SY5Y cells transfected with anti-miR-19b or si-HAPLN4 using Lipofectamine RNAiMAX Transfection Reagent (13778030, Thermo Fisher Scientific), respectively.2.3. Real-time quantitative PCR
Total RNA was isolated from cells with miRNeasy mini kit (217004, Qiagen). After extraction, cDNA was synthesized using M-MLV reverse transcriptase (Promega, Madison, WI, USA). The expression levels of miR-19b and HAPLN4 were measured by using real-time quantitative PCR (RT-qPCR) with a SYBR Green PCR master mix (A25776, Thermo Fisher Scientific) on a 7900HT fast real-time PCR detection system (4365542, Thermo Fisher Scientific). GAPDH was used as a housekeeping gene, and U6 was used as an internal reference for miRNA. The relative expression was calculated with 2−ΔΔCt method.2.4. Cell viability assay
3-(4,5-Dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was applied to measure cell viability. Briefly, at indicated time point, 20 μl of MTT solution (M6494, Thermo Fisher Scientific) was added to each cell culture medium for 4 h. Then, the medium was discarded, and the formazan crystal was dissolved by adding 200 μl dimethyl sulfoxide (156914, Sigma-Aldrich, St. Louis, MO, USA). The absorbance was measured at 490 nm by using a microplate reader (SpectraMax M5, Molecular Devices, Sunnyvale, CA, USA).2.5. Flow cytometry
Cell apoptosis was measured using flow cytometry with Annexin V-FLUOS Staining (AV-PI) kit (11858777001, Sigma-Aldrich) according to the manufacturer's protocol. Briefly, cells were harvested, washed three times with PBS, and resuspended with 300 μl of buffer containing 5 μl of FITC-conjugated annexin V. Then the mixture was incubated in the dark for 10 min and 5 ml of PI was added. Lastly, cell apoptosis was analyzed by flow cytometry (343098, FACSCalibur, BD Biosciences, Franklin Lakes, NJ, USA) with CellQuest software.2.6. ELISA detection
LDH release was measured with LDH Cytotoxicity Detection kit (11644793001, Sigma-Aldrich) according to the manufacturer's protocol. Briefly, at indicated time point, 50 μl of medium containing the released LDH was added to each cell culture medium and mixed with 50 μl reaction mixture for 30 min. Then, 50 μl of stop solution was added to terminate the reactions. The absorbance was measured using a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA) at 490 and 680 nm.Caspase-3 activity was measured by using the caspase-3 activity kit (E13183, Thermo Fisher Scientific). Briefly, cells were homogenized in reaction buffer and 2 mM caspase-3 substrate. Then, the mixture was incubated for 2 h at 37 °C, following the detection of absorbance at 405 nm using the microplate reader. Similarly, SOD expression, ROS production, TNF-α and IL-1β secretion were measured with the corresponding commercially available ELISA kits (Thermo Fisher Scientific), according to the instructions of manufacturer.
2.7. Dual-luciferase reporter assay
A fragment of 3′-UTR of HAPLN4 containing the target sequence (UUUGCAC) of miR-19b was amplified by PCR, and then the produces were inserted into psiCHECK-2 vectors (C8011, Promega) to construct HAPLN4 wild-type luciferase reporter vector (HAPLN4-WT). The target seed sequence (UUUGCAC) was modified using the QuickChange II Site-Directed Mutagenesis kit (200518, Stratagene, La Jolla, CA, USA) to generate HAPLN4 mutant-type luciferase reporter vector (HAPLN4-Mu). Then, SH-SY5Y cells were transfected with HAPLN4-WT or HAPLN4-Mu alone, or cotransfected with miR-19b mimics or anti-miR-19b for 24 h, following the detection of luciferase activity by SteadyGlo Luciferase Assay System (E2510, Promega).2.8. Western blot assay
Total protein was extracted from cells using RIPA lysis buffer (P0013C, Beyotime, Nantong, China) and the protein concentration was measured using BCA Protein Assay Kit (23225, Thermo Fisher Scientific). An equal amount of protein (50 μg) was separated by 12% SDS-PAGE, and then transferred to PVDF membrane (Millipore, Billerica, MA, USA). After blocked with TBS containing 5% defatted milk and 0.5% Tween-20 for 2 h at room temperature, the PVDF membranes were incubated with anti-HAPLN4 (ab188148, Abcam), anti-p-ERK (ab65142, Abcam), anti-ERK (ab54230, Abcam), anti-p-JNK (ab207477, Abcam), anti-JNK (ab176662, Abcam), anti-p-p38 (4511, Cell signaling Technology, Danvers, MA, USA), anti-p38 (ab170099, Abcam), anti-β-actin (4970, Cell signaling Technology) overnight at 4 °C, followed by incubated with HRP-conjugated secondary antibodies (7074, Cell signaling Technology). Protein blot images were quantized by ImageJ2x software (National Institutes of Health, Bethesda, MD, USA).2.9. Statistical analysis
All data were analyzed by Student's t tests and one-way ANOVAs with SPSS version 13.0 (SPSS Inc., IL, USA). The data in this study were expressed as mean ± standard deviation (SD). P < 0.05 was considered statistically significant.3. Results
3.1. MiR-19b reversed MPP+-induced suppression of cell viability and promotion of apoptosis in SH-SY5Y cells
To investigate the function of miR-19b in PD, RT-qPCR assay was first applied to detect the expression pattern of miR-19b in MPP+-induced SH-SY5Y cells. The data revealed that after being treated with 2 mM MPP+ for 12 h, miR-19b level was strikingly inhibited compared with the control group (Fig. 1A). Further, miR-19b mimics and inhibitors were obtained and then transfected into SH-SY5Y cells to examine its efficiency. As displayed in Fig. 1B, the expression of miR-19b was significantly enhanced after transfection with miR-19b mimics, while miR-19b level was decreased following introduction with anti-miR-19b, compared with the control group in SH-SY5Y cells. Then, gain- and loss-of-function experiments were performed to further explore the effect of miR-19b on the viability of MPP+-induced SH-SY5Y cells. After the transfected SH-SY5Y cells were treated with 0–4 mM MPP+ for 24 h or 2 mM for 0–24 h, cell viability was detected by MTT assay. As expected, MPP+ treatment reduced the cell viability at a dose- or time-dependent manner (Fig. 1C–F). However, MPP+-induced viability suppression at 2 and 4 mM for 24 h (Fig. 1C) or 2 mM for 12 h and 24 h (Fig. 1D) was evidently relieved following miR-19b upregulation. On the contrary, MPP+-induced viability suppression was markedly aggravated by the introduction with anti-miR-19b in SH-SY5Y cells compared with corresponding control (Fig. 1E and F).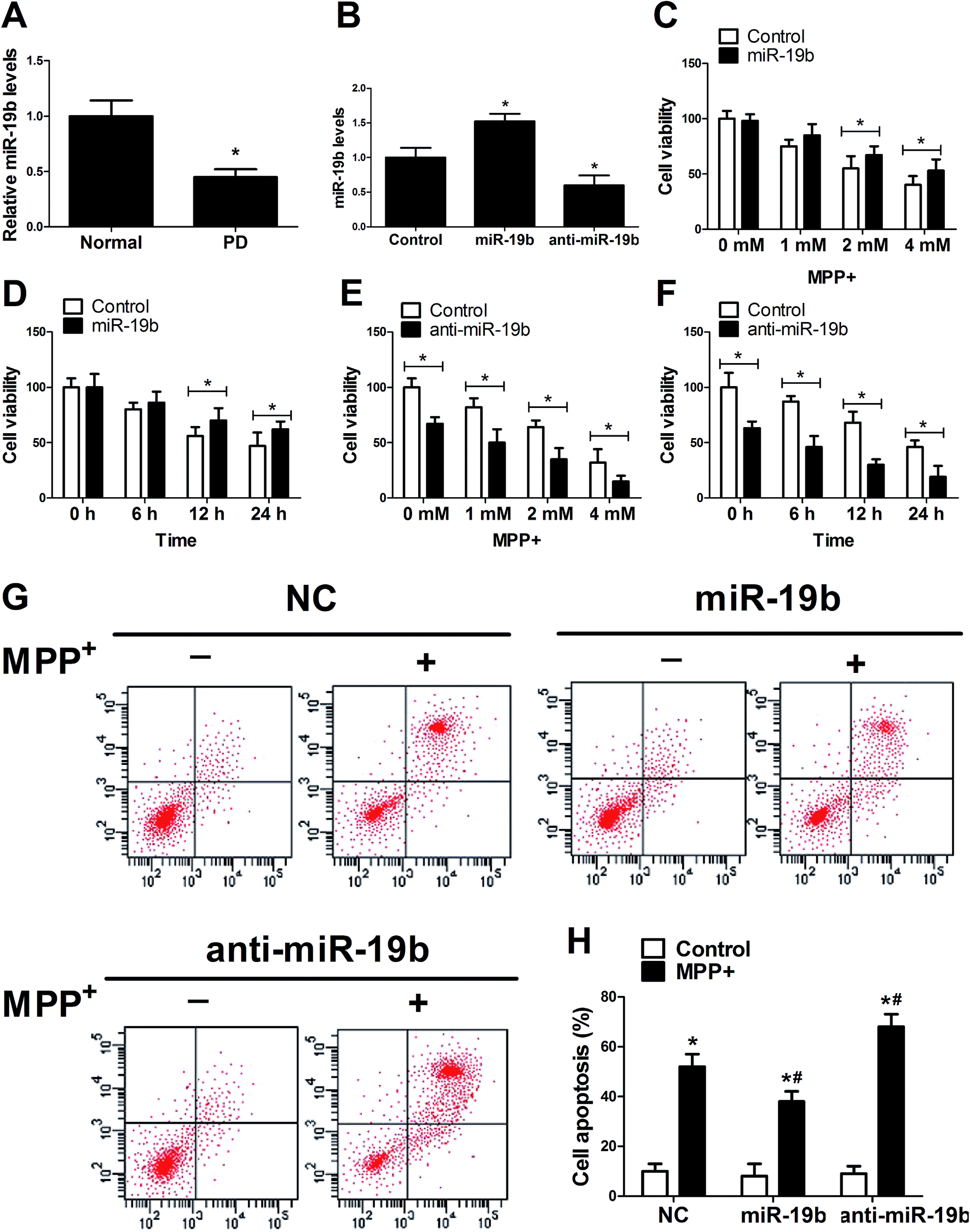 | ||
| Fig. 1 MiR-19 reversed MPP+-induced suppression of cell viability and promotion of cell apoptosis in SH-SY5Y cells. (A) SH-SY5Y cells were treated with 2 mM of MPP+ for 12 h and then the expression pattern of miR-19 was assessed by RT-qPCR assay. (B) SH-SY5Y cells were transfected with miR-19 mimics or anti-miR-19b for 24 h, followed by the detection of miR-19 level. (C–F) SH-SY5Y transfected with miR-19 mimics or anti-miR-19 were treated with various concentrations (0, 1, 2, and 4 mM) of MPP+ for 24 h or with 2 mM of MPP+ for different times (0, 6, 12, and 24 h), followed by the determination of cell viability by MTT assay. (G and H) SH-SY5Y cells transfected with miR-19 mimics or anti-miR-19 were treated with or without 2 mM MPP+ for 12 h, followed by the analysis of cell apoptosis by flow cytometry. *P < 0.05 vs. respective control. | ||
Subsequently, we explored the effect of miR-19b on apoptosis of MPP+-treated SH-SY5Y cells. SH-SY5Y cells transfected with miR-19b mimics or anti-miR-19b were exposed to 2 mM MPP+ for 12 h, followed by the detection of cell apoptosis by flow cytometry. The data indicated that cell apoptosis was remarkably facilitated by the treatment with MPP+. Moreover, MPP+-induced apoptosis was significantly attenuated by the introduction with miR-19b mimics, while miR-19b downregulation strikingly enhanced MPP+-induced apoptosis in SH-SY5Y cells (Fig. 1G and H). All the data suggested that miR-19b played a protective role in MPP+-treated SH-SY5Y cells.
3.2. MiR-19b alleviated MPP+-induced cytotoxicity in SH-SY5Y cells
Activation of apoptotic cascade, oxidative stress and neuroinflammatory response have been reported to be closely associated with the development of PD.15 Hence, to further investigate the effect of miR-19b on PD, SH-SY5Y cells transfected with miR-19b mimics or anti-miR-19b were exposed to 2 mM MPP+ for 12 h, followed by the assessment of LDH release, caspase 3 activity, SOD production, ROS generation, IL-1β level and TNF-α expression. These results displayed that MPP+ treatment led to a dramatic promotion in LDH release (Fig. 2A), caspase 3 activity (Fig. 2B), ROS generation (Fig. 2D), TNF-α (Fig. 2E) and IL-1β (Fig. 2F) expressions, as well as a significant inhibition in SOD production (Fig. 2C). Nevertheless, MPP+-induced cytotoxicity effect was markedly alleviated after miR-19b upregulation, while it was evidently enhanced when introduction with anti-miR-19b in SH-SY5Y cells compared with control group. All these results implied that overexpression of miR-19b alleviated MPP+-induced cell cytotoxicity in SH-SY5Y cells.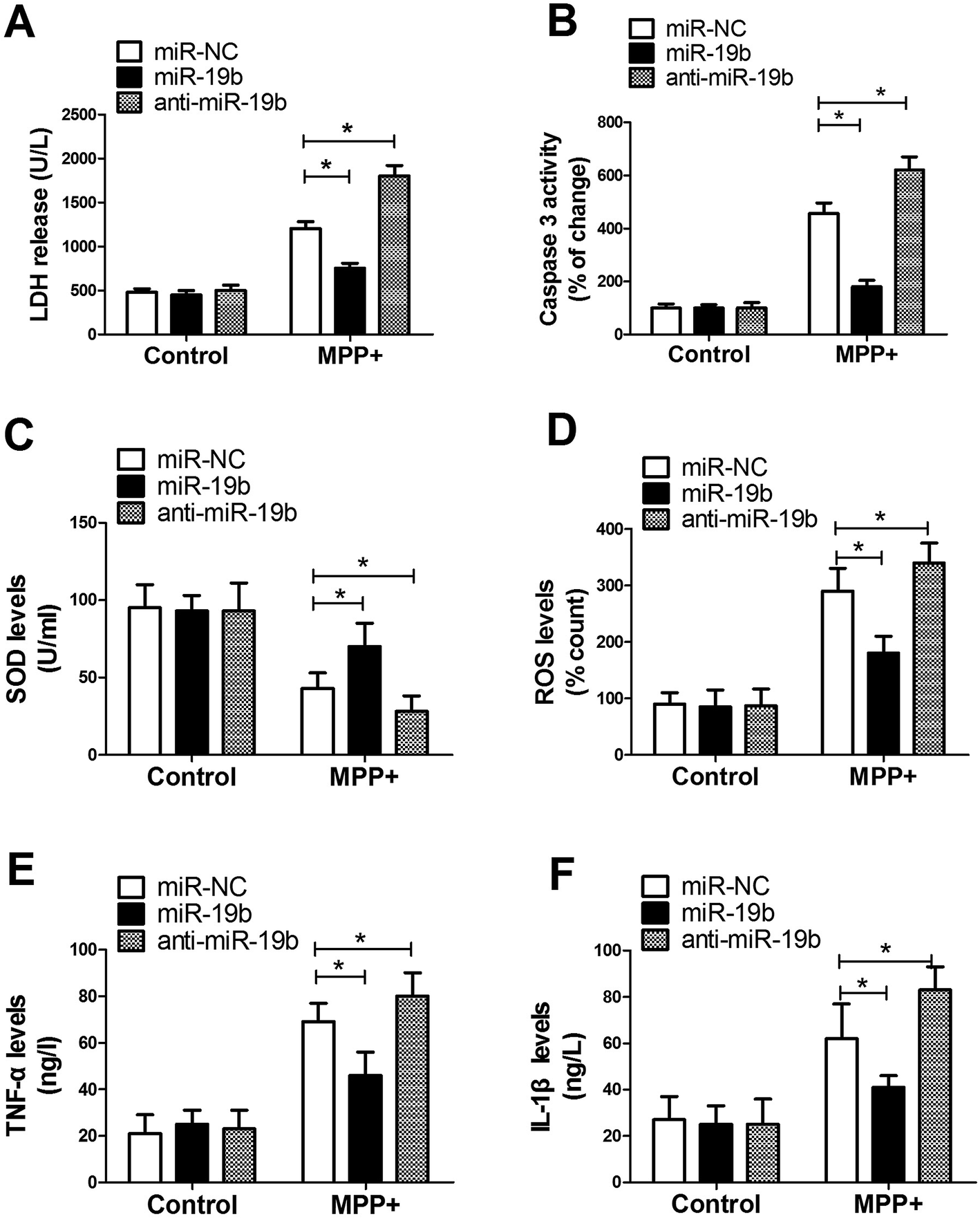 | ||
| Fig. 2 MiR-19 alleviated MPP+-induced cytotoxicity of SH-SY5Y cells. SH-SY5Y cells transfected with miR-19 mimics or anti-miR-19 were treated with or without 2 mM MPP+ for 12 h, followed by the assessment of LDH release (A), caspase 3 activity (B), SOD levels (C), ROS generation (D), TNF-α (E) and IL-1β secretion (F) by ELISA assays. *P < 0.05 vs. corresponding control. | ||
3.3. HAPLN4 was a direct target of miR-19b
To further explore the underlying mechanism of miR-19b in protecting SH-SY5Y cells against MPP+-induced cytotoxicity, the online software algorithms were used to search for the candidate target gene of miR-19b. Interestingly, the complementary sequences of miR-19b were found in the 3′-UTR of HAPLN4 mRNA (Fig. 3A). In order to verify the binding between miR-19b and HAPLN4, dual-luciferase reporter assay was performed in SH-SY5Y cells by transfecting HAPLN4 wild-type luciferase reporter vector (HAPLN4-WT) or HAPLN4 mutant-type luciferase reporter vector (HAPLN4-Mu) alone, or cotransfected with miR-19b mimics or anti-miR-19b. As shown in Fig. 3B, the luciferase activity of HAPLN4-WT was markedly attenuated following miR-19b upregulation, while it was markedly promoted by the introduction of anti-miR-19b. However, mutant of putative sites in HAPLN4 reporter vector had little effect in luciferase activity following miR-19b increase or reduction (Fig. 3B). To further observe the regulatory effect of miR-19b on HAPLN4, miR-19b mimics and anti-miR-19b were transfected into SH-SY5Y cells respectively. RT-qPCR assay revealed that the mRNA level of HAPLN4 was not changed by the introduction with miR-19b mimics or anti-miR-19b when compared with the control group (Fig. 3C). Western blot analysis demonstrated that HAPLN4 expression was significantly repressed by miR-19b upregulation, whereas the level of HAPLN4 was strikingly promoted following miR-19b downregulation (Fig. 3D). These results confirmed miR-19b repressed HAPLN4 expression in a post-transcriptional manner. All the data indicated that HAPLN4 was a direct target of miR-19b in SH-SY5Y cells.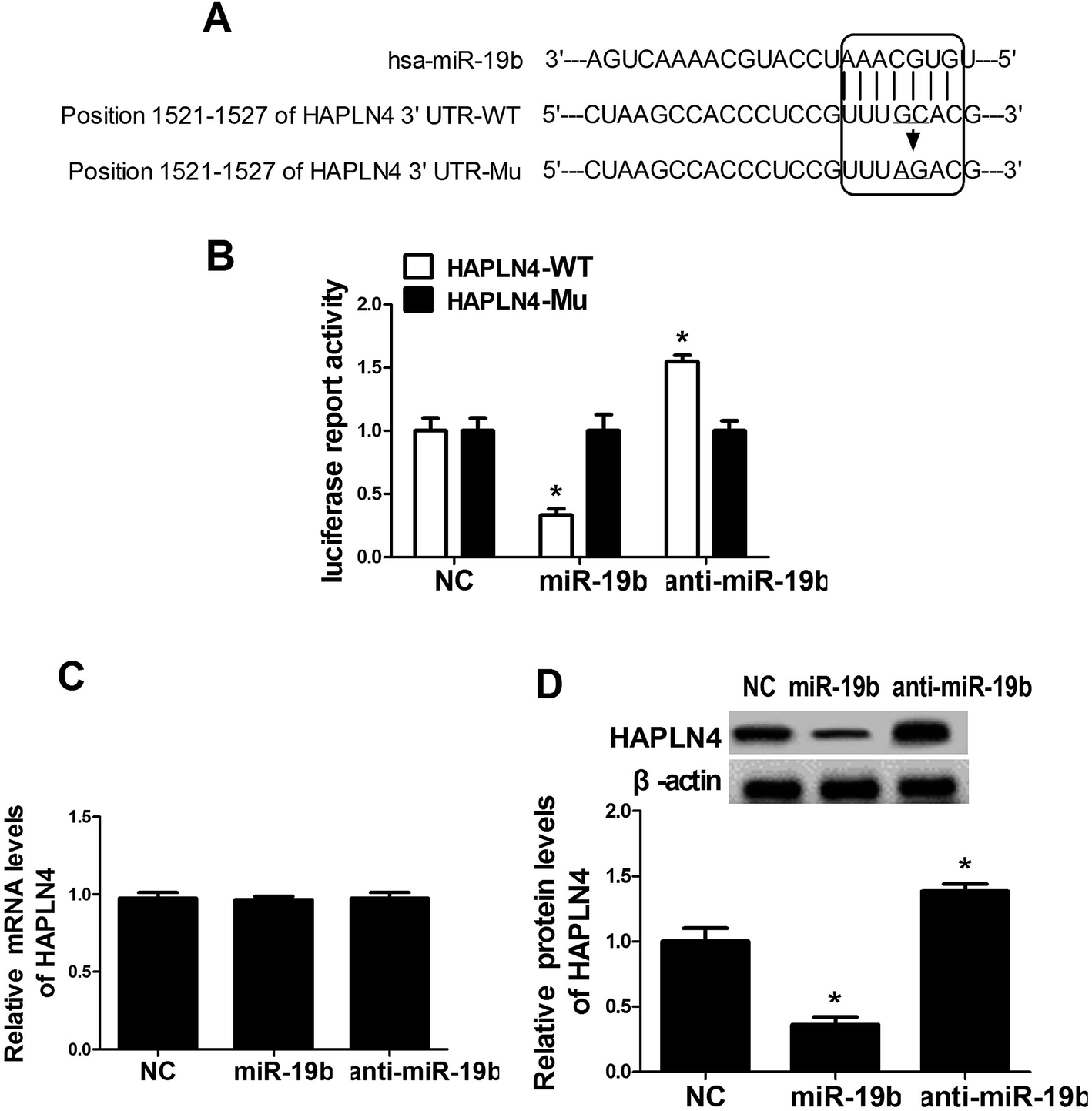 | ||
| Fig. 3 HAPLN4 was a direct target of miR-19. (A) Sequence alignment between miR-19 and the 3′-UTR of HAPLN4. (B) The interplay between miR-19 and HAPLN4 was detected by dual-luciferase reporter assay. HAPLN4 wild-type luciferase reporter plasmid (HAPLN4-WT) and HAPLN4 mutant-type luciferase reporter plasmid (HAPLN4-Mu) were constructed and transfected into SH-SY5Y cells together with miR-19 mimics or anti-miR-19. (C and D) SH-SY5Y cells were transfected with miR-19 mimics or anti-miR-19, followed by the detection of HAPLN4 mRNA level by RT-qPCR assay (C) and HAPLN4 protein expression by western blot with β-actin as internal reference (D). *P < 0.05 vs. respective control. | ||
3.4. MiR-19b-mediated anti-apoptosis effect was abated following HAPLN4 expression restoration in MPP+-induced SH-SY5Y cells
Then, siRNA of HAPLN4 (si-HAPLN4) and HAPLN4 overexpression plasmid (pcDNA-HAPLN4) were transfected into SH-SY5Y cells to regulate HAPLN4 expression. As displayed in Fig. 4A, HAPLN4 expression was significantly repressed by the introduction with si-HAPLN4, while it was strikingly facilitated after transfection with HAPLN4. Further, transfected SH-SY5Y cells were exposed to 2 mM MPP+ for 12 h, followed by the evaluation of cell apoptosis. The data showed that MPP+-elicited apoptosis was strikingly inhibited when HAPLN4 was downregulated, whereas overexpression of HAPLN4 dramatically accelerated MPP+-induced apoptosis in SH-SY5Y cells (Fig. 4B and C). These data indicated that HAPLN4 played vital roles in the pathogenesis of PD. Subsequently, to further explore whether the protection effect of miR-19b was mediated by HAPLN4, SH-SY5Y cells were transfected with miR-19b mimics alone, or in combination with pcDNA-HAPLN4 prior to treatment with MPP+. Western blot analysis displayed that compared with the control group, miR-19b-induced decrease of HAPLN4 expression was dramatically restored by the cotransfection with HAPLN4 (Fig. 4D). Moreover, the restoration of HAPLN4 expression significantly reversed the inhibitory effect of miR-19b on apoptosis effect in MPP+-induced SH-SY5Y cells (Fig. 4E and F). All these results suggested that miR-19b exerted neuroprotective function by targeting HAPLN4 in MPP+-induced SH-SY5Y cells.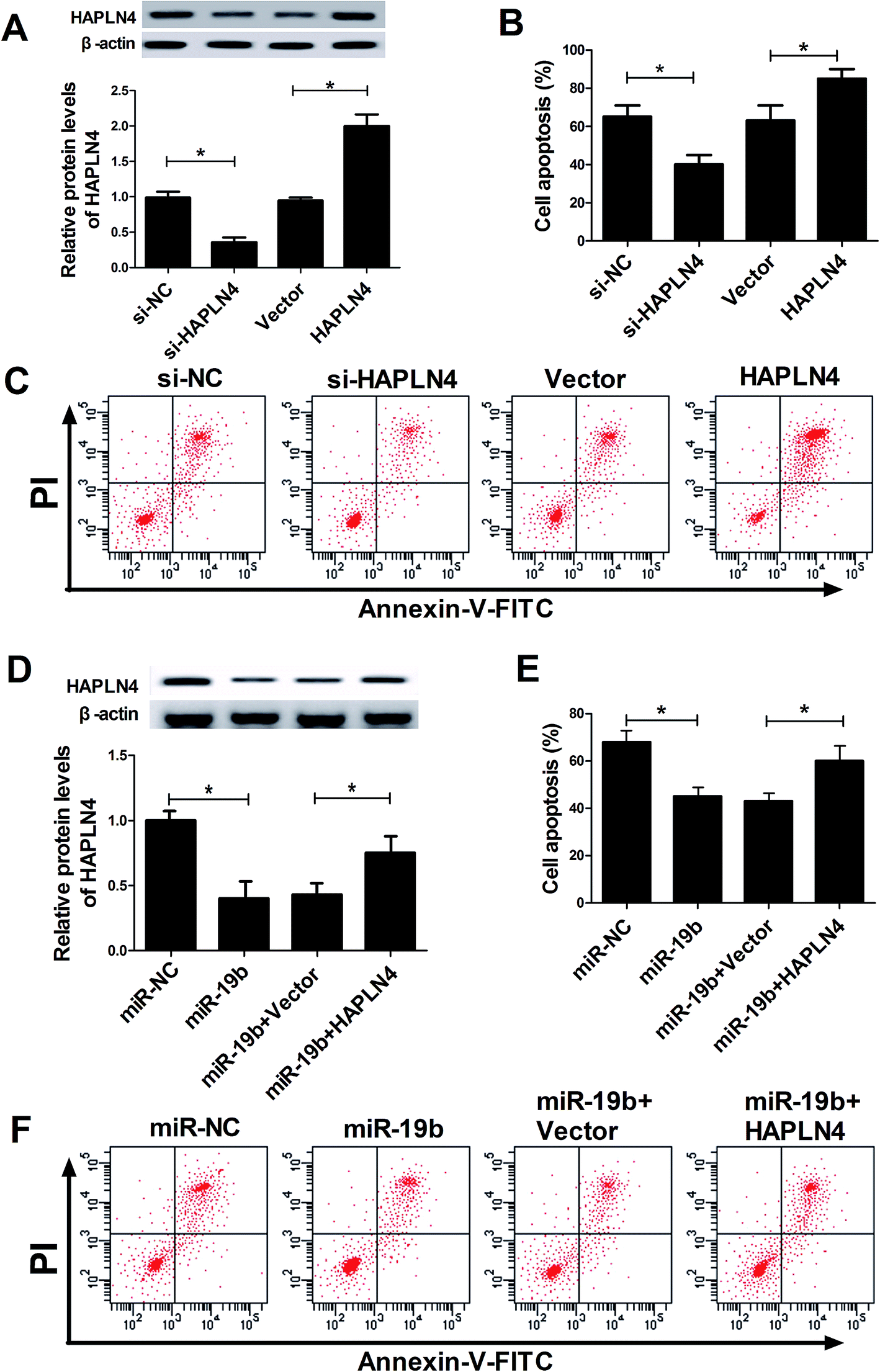 | ||
| Fig. 4 MiR-19-mediacted anti-apoptosis effect was abated following HAPLN4 expression restoration in SH-SY5Y cells. SH-SY5Y cells transfected with si-NC, si-HAPLN4, Vector, or HAPLN4 overexpression plasmid (pcDNA-HAPLN4) were exposed to 2 mM MPP+ for 12 h, followed by the detection of HAPLN4 level (A) and cell apoptosis (B and C). SH-SY5Y cells were transfected with miR-NC, miR-19 mimics, miR-19 mimics + vector, miR-19 mimics + pcDNA-HAPLN4 prior to treatment with MPP+, followed by the determination of HAPLN4 expression (D) and cell apoptosis (E and F). *P < 0.05 vs. corresponding control. | ||
3.5. MAPK signaling pathway was involved in miR-19b/HAPLN4-mediated regulation in MPP+-treated SH-SY5Y cells
MAPK family, consisting of extracellular signal-regulated kinase (ERK), p38 and c-JunNH2-terminal kinase (JNK), are serine–threonine kinases that mediate intracellular signaling associated with a variety of cellular activities including cell proliferation, survival, death and transformation.16 Therefore, we further observed whether miR-19b/HAPLN4 could regulate the MAPK signaling in MPP+-treated SH-SY5Y cells, by analyzing the phosphorylation levels of MAPK signaling molecules after transfected with pcDNA-HAPLN4 and miR-19b mimics + pcDNA-HAPLN4. The data presented that HAPLN4 activated MAPK signaling pathway, revealed by the increase of p-ERK, p-JNK and p-p38 levels following HAPLN4 upregulation (Fig. 5A–D). Whereas, HAPLN4-triggered MAPK signaling was markedly blocked by cotransfection with miR-19b mimics (Fig. 5A–D). All these results hinted that miR-19b abated MPP+-induced injury of SH-SY5Y cells possibly by inactivating MAPK signaling pathway via HAPLN4.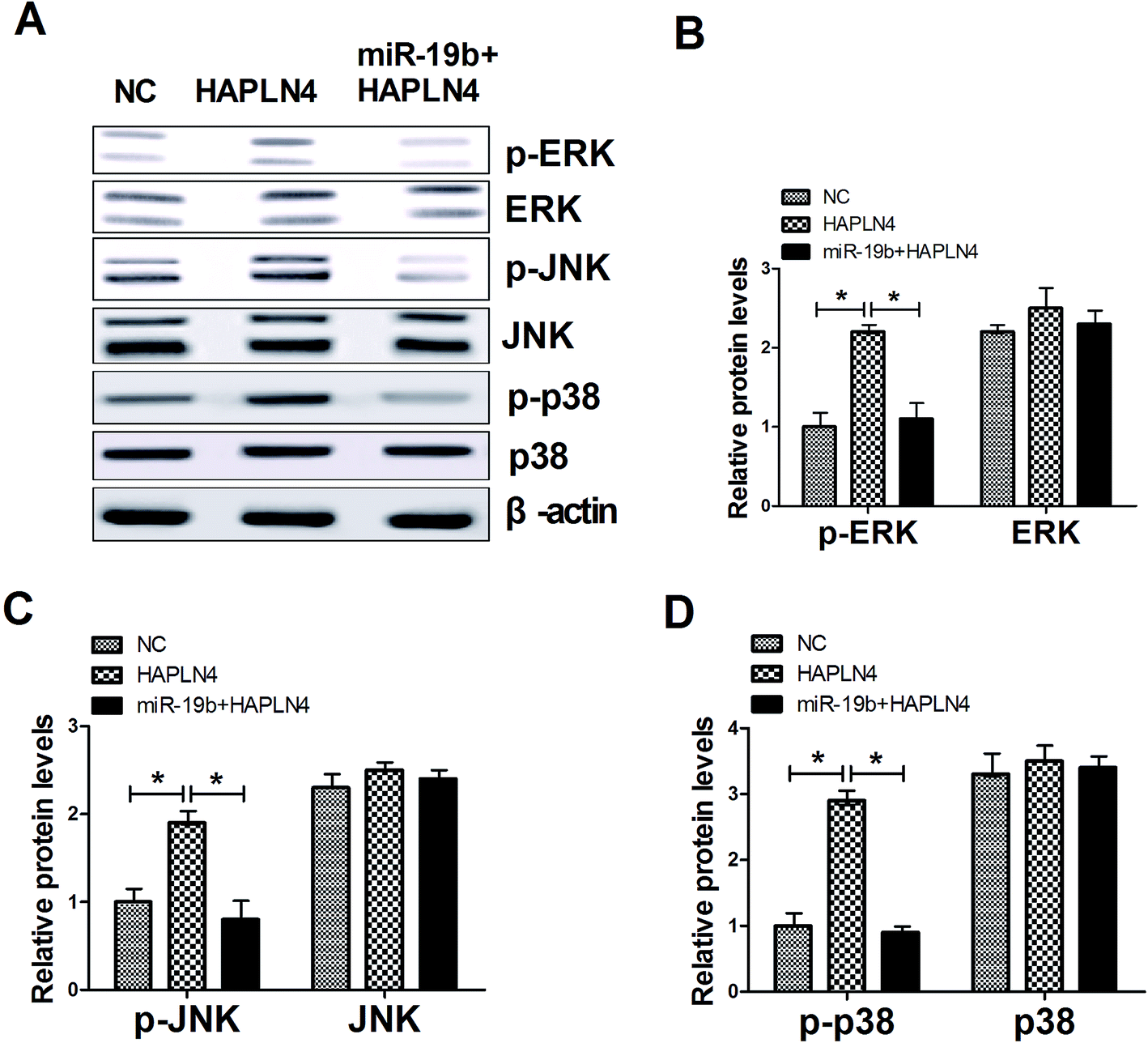 | ||
| Fig. 5 MAPK signaling pathway was involved in miR-19/HAPLN4-mediated regulation in MPP+-treated SH-SY5Y cells. Western blot assay of p-ERK (ERK), p-JNK (JNK) and p-p38 (p38) in MPP+-induced SH-SY5Y cells transfected with pcDNA-HAPLN4 or miR-19 mimics + pcDNA-HAPLN4. β-actin was used as the internal reference (A–D). *P < 0.05 vs. respective control. | ||
4. Discussion
Neuroinflammation, which results primarily from the presence of chronically activated glial cells in the brain, is a common feature in the pathology of PD.17 Oxidative stress may damage dopaminergic neurons and give rise to the symptoms of neurodegenerative diseases including PD.18 Antioxidants such as SOD, as scavenger of ROS and free radicals, are critical for the prevention of PD.19 Neuroinflammatory mediators such as IL-1β and TNF-α, also play vital roles in progression of PD.20 In this study, SH-SY5Y cells were treated with MPP+ to construct the model of PD in vitro. The data showed that MPP+ treatment induced cell injury at a dose- or time-dependent manner. Moreover, in accordance with previous report,21 MPP+ treatment led to a dramatic promotion in LDH release, caspase 3 activity, oxidative stress production and neuroinflammatory response generation in SH-SY5Y cells. Taken together, these results implied that MPP+ treatment induced cell injury in SH-SY5Y cells.MiRNAs, the post-transcriptional gene expression regulators, play key roles in neurodegenerative diseases such as PD.22 For example, Li et al.23 found that miR-7 suppressed neuronal apoptosis through targeting Bax and Sirt2 in MPP+-triggered SH-SY5Y cells. Cho et al.24 demonstrated that miR-205 expression was significantly downregulated in the brains of PD patients, and overexpression of miR-205 might provide an applicable therapeutic strategy through suppressing leucine-rich repeat kinase 2 protein (LRRK2) expression in PD. It has been shown that miR-124 regulated cell apoptosis and autophagy via targeting a BH3-only protein (Bim) in MPTP-treated mice and MPP+-treated SH-SY5Y cells.25 MiR-19b, a key member of the miR-17/92 cluster, is pointed to be involved in a lot of neurodegenerative diseases. For instance, miR-19/miR-101/miR-130 modified the penetrance of spinocerebellar ataxia type 1 via the co-regulation of ataxin 1 levels, providing a new candidate mechanism for modulating the pathogenesis of neurodegenerative diseases.26 A recent report revealed that miR-19 was vital for aluminum (Al)-induced neural cell apoptosis, a key event in the process of neurodegenerative diseases, particularly in AD.27 Also, miR-19a and miR-19b-1 played important roles in the progression of AD via targeting gene phosphatase and tensin homolog (PTEN).12 Our present study demonstrated that overexpression of miR-19b markedly reversed MPP+-induced suppression of cell viability and MPP+-triggered promotion of apoptosis in SH-SY5Y cells. Moreover, miR-19b alleviated MPP+-induced cytotoxicity of SH-SY5Y cells, revealed by the decrease of caspase-3 activity, LDH release, oxidative stress and inflammation response. All these results suggested that miR-19b played a protective role against MPP+-induced cytotoxicity of SH-SY5Y cells.
Subsequently, online software algorithms were applied to search for the candidate target gene of miR-19b. Of interest, the predicted data displayed that HAPLN4 might be a target gene of miR-19b and the complementary sequences of miR-19b were located in the 3′-UTR of HAPLN4. Furthermore, we validated that HAPLN4 was a direct target of miR-19b and miR-19b suppressed HAPLN4 expression by binding to its 3′-UTR in a post-transcriptional manner. HAPLN4, also known as brain-specific link protein (Bral-2), has been pointed to be restricted in expression to the brain/central nervous system and be implicated in multiple brain injury disease.28 In the present study, we found that overexpression of HAPLN4 dramatically accelerated cell apoptosis of MPP+-treated SH-SY5Y cells. Moreover, the restoration of HAPLN4 expression significantly reversed miR-19b-triggered anti-apoptosis effect in MPP+-induced SH-SY5Y cells. Accumulation of misfolded proteins aggregates, failure of protein clearance pathways, mitochondrial damage, oxidative stress, excitotoxicity, neuroinflammation, and genetic mutations have been found to be associated with the apoptosis of neurons.15 Considering the regulatory effects of miR-19b on HAPLN4 expression, we conclude that HAPLN4 enhanced MPP+-induced apoptosis possibly via increasing caspase 3 activity, ROS production and inflammatory responses. However, the detailed molecular mechanisms of HAPLN4-mediated apoptosis in neuronal cells are still need to be explored in our further research. A recent report demonstrated that HAPLN4 showed a high expression following an injury of the brain.28 Also, the mutations or variants of gene HAPLN4 was reported to be associated with the pathophysiological process of essential tremor (ET).29 HAPLN2 and HAPLN4 were the members of the hyaluronan and proteoglycan binding link proteins.30 Similarly, a recent document displayed that HAPLN2 was markedly upregulated in the substantia nigra of PD patients, and overexpression of HAPLN2 increased vulnerability of a dopaminergic cell line (MES23.5 cells), indicating HAPLN2 as a novel factor contributing to neurodegeneration in PD.31 All of these research further confirm the reliability of our results.
MAPK signaling pathway has been demonstrated to lead to a progressive dysfunction in multiple neurodegenerative diseases, such as AD, PD, Huntington's disease, prion disease.32 Our current study revealed that HAPLN4 activated MAPK signaling pathway via enhancing the phosphorylation levels of ERK, JNK and p38, and HAPLN4-triggered MAPK signaling was markedly reversed by regaining of miR-19b expression. All these data implied that miR-19b abated MPP+-induced injury of SH-SY5Y cells by blocking MAPK signaling pathway through targeting HAPLN4. Similar with our findings, Musgrove et al.33 discovered that α-synuclein protected neurons from apoptosis via inhibition of MAPK signaling pathway.
5. Conclusion
In conclusion, miR-19b overexpression alleviated MPP+-induced cytotoxicity via regulating HAPLN4/MAPK signaling pathway in SH-SY5Y cells. This study contributes to the increasing understanding of the role and mechanism of miR-19b in PD, providing a novel biomarker and therapeutic target for PD.Authors' contribution
Lijiao Geng and Wei Liu designed the concept, performed the experiments and prepared the manuscript. Yong Chen analyzed the data and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.References
- J. Pagonabarraga, J. Kulisevsky, A. P. Strafella and P. Krack, Lancet Neurol., 2015, 14, 518–531 CrossRef PubMed.
- A. Antonini, P. Barone, R. Marconi, L. Morgante, S. Zappulla, F. E. Pontieri, S. Ramat, M. G. Ceravolo, G. Meco and G. Cicarelli, J. Neurol., 2012, 259, 2621–2631 CrossRef PubMed.
- T. Jiang, Q. Sun and S. Chen, Prog. Neurobiol., 2016, 147, 1–19 CrossRef CAS PubMed.
- E. C. Hirsch, S. Vyas and S. Hunot, Parkinsonism Relat. Disord., 2012, 18, S210–S212 CrossRef PubMed.
- L. V. Kalia, S. K. Kalia and A. E. Lang, Mov. Disord., 2015, 30, 1442–1450 CrossRef CAS PubMed.
- G. A. Calin and C. M. Croce, J. Clin. Invest., 2007, 117, 2059–2066 CrossRef CAS PubMed.
- D. D. Cao, L. Li and W. Y. Chan, Int. J. Mol. Sci., 2016, 17, E842 CrossRef PubMed.
- Z. Yan, L. Ming, R. H. Du, Q. Chen, C. Y. Jiang, K. Z. Zhang, J. H. Ding and H. Gang, Mol. Neurodegener., 2016, 11, 28 CrossRef PubMed.
- P. Y. Chao, H. Z. Zhen, H. Z. Li and C. R. Han, J. Mol. Neurosci., 2016, 60, 1–8 CrossRef PubMed.
- A. D. Thome, A. S. Harms, L. A. Volpicellidaley and D. G. Standaert, J. Neurosci., 2016, 36, 2383–2390 CrossRef CAS PubMed.
- J. Han, H. J. Kim, S. T. Schafer, A. Paquola, G. D. Clemenson, T. Toda, J. Oh, A. R. Pankonin, B. S. Lee and S. T. Johnston, Neuron, 2016, 91, 79–89 CrossRef CAS PubMed.
- E. Mogilyansky and I. Rigoutsos, Cell Death Differ., 2013, 20, 1603–1614 CrossRef CAS PubMed.
- Y. X. Gui, H. Liu, L. S. Zhang, W. Lv and X. Y. Hu, OncoTargets Ther., 2015, 6, 37043–37053 Search PubMed.
- M. Zhu, B. Li, X. Ma, C. Huang, R. Wu, W. Zhu, X. Li, Z. Liang, F. Deng and J. Zhu, Neurochem. Res., 2016, 41, 2110–2118 CrossRef CAS PubMed.
- P. Maiti, J. Manna and G. L. Dunbar, Transl. Neurodegener., 2017, 6, 28 CrossRef PubMed.
- A. S. Dhillon, S. Hagan, O. Rath and W. Kolch, Oncogene, 2007, 26, 3279–3290 CrossRef CAS PubMed.
- L. Qian, P. M. Flood and J. S. Hong, J. Neural Transm., 2010, 117, 971–979 CrossRef CAS PubMed.
- A. Sarrafchi, M. Bahmani, H. Shirzad and M. Rafieiankopaei, Curr. Pharm. Des., 2016, 22, 238–246 CrossRef PubMed.
- X. X. Xie, S. T. Kou, Z. H. Pu, C. Y. Hou and Y. P. Tian, Zhongguo Zhenjiu, 2007, 27, 753–756 CAS.
- T. Nagatsu and M. Sawada, Curr. Pharm. Des., 2005, 11, 999–1016 CrossRef CAS PubMed.
- P. Gong, F. Deng, W. Zhang, J. Ji, J. Liu, Y. Sun and J. Hu, Exp. Ther. Med., 2017, 14, 4431–4437 Search PubMed.
- S. S. Hébert and S. B. De, Trends Neurosci., 2009, 32, 199–206 CrossRef PubMed.
- S. Li, X. Lv, K. Zhai, R. Xu, Y. Zhang, S. Zhao, X. Qin, L. Yin and J. Lou, Am. J. Transl. Res., 2016, 8, 993–1004 Search PubMed.
- H. J. Cho, G. Liu, S. M. Jin, L. Parisiadou, C. Xie, J. Yu, L. Sun, B. Ma, J. Ding and R. Vancraenenbroeck, Hum. Mol. Genet., 2013, 22, 608–620 CrossRef CAS PubMed.
- H. Wang, Y. Ye, Z. Zhu, L. Mo, C. Lin, Q. Wang, H. Wang, X. Gong, X. He and G. Lu, Brain Pathol., 2016, 26, 167–176 CrossRef CAS PubMed.
- Y. Lee, R. C. Samaco, J. R. Gatchel, C. Thaller, H. T. Orr and H. Y. Zoghbi, Nat. Neurosci., 2008, 11, 1137–1139 CrossRef CAS PubMed.
- M. Zhu, C. Huang, X. Ma, R. Wu, W. Zhu, X. Li, Z. Liang, F. Deng, J. Zhu and W. Xie, J. Alzheimer's Dis., 2016, 50, 1149–1162 CrossRef CAS PubMed.
- A. P. Mann, P. Scodeller, S. Hussain, J. Joo, E. Kwon, G. B. Braun, T. Mölder, Z. G. She, V. R. Kotamraju and B. Ranscht, Nat. Commun., 2016, 7, 11980 CrossRef CAS PubMed.
- X. Liu, N. Hernandez, S. Kisselev, A. Floratos, A. Sawle, I. Ionitalaza, R. Ottman, E. D. Louis and L. N. Clark, Eur. J. Hum. Genet., 2015, 24, 1009–1015 Search PubMed.
- A. P. Spicer, A. Joo and B. R. Jr, J. Biol. Chem., 2003, 278, 21083–21091 CrossRef CAS PubMed.
- Q. Wang, Q. Zhou, S. Zhang, W. Shao, Y. Yin, Y. Li, J. Hou, X. Zhang, Y. Guo and X. Wang, Front. Aging Neurosci., 2016, 8, 197 Search PubMed.
- E. K. Kim and E. J. Choi, Biochim. Biophys. Acta, 2010, 1802, 396–405 CrossRef CAS PubMed.
- R. E. Musgrove, A. E. King and T. C. Dickson, Neurotoxic. Res., 2013, 23, 358–369 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |