
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation and properties of a novel covalently bonded energetic boron powder and its composite
Baohui Zheng ,
Tao Liu,
Dayuan Gao,
Qing Zhu,
Shangbin Li,
Guan Luo* and
Xia Gao
,
Tao Liu,
Dayuan Gao,
Qing Zhu,
Shangbin Li,
Guan Luo* and
Xia Gao
Institute of Chemical Materials, China Academy of Engineering Physics, Mianyang 621900, China. E-mail: luoguan@caep.cn; Tel: +86-816-2487915
First published on 22nd March 2018
Abstract
Promoting the ignition of boron powders in propellants, explosives and pyrotechnics has been a promising research direction. In this paper, a new strategy for covalently bonded energetic boron powders was designed. Specifically, 2,4-toluene diisocyanate (TDI) and 3-amino-1,2,4-triazole (ATZ) were used as grafting molecules, and then acidification, carbamation and ureylene addition were performed serially on the surface of the boron particles. The reaction conditions were optimized using infrared (IR) spectroscopy and X-ray photoelectron spectroscopy (XPS). At lower temperatures, the addition of carbamation or ureylene can improve the yield and stability of isocyanate and urea groups. The chemical composition, microstructure and surface properties of the boron powders were analyzed with nuclear magnetic resonance (NMR) spectroscopy, scanning electron microscopy (SEM), transmission electron microscopy (TEM) and contact angle (CA) analysis, respectively. The covalent bonding type was confirmed by the typical peaks at 155.7 and 157.5 ppm in the 13C NMR spectra of the intermediate isocyanate-grafting boron powders (TB) and target product triazole-grafting boron powders (TTB). The static water contact angles on the surfaces of TB and TTB were 148.3° and 37.0°, respectively. Influences of surface modification of boron powders on the rheological properties of boron/hydroxyl-terminated polybutadiene (HTPB) composites were investigated. Moreover, the prepared samples were characterized by thermogravimetry (TG) and differential scanning calorimetry (DSC) to investigate the thermal stability and reaction activity, and the results showed that this grafting strategy could significantly reduce the critical reaction temperature of B/KNO3. Consequently, it is anticipated that the modified boron powders can potentially be used in propellants, explosives and pyrotechnics with high impulses.
1. Introduction
The improvement of impulse properties, which leads to greater cratering and fragmentation effects, requires the addition of metal or metal-like powders in propellants, explosives and pyrotechnics.1 A variety of reactive metal or metal-like powders (Al, B, Mg and Zr) have been utilized, such as boron-based solid propellants,2 aluminum-based high energy explosives3 and ignition powders containing boron, magnesium and zirconium.4 Among them, boron has higher gravimetric and volumetric heat in comparison with aluminum and many other metal fuels.5 Boron has a diverse range of industrial applications in areas such as semiconductors, protective coatings, pyrotechnic flares, high-density fuels, and refractory materials.6 Research has shown that compared with aluminum-containing compositions, such as Al/octogen (HMX) and hexogen (RDX)/Al/ammonium perchlorate (AP), boron-containing ones, B/HMX and RDX/Al/B/AP, have a higher energy.7 This indicates that boron is a potential candidate for use as a fuel additive in almost all energetic materials. Nevertheless, the elemental boron particles have a tight coating layer in the form of oxides and hydrates, which reduces the performance as a fuel and leads to inefficient burning.8 Moreover, it is extremely difficult for the elemental boron particle to efficiently burn in explosives due to energetic materials such as RDX and HMX having only nanoseconds to detonate, which works against the heat and mass transfer.9Many methods have attempted to promote the ignition and combustion of boron powders, including ultrafine processing,10 adding easily combustible metal powders (Li, Mg, Ti and Zr),11 coating with boron carbide (BC), silicane, lithium fluoride (LiF), glycidyl azide polymer (GAP), and others.12 In general, easily combustible metal powders also possess more active property, which results in potential process complexity and security hazards in the preparation and usage of propellants, explosives and pyrotechnics. In addition, the outermost shell is usually separated from the boron core using the adopted coating method, and the particle properties tightly depend on the integrity and structure strength of the coating layer. In view of the above, an intensive study of covalent bonding and chemically stable boron powders is absolutely imperative.
Currently, covalent bonds linking many kinds of metals (Al),13 metal oxides (SiO2,14 Al2O3 (ref. 15)) and metal nitrides (Si3N4,16 AlN (ref. 17)) have been successfully created. Through a similar measurement, the acid reactive sites on the surface of boron particles can be utilized as the covalent bonding reactive center. It has been reported that surface hydroxyl groups of the boron powders could react with maleic anhydride (MAH),18 undecylenic acid19 and fatty alcohol.20 Thus, through the method of organically modifying the boron surface with some small energetic molecule, it is possible to enable the boron powders with improved ignition properties.
In this article, a novel covalently bonded, energetic boron powder was prepared by acidification, carbamation and ureylene addition reactions. Among the grafting molecules, ATZ is used as the energetic gas generator.21 After optimizing the reaction conditions, the properties of the modified boron powders and their composites with potassium nitrate (KNO3) were investigated by infrared (IR) spectroscopy, X-ray photoelectron spectroscopy (XPS), solid-state nuclear magnetic resonance (NMR) spectroscopy, scanning electron microscopy (SEM), transmission electron microscopy (TEM), contact angle (CA) analysis, thermogravimetry (TG), and differential scanning calorimetry (DSC).
2. Experimental section
2.1 Materials
Elemental boron powder (99 wt%) was purchased from Baoding Pengda Chemical Industry Co., Ltd. (Baoding, China). Nitric acid (65–68 wt%), dibutyltin dilaurate (DBTL), 2,4-toluene diisocyanate, 3-amino-1,2,4-triazole and potassium nitrate were of analytical purity and obtained from Sinopharm Chemical Reagent Co., Ltd. Acetone containing water at 0.3 wt% and other reagents used in the presented study were commercially purchased.2.2 Characterization
Infrared (IR) spectra were collected with a Nicolet 6700 FT-IR spectrophotometer using KBr pellets. X-ray photoelectron spectroscopy (XPS) analysis was conducted with a VG ESCALAB 250 instrument using Mg-Kα radiation at 200 W. Solid-state nuclear magnetic resonance spectroscopy (NMR) tests were performed on a Brucker Advance III 400 MHz. Scanning electron microscopy (SEM) measurements were performed with a ZEISS Ultra 55 scanning electron microscope at an operating voltage of 20 kV. Transmission electron microscopy (TEM) analyses were performed on a Zeiss Libra 200 FE (Germany) TEM, with an accelerating voltage of 200 kV. The particle size distribution was characterized using a Beckman-Coulter LS 230 laser particle size analyzer. The rheological parameters of B/HTPB were characterized using an Anton Paar MCR 102 rheometer and parallel plate geometry (PP25, plate diameter 24.978 mm, plate distance 1 mm). Thermogravimetry (TG) curves were recorded with a TG-DTA 6200 LAB SYS thermal gravimetric analyzer, and differential scanning calorimetry (DSC) was recorded with a TA Q200 instrument from room temperature to 1200 °C in a nitrogen atmosphere at a heating rate of 20 °C min−1.2.3 Preparation of covalent bonded energetic boron powders
Grafting on the surface of boron particles was conducted using consecutive condensation reactions between –OH, –NH– and –NCO groups, as shown in Scheme 1. To endow more hydroxyl groups on the surface of the boron particles, the raw boron particles were treated with concentrated nitric acid in the solvent acetone. The secondary amine in the triazole of ATZ is more active than the primary amine, which was proved by numerous researches, especially in the reaction between 3-amino-1,2,4-triazole or its analogs and isocyanates or isothiocyanates.22,23 Thus, the intermediate TB reacted with 1 equiv of ATZ and generated the target material TTB with a structure as shown in Scheme 1. In all three steps, the purification of boron powders was only conducted with filtration in this conveniently synthetic route.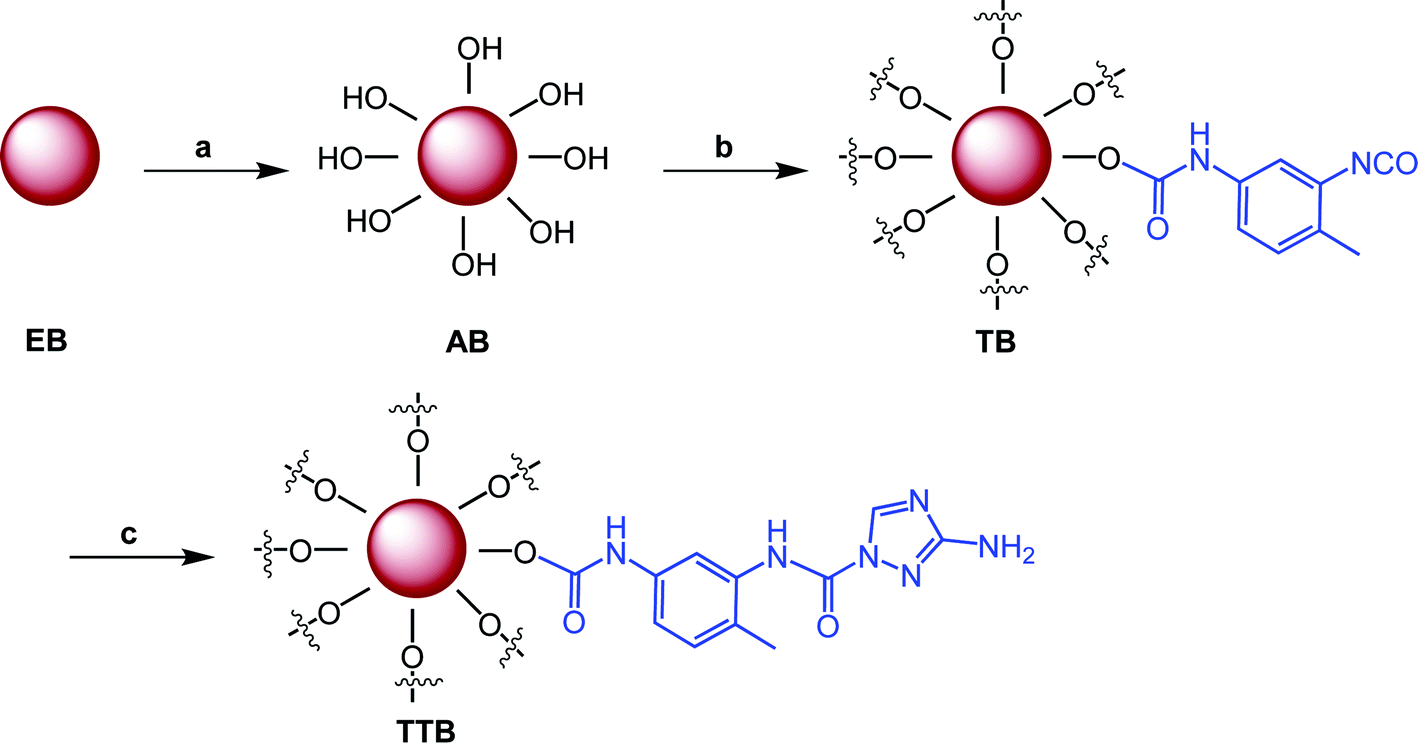 | ||
| Scheme 1 Synthetic route of the target material with the conditions: (a) HNO3, acetone, r.t., 24 h; (b) TDI, toluene, DBTL, 50 °C, 5 h; and (c) ATZ, acetonitrile, 40 °C, 5 h. | ||
In a typical experiment, 20 g of boron powder and 200 mL of acetone were placed in a round-bottom flask. After 1.5 mL of nitric acid was added dropwise, the mixture was stirred for another 24 h at room temperature. The resultant mixture was filtered and washed twice with 150 mL acetone, and then approximately 20 g of the acidified boron powder (AB) was obtained after vacuum drying overnight at room temperature.
Then, 2.0 g acidified boron powder (AB) was dispersed with 200 mL anhydrous toluene in a three-neck flask equipped with a condenser and a magnetic stirrer. After that, 0.73 g of TDI and two drops of DBTL were added into the reaction mixture and left to stir at 50 °C for 5 h. Finally, the mixture was cooled to room temperature, and treated by filtration through Buchner funnel. The residue was washed with 150 mL ethyl acetate three times. Approximately 2.20 g TDI-grafting boron powders (TB) were obtained after vacuum drying overnight at room temperature.
TB (0.50 g), ATZ (0.12 g), two drops of DBTL and 50 mL anhydrous acetonitrile were placed in a three-neck flask equipped with a condenser. The reaction was carried out under vigorous stirring at 40 °C for 5 h. The mixture was filtered during the warm state, and the residue was washed with 60 mL ethyl alcohol three times. Then, it was dried under vacuum overnight at room temperature to give approximately 0.48 g of further ureylene-based product TTB.
2.4 Preparation of B/KNO3 composites
First, 0.50 g of boron powder, 0.75 g of potassium nitrate and an additional 0.5 wt% phenol formaldehyde resin were mixed in a weight ratio of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60:0.5 with 20 mL ethyl alcohol in a beaker. After ultrasonic processing for 20 minutes, the mixture was evaporated at 50 °C while stirring for 5 h. Then, the sample was vacuum dried overnight at room temperature, and the B/KNO3 composites were obtained.
:60:0.5 with 20 mL ethyl alcohol in a beaker. After ultrasonic processing for 20 minutes, the mixture was evaporated at 50 °C while stirring for 5 h. Then, the sample was vacuum dried overnight at room temperature, and the B/KNO3 composites were obtained.
3. Results and discussion
3.1 Preparation of TB and TTB
The hydroxyl and isocyanate groups are crucial reactive sites for the chemical bonding of TDI and ATZ. To confirm that the modification of the boron powders is successful, IR spectra analyses of the samples, including the raw material EB, intermediates AB and TB, and the target material TTB, were performed, as shown in Fig. 1. According to the above test results, the following can be observed: (1) during the acidification process, the absorption at 3440 cm−1 in the IR spectrum of EB corresponding to the B–B weakened, and the 3410 cm−1 band caused by O–H appeared possibly due to the surface B2O3 reacting with water in the nitric acid and acetone solvent. (2) After the carbamation of AB at 50 °C for 5 h, the reactive site –OH reacted with the –NCO group from the TDI molecule, generating a new group –NH–COO– and an unreacted –NCO group. Therefore, in the IR spectrum of TB, the band at 3410 cm−1 caused by stretching vibrations of the O–H disappeared, and a prominent peak at 2276 cm−1 caused by antisymmetric stretching vibrations of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO appeared.22 The bands at 3305, 1647, and 1499–1618 cm−1 could be attributed to the stretching vibrations of N–H, CO in –NH–COO– group and the benzene framework vibration, respectively.22 (3) In the last ureylene addition step at 40 °C for 5 h, the active sites of NCO in the TB reacted with the –NH– group in the triazole ring of the ATZ, generating a urea group –NH–CO–N
CO appeared.22 The bands at 3305, 1647, and 1499–1618 cm−1 could be attributed to the stretching vibrations of N–H, CO in –NH–COO– group and the benzene framework vibration, respectively.22 (3) In the last ureylene addition step at 40 °C for 5 h, the active sites of NCO in the TB reacted with the –NH– group in the triazole ring of the ATZ, generating a urea group –NH–CO–N![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) . In the IR spectrum of the TTB, the peak at 2276 cm−1 corresponding to the NCO disappeared, and the emerged peaks at 3364 and 1740 cm−1 could be attributed to the stretching vibrations of N–H and CO in the –NH–CO–N group, respectively.
. In the IR spectrum of the TTB, the peak at 2276 cm−1 corresponding to the NCO disappeared, and the emerged peaks at 3364 and 1740 cm−1 could be attributed to the stretching vibrations of N–H and CO in the –NH–CO–N group, respectively.
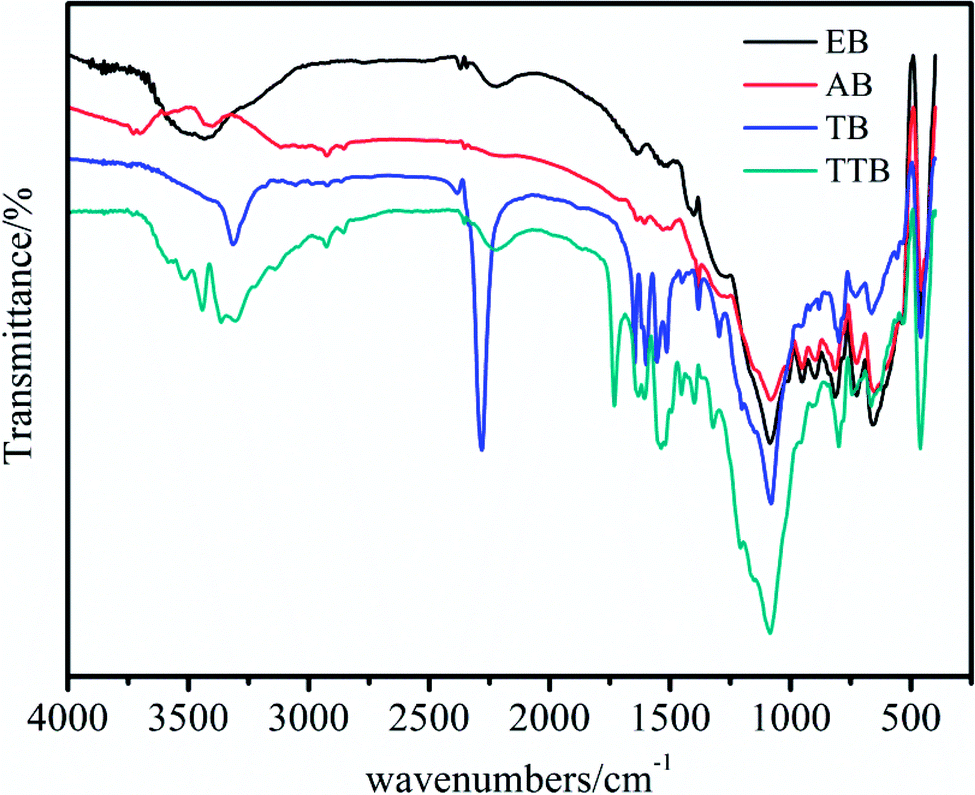 | ||
| Fig. 1 The IR spectra of the EB, AB, TB and TTB. | ||
Due to the importance of maintaining the active isocyanate in the intermediate TB, the reaction conditions in the carbamation and ureylene addition steps are essential to optimize. Herein, the IR and XPS spectra were applied as qualitative and semiquantitative research methods to attain better reaction conditions. In the preparation of TB, the effects of the reaction temperature at 50, 70, and 90 °C and the reaction time for 5, 10, and 15 h on the structure of TB were studied. The reaction temperatures of 40, 60, and 80 °C and reaction times of 5, 10, and 15 h in the preparation of TTB were also investigated.
The IR spectra of TB and TTB samples at various conditions are shown in Fig. 2. It was found that at both 50 °C and 70 °C, the carbamation was successfully processed because of the predominant peaks attributed to the NCO and benzene ring. However, at 90 °C, none of the above-mentioned peaks were found in the IR spectrum, and an additionally peak at 3448 cm−1 was strengthened, which can infer that the introduced TDI on the boron particle surface was thermally decomposed. For the preparation of TTB at different temperatures, the typical absorption characteristic peak at 2276 cm−1 corresponding to the NCO disappeared, and the typical absorption peaks at 3364 and 1740 cm−1 were attributed to the N–H and CO of the –NH–CO–N group emerged. However, the absorption strength assigned to the CO of the –NH–CO–N relative to the benzene ring tended to decrease at higher temperatures. Therefore, the lower temperature is beneficial to both the carbamation and the ureylene addition reactions.
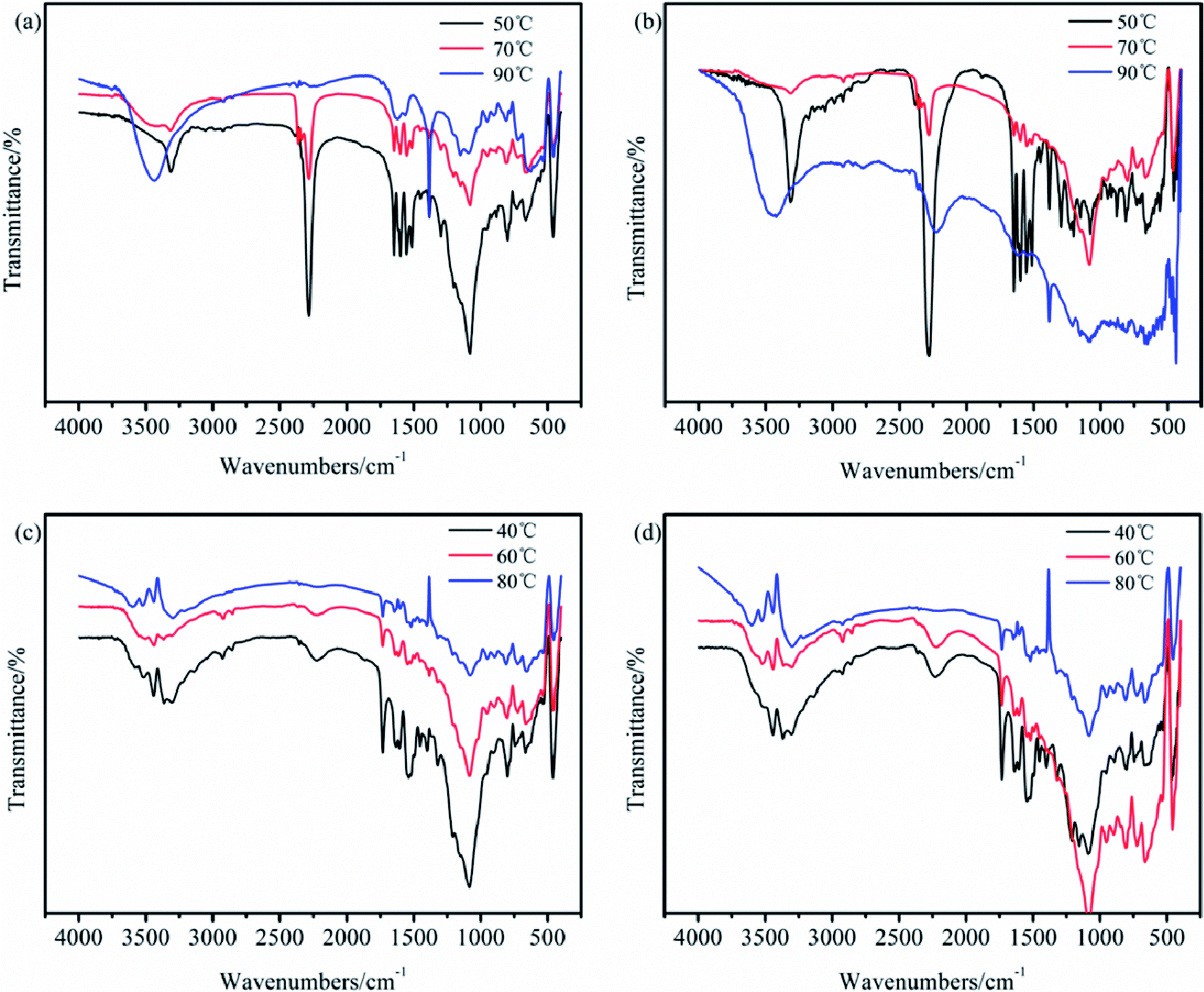 | ||
| Fig. 2 IR spectra of the TB prepared for (a) 5 h and (b) 10 h, and the TTB prepared for (c) 5 h and (d) 10 h at different temperatures. | ||
The elemental analysis of the particle surface was used to evaluate the reaction conditions. The XPS spectra of the TB prepared at 50, 70, and 90 °C, and TTB prepared at 40, 60, and 80 °C for 10 h were shown in Fig. 3. And the elemental content of the samples were given in Table 1, which was gained by normalization processing the ratio of characteristic peak area to the sensitivity factor. In Fig. 3, peak from 392 to 402 eV was displayed in the XPS pattern of the N 1s state, which verified the incorporation of N from the TDI into the TB particles. In the enlarged XPS spectrum of B 1s in Fig. 4, the main component of EB at 188.0 eV corresponded to the B–B bond,24 and the component at 189.4 eV and 193.2 eV was assigned to the B–O bond in the suboxide (Bx–O) and oxide (B2O3) compounds, respectively.25 After acidification, in the enlarged XPS spectrum of B 1s in AB, the component at 192.6 eV was assigned to the H3BO3,26 and the typical peak at 189.4 eV was assigned to the B–O bond in the suboxide (Bx–O) strengthened, resulting from the acidification and oxidation with nitric acid. In Table 1, in the XPS spectrum of the TTB, the N content was higher than that of the TB, which was caused by the incorporation of N from the ATZ into the TTB particles. The N content of TB prepared at 50 °C was obviously higher than that prepared at 70 °C and 90 °C, and the N content of TTB prepared at 40 °C was also obviously higher than that prepared at 60 °C and 80 °C. Thus, the reactions are sensitive to the temperature conditions, and 50 °C and 40 °C are appropriate to the carbamation and the ureylene addition reactions, respectively.
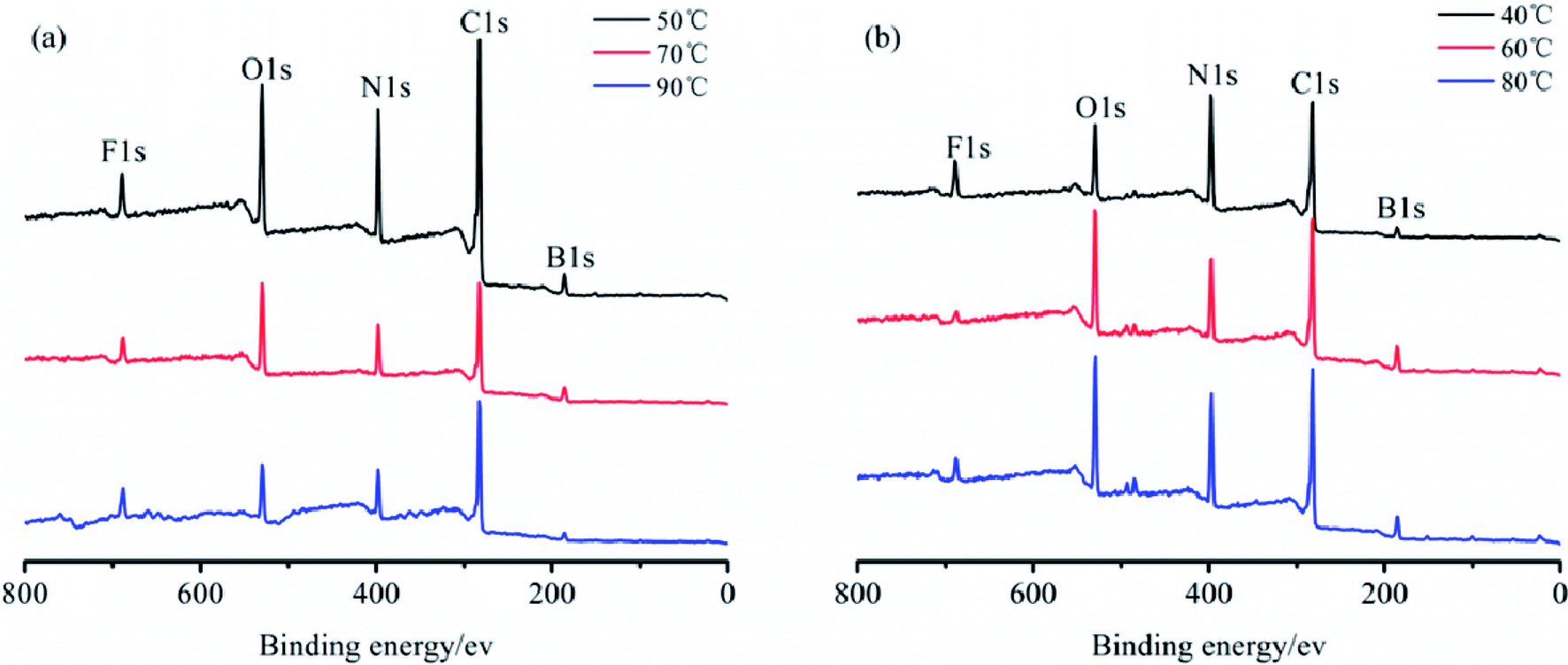 | ||
| Fig. 3 XPS spectra of (a) TB and (b) TTB prepared for 10 h at different temperatures. | ||
| State | Content in TB/% | Content in TTB/% | ||||
|---|---|---|---|---|---|---|
| 50 °C | 70 °C | 90 °C | 40 °C | 60 °C | 80 °C | |
| C1s | 57.07 | 48.11 | 59.88 | 46.28 | 46.06 | 43.55 |
| N1s | 17.28 | 13.62 | 12.26 | 26.03 | 14.45 | 18.71 |
| O1s | 9.25 | 14.23 | 11.26 | 10.75 | 11.53 | 13.32 |
| B1s | 13.66 | 20.13 | 10.81 | 11.86 | 25.71 | 21.47 |
| Other | 2.74 | 3.91 | 5.7 | 5.08 | 2.26 | 2.96 |
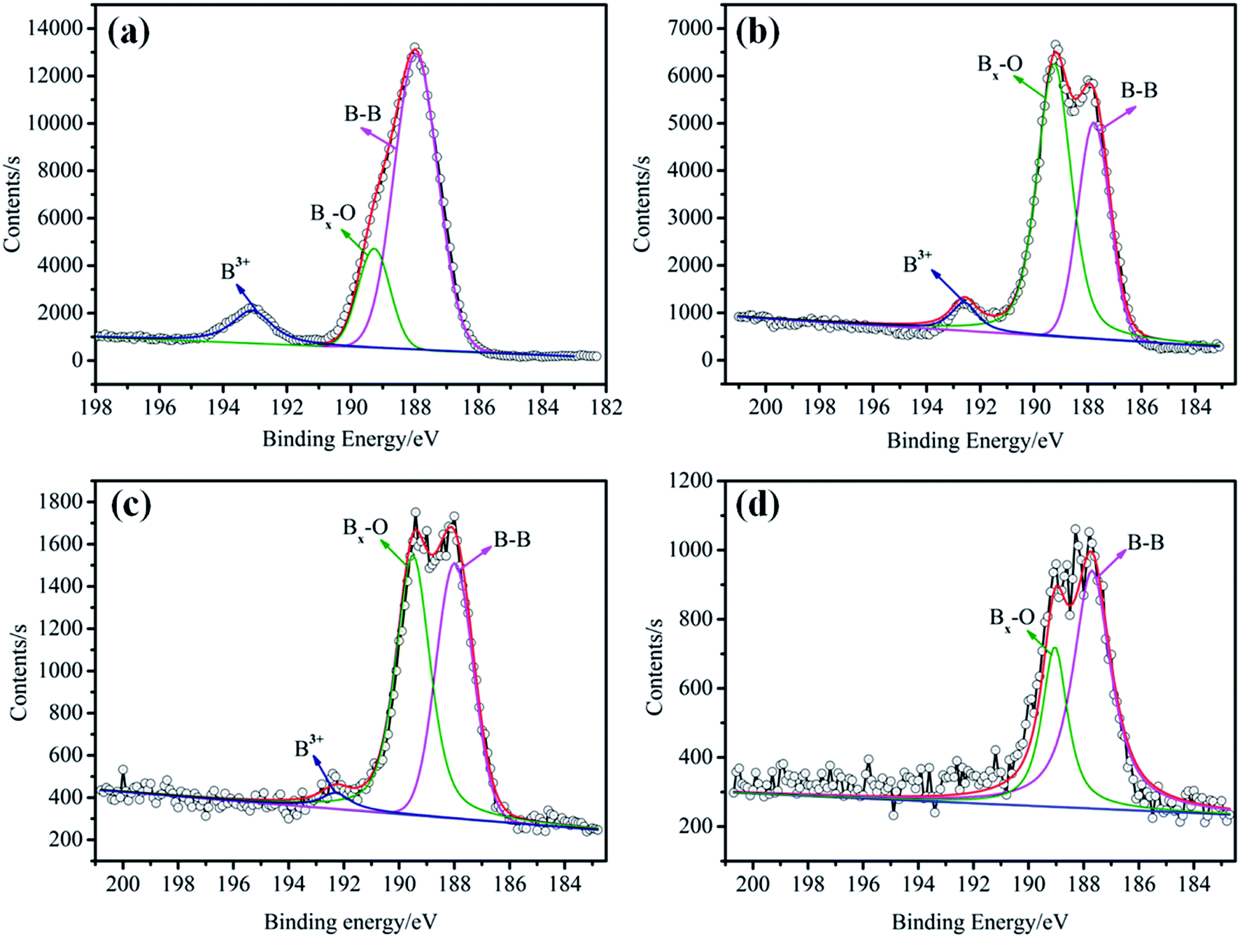 | ||
| Fig. 4 Enlarged XPS spectra of B 1s for (a) EB, (b) AB, (c) TB and (d) TTB. | ||
Herein, the reaction time was further investigated using IR spectroscopy to confirm the compositions of TB and TTB. The IR spectra of TB prepared at 50 °C for 5, 10, and 15 h and TTB prepared at 40 °C with TB prepared at 50 °C for 10 h as the raw material are shown in Fig. 5. The bands at 3305, 2276, 1740 and 1596 cm−1 corresponding to the N–H of–NH–COO–, NCO, CO of –NH–CO–N and benzene ring were easily distinguished. The absorption frequencies from the reaction times of 5, 10, and 15 h were nearly equal. This indicates that along with the extended reaction time, the –NH–COO–, NCO and –NH–CO–N groups are chemically stable, so a 5 h reaction time is appropriate for the preparation of both TB and TTB.
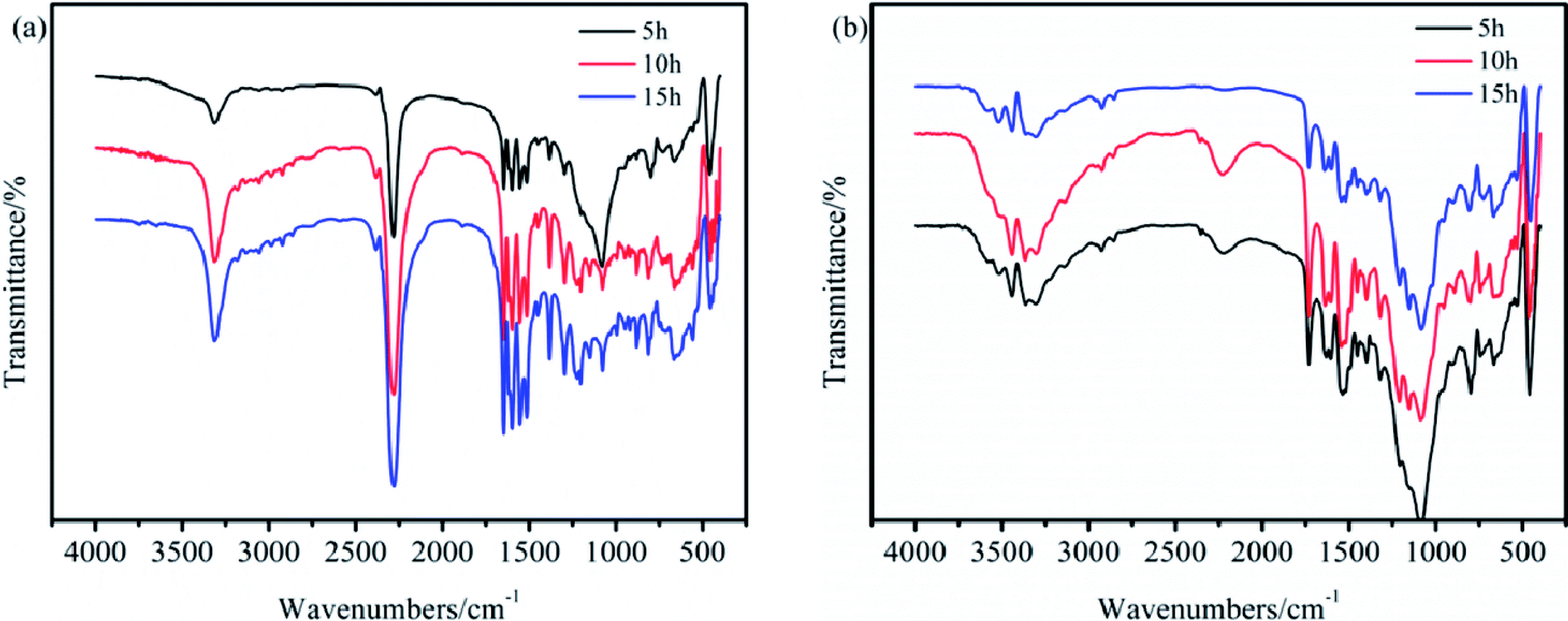 | ||
| Fig. 5 IR spectra of (a) TB prepared at 50 °C and (b) TTB prepared at 40 °C for different times. | ||
After optimizing the reaction conditions, conditions of temperature of 50 °C for 5 h in the preparation of TB, and 40 °C for 5 h in the preparation of TTB were proven as the improved reaction conditions. Finally, the target material TTB was prepared at the optimized conditions mentioned above, and the resulting XPS spectrum and elemental content are shown in Fig. 6. The N content of the particle surface is 52.2% in the target material TTB, which is likely beneficial to improve the impulse of the energetic boron powder and its composites.
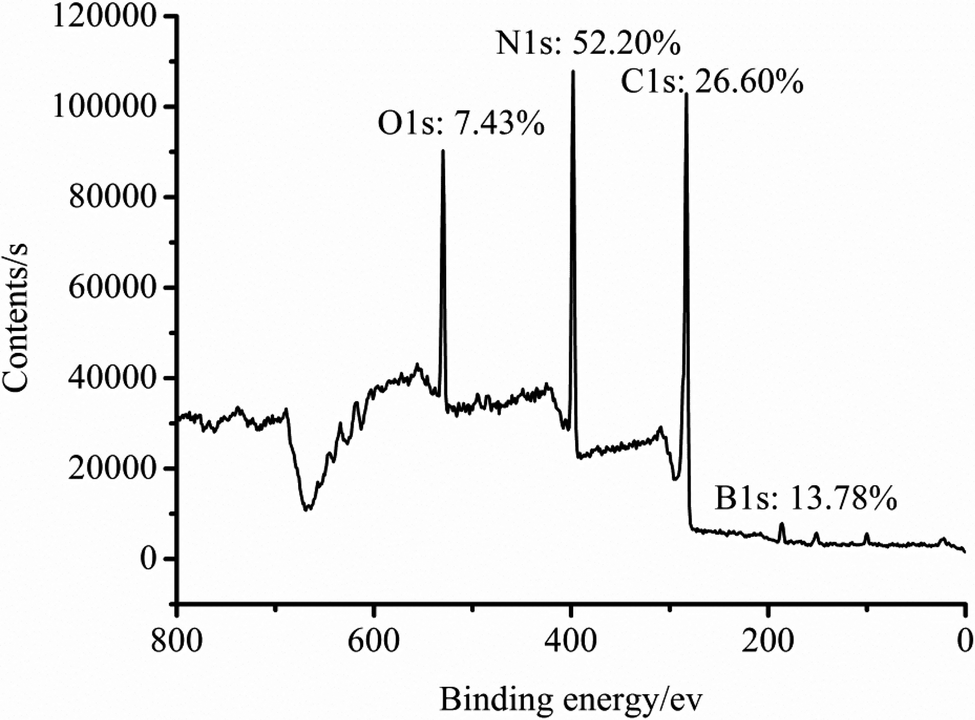 | ||
| Fig. 6 XPS spectrum of the TTB prepared at the optimized reaction conditions. | ||
3.2 Solid-state 13C-NMR analysis
Solid-state 13C-NMR spectrometry was carried out, and the results for the TB and TTB are shown in Fig. 7. In Fig. 7a, the peak signals at 17.4, 134.1, and 155.7 ppm came from the –CH3 (C-3), unreacted –NCO (C-1) and the carbamate group (–NH–COO–, C-2) of TB molecule, respectively.22 The peaks at 137.1, 136.4, 130.7, 119.8, 118.4, and 115.9 ppm could be respectively assigned to the C-7, 5, 9, 4, 8, and 6 in the benzene ring of TB molecule as shown in the insert figure of Fig. 7a.22 In Fig. 7b, the peak at 19.6 ppm was the signal of the –CH3 (C-3) of TTB molecule. The signals from the carbon adjacent to the carbamate group (–NH–COO–, C-2) and urea group (–NH–CO–N, C-1) both appeared approximately at 157.5 ppm. The peak at 147.5 ppm was the signal of ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) N–CHN– carbon in the triazole of the ATZ,23 indicating that the ATZ molecule has been successfully introduced to the modified boron powders. The peak at 135.8 ppm was assigned to the C-7 and 5 in the benzene ring linked with the carbamate and urea groups of TTB molecule. The other peaks at 130.6, 128.0, 122.9, and 119.8 ppm were the signals of C-9, 4, 8, and 6 in the benzene ring. There was no additional peak attributed to the quaternary carbon atom of C–NH2 in ATZ, possibly because its longer relaxation time or change of electronic state produced by charge transfer made the signal missing.27,28 The IR spectra and NMR spectra analyses for the TB and TTB illustrate that a novel covalently bonded energetic boron powder has been successfully prepared.
N–CHN– carbon in the triazole of the ATZ,23 indicating that the ATZ molecule has been successfully introduced to the modified boron powders. The peak at 135.8 ppm was assigned to the C-7 and 5 in the benzene ring linked with the carbamate and urea groups of TTB molecule. The other peaks at 130.6, 128.0, 122.9, and 119.8 ppm were the signals of C-9, 4, 8, and 6 in the benzene ring. There was no additional peak attributed to the quaternary carbon atom of C–NH2 in ATZ, possibly because its longer relaxation time or change of electronic state produced by charge transfer made the signal missing.27,28 The IR spectra and NMR spectra analyses for the TB and TTB illustrate that a novel covalently bonded energetic boron powder has been successfully prepared.
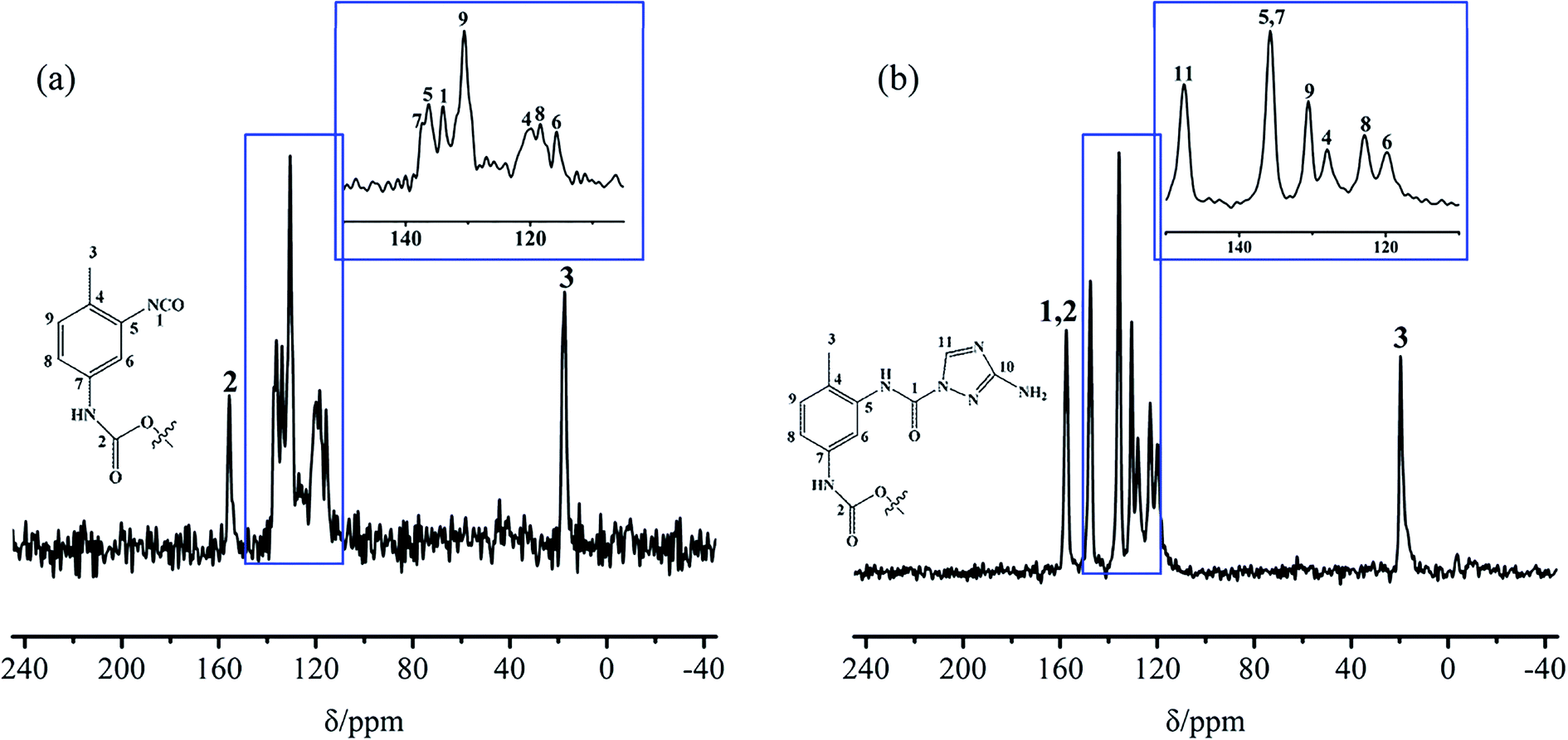 | ||
| Fig. 7 13C-NMR of (a) TB and (b) TTB. | ||
3.3 SEM and TEM micrographs of the samples
The surface morphology of EB, AB, TB and TTB was studied with SEM and shown in Fig. 8. As seen from Fig. 8a–b, d–e, the particle surface of the raw material EB and intermediate AB is glossy. After carbamation of AB, the particle surface of the intermediate TB became coarse with some flake substance adhering to the surface, which was caused by the grafted TDI molecules. In the last modification step, the TDI molecules on the surface reacted with ATZ, so the former flake substance was transferred to tiny particles to form a tight coating layer.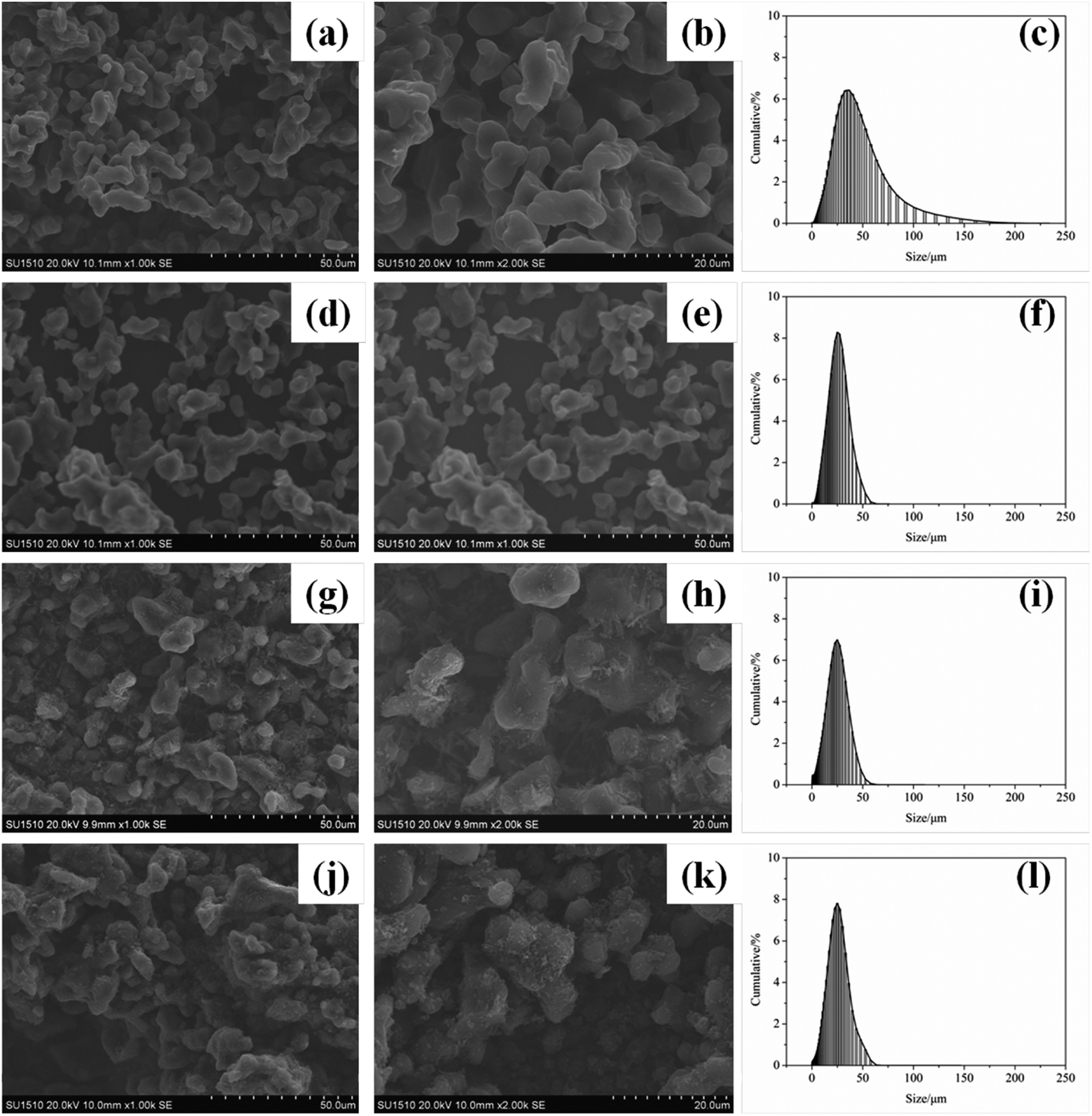 | ||
| Fig. 8 SEM images and size distribution for (a–c) EB, (d–f) AB, (g–i) TB, and (j–l) TTB. | ||
The particle size distribution of the four samples was also presented in Fig. 8, and the median particle size of EB, AB, TB and TTB was 33.3, 22.6, 19.7 and 21.5 μm, respectively. After acidification, the particle size significantly reduced, because the surface B2O3 reacted with water in the nitric acid and acetone solvent. The particle size of TB and TTB was a little smaller than that of AB, possibly owing to the mass loss of ultrafine particles in the filtration. Different from the particle size by laser particle size analysis, the particle size of TB in SEM micrographs seemed larger than others, because the unreacted –NCO groups on the surface have strong interaction with other particles, and then TB particles tended to reunite.
To further verify the morphological change before and after the chemical modification, the morphologies of the EB and TTB were studied using TEM. As is shown in Fig. 9, it is obvious that the surface of the TTB particles is rougher, which is probably caused by the organic coating layer.
 | ||
| Fig. 9 TEM images of the boron powders for (a) EB and (b) TTB. | ||
3.4 Static water contact angle on the boron surface
The changes in the surface properties of the boron powders after chemical modification are also accompanied by changes in the surface energy. The static water contact angle on the surface of the EB, AB, TB and TTB were measured, and the results are given in Fig. 10. It is clearly seen from Fig. 10a–b that water can easily permeate the surface of the boron powders EB and AB, giving a static water contact angle of 0° for both. After the surface was grafted with TDI molecules, the water drop could steadily stay on the surface of the TB pellet, as shown in Fig. 10c, with a static water contact angle of 148.3°. In conclusion, the surface properties of the boron powders are significantly changed from hydrophilic to hydrophobic after grafting with the nonpolar TDI molecules owing to the highly nonpolar benzene ring and the methyl group. After the ureylene addition between the TB and the polar molecule ATZ due to the numerous N atoms and amino groups, the surface properties of the TTB boron powders were changed to be more polar than the TB, and the static water contact angle was 37.0°. | ||
| Fig. 10 Images of a water droplet sitting on the surface of the (a) EB, (b) AB, (c) TB, and (d) TTB. | ||
3.5 Rheological property
Rheological characterization is important for boron powders in hydroxyl-terminated polybutadiene (HTPB), a widely used binder of solid propellants and cast cured explosives.29 In our work, the rheological property of HTPB containing 30% boron powers heating at 70 °C for different time were tested by a rheometer. Fig. 11a showed the relationship between the viscosity and shear rate at the beginning of heating. The results showed that the viscosity of EB/HTPB was nearly constant at 6.7–9.3 Pa s with an increase in shear rate, but the viscosity of TTB/HTPB decreased from 163 Pa s to 5.8 Pa s. As shown in Fig. 11b, after heating at 70 °C for 10 hours, the viscosity of EB/HTPB was also constant at 6.8–9.3 Pa s, which was equal with that before heating. While the viscosity of TTB/HTPB sustained at 4.5–5.1 Pa s, indicating that the mixture became to a Newtonian fluid. It was inferred that, at the beginning of heating, TTB/HTPB owned a higher viscosity, due to the ragged coating layer of TTB, and after heating for a long time, the viscosity of TTB/HTPB decreased because of the excellent infiltration between the polar grafting molecular ATZ with HTPB. Therefore, the energetic boron powders with a strategy of covalent bonding on particle surface, would not deteriorate the dispersion and processing properties.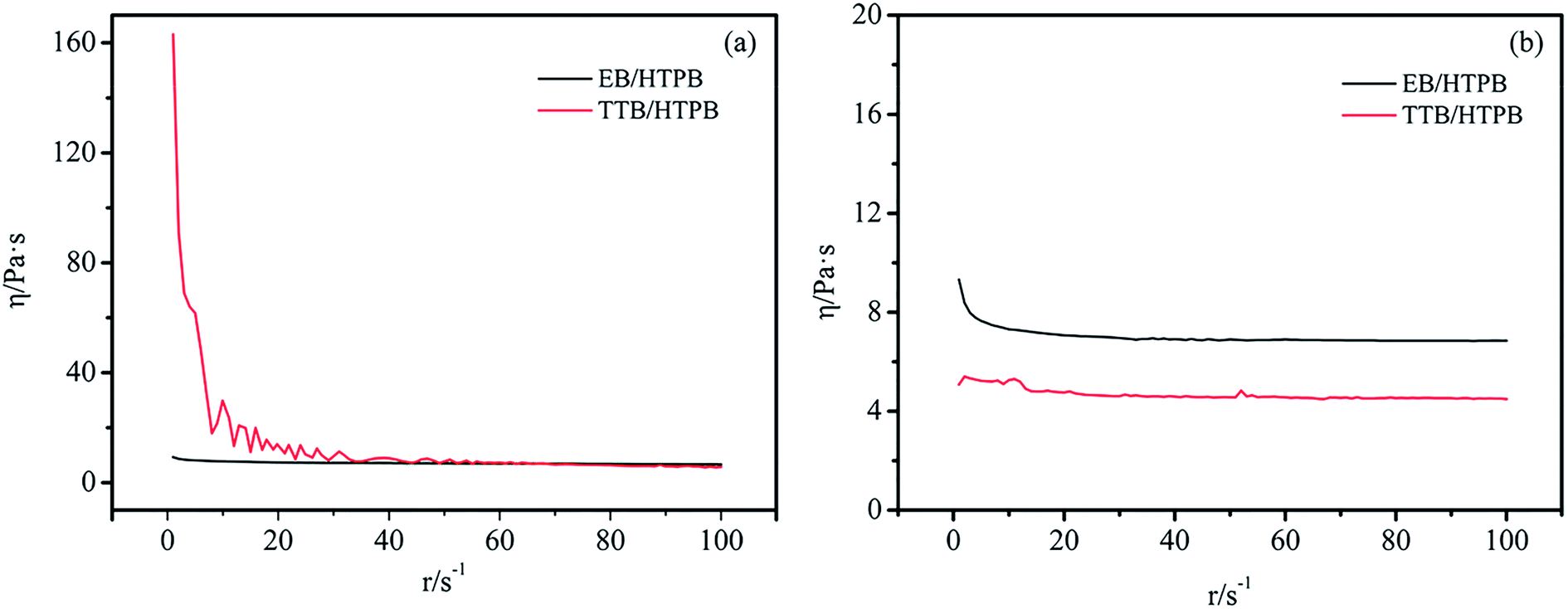 | ||
| Fig. 11 Viscosity of B/HTPB heating at 70 °C for (a) 0 h, (b) 10 h. | ||
3.6 Thermal stability of TB and TTB
To determine the thermal stability of TB and TTB in a nitrogen atmosphere from room temperature to 1200 °C, simultaneous thermogravimetry/differential scanning calorimetry (TG/DSC) measurements were taken, and the results are given in Fig. 12. As shown in Fig. 12a, there are two weak endothermic peaks appearing at 192 °C and 304 °C in the DSC curve, and the weight loss of TB began from 189 °C and ended at 342 °C, with a total weight loss of 7.1%. This process can be assigned to the decomposition of the TDI group on the surface of the TB particles. In Fig. 12b, three weak endothermic peaks located at 245, 283 and 329 °C can be found in the DSC curve. The weight loss of the TTB began from 198 °C to 375 °C with a total weight loss 8.4%. The TTB has a higher weight loss ratio due to the introduction of the ATZ molecule based on the TB powders. The results show that both TB and TTB are thermally stable, which is convenient for the preparation, storage and usage of the target material TTB and its key intermediate product TB.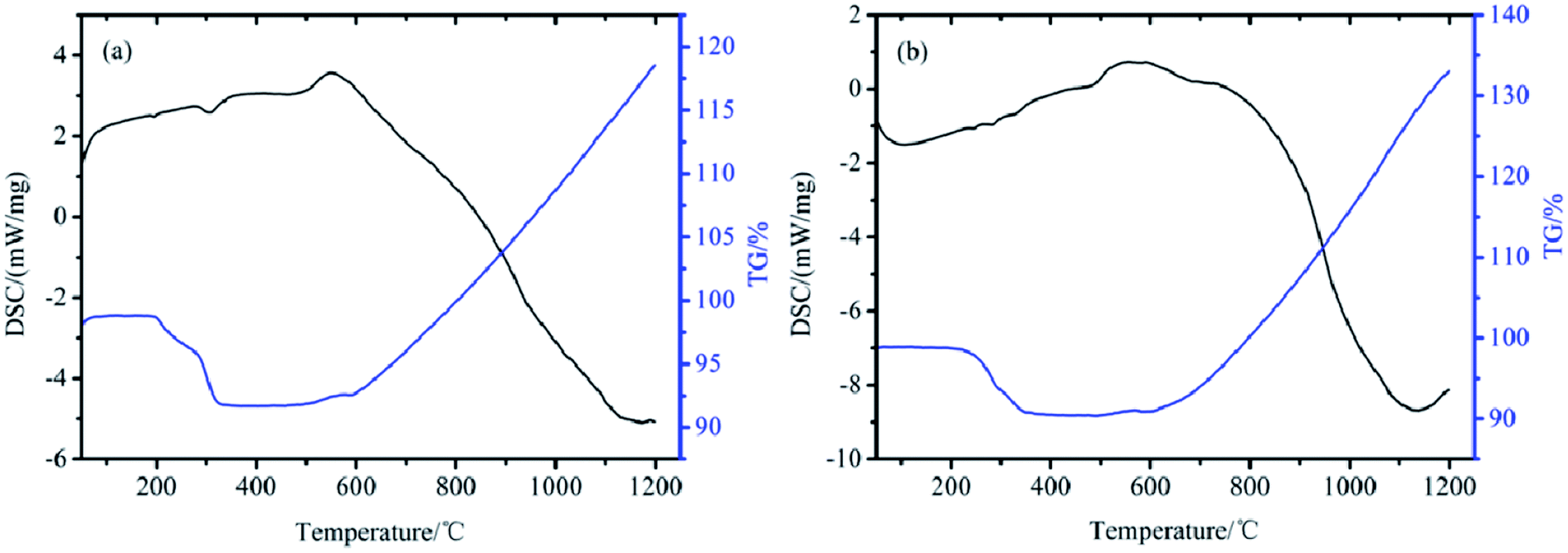 | ||
| Fig. 12 TG/DSC curves of (a) TB and (b) TTB. | ||
3.7 Thermal reaction of the B/KNO3 composites
To investigate the reaction activities of boron powders before and after the modification, the composites EB/KNO3 and TTB/KNO3 were prepared as well-known pyrotechnics. TG/DSC was used to characterize their thermo-reaction properties. From the DSC curves shown in Fig. 13, the heat releasing reaction of the TTB/KNO3 mainly occurred at the peak temperature of 450 °C and was assigned to their oxidation–reduction reaction, which occurred at 30 °C earlier than that of EB/KNO3 with a predominant exothermic peak at 480 °C. In the TG curves, the tiny endothermic peaks at 138 °C and 338 °C were assigned to the rhombic to trigonal crystalline phase transition and melting of KNO3.30 The weight loss began at 430 °C for the EB/KNO3 and 200 °C for the TTB/KNO3, which can be attributed to the continuous decomposition of the organic layer and the oxidation–reduction reaction between the boron and potassium nitrate starting from 400 °C. Both the TG and DSC curves indicate that the usage of the covalent bonded energetic boron powders is beneficial to reduce the critical reaction temperature of the pyrotechnics B/KNO3 composites and improve the ignition of the boron powders. In our work, the grafting modification on the boron particle surface with energetic molecule was more efficient to reduce the critical reaction temperature of boron–potassium nitrate pyrotechnic charge than the previous research, in which cellulosic binders, phenolic resin and RDX or HMX were mixed physically.31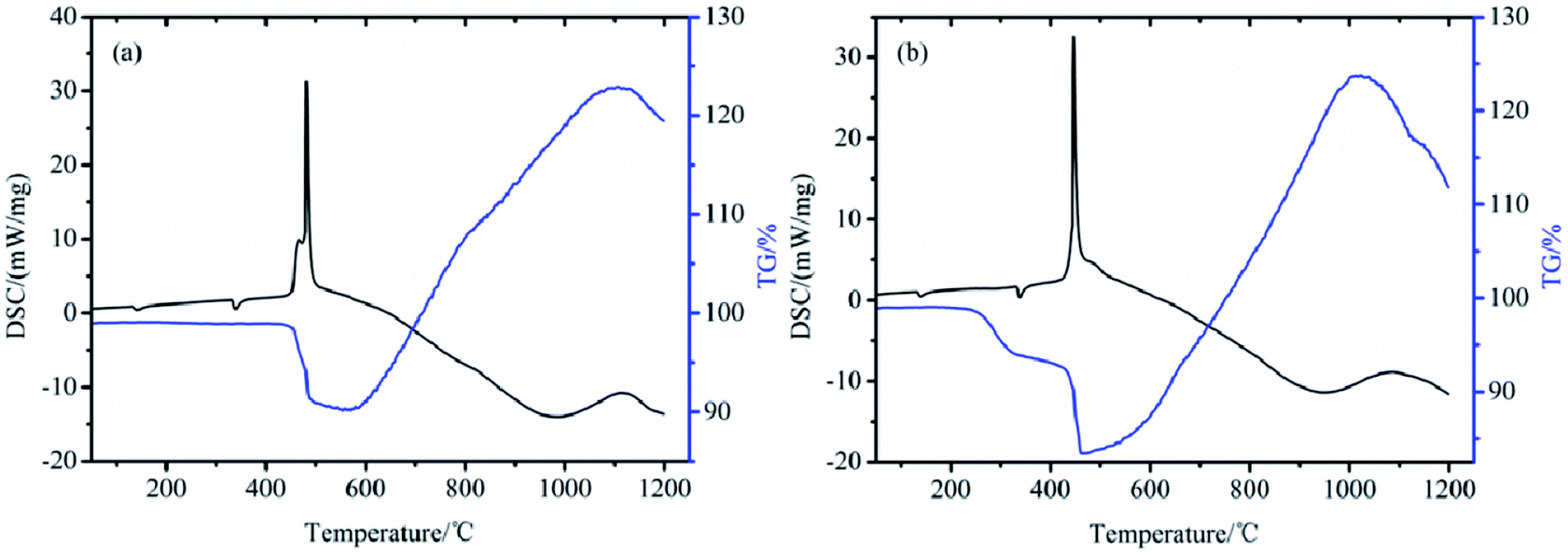 | ||
| Fig. 13 TG/DSC curves of (a) EB/KNO3 and (b) TTB/KNO3. | ||
4. Conclusion
In conclusion, we developed a new kind of covalently bonded energetic boron powder with improved ignition properties in B/KNO3 used as pyrotechnics. The reaction conditions predominantly affect the chemical structure of the modified boron powders and the stability of the isocyanate reaction sites. The small organic molecules are grafted on the boron surface through covalent bonds. The covalently bonded energetic boron powders enable the pyrotechnic B/KNO3 to react at a lower critical temperature. This approach gives a new method to prepare highly energetic boron powders, which can be widely used in propellants, explosives and pyrotechnics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 11502249, 11572359, 11502248) is gratefully acknowledged.References
- (a) V. Pepekin, Int. Annu. Conf. ICT, 1996, vol. V19 Search PubMed; (b) A. Maranda, Propellants, Explos., Pyrotech., 1990, 4, 161–165 CrossRef; (c) H. Ghassemi and H. F. Fasih, Aerosp. Sci. Technol., 2013, 1, 1–8 CrossRef.
- (a) Z. Chen and R. Yang, J. Propul. Technol., 1998, 1, 102–106 Search PubMed; (b) W. Pang and X. Fan, Chem. Propellants Polym. Mater., 2009, 2, 1–5 Search PubMed.
- N. H. Yen and L. Y. Wang, Propellants, Explos., Pyrotech., 2012, 2, 143–155 CrossRef.
- C. Rossi, K. Zhang, D. Estève, P. Alphonse, P. Tailhades and C. Vahlas, J. Microelectromech. Syst., 2007, 4, 919–931 CrossRef.
- J. Xi, J. Liu, H. Li, Y. Wang, Y. Zhang, J. Zhou and K. Cen, Chin. J. Energ. Mater., 2013, 4, 533–538 Search PubMed.
- R. M. Adams, J. Am. Chem. Soc., 1965, 15, 3535–3536 Search PubMed.
- (a) M. N. Makhov, Russ. J. Phys. Chem. B, 2015, 1, 50–55 CrossRef; (b) S. Xu, Yu. Chen, X. Chen, D. Wu and D. Liu, Combust., Expl., Shock Waves, 2016, vol. 3, pp. 342–349 Search PubMed.
- (a) W. Zhang, H. Zhu and D. Fang, Chin. J. Energ. Mater., 1998, 4, 179–186 Search PubMed; (b) R. W. Conner and D. D. Dlott, J. Phys. Chem. C, 2012, 116, 2751–2760 CrossRef CAS.
- (a) D. J. Erskine, L. Green and C. Tarver, The American physical society topical conference on shock compression of condensed matter, Albuquerque, 1989 Search PubMed; (b) C. M. Tarver, 14th APS Topical Conference on SCCM, 2005 Search PubMed.
- (a) A. Macek and J. Semple, Combust. Sci. Technol., 1969, 3, 181–191 CrossRef; (b) C. L. Yeh and K. K. Kuo, Prog. Energy Combust. Sci., 1996, 6, 511–541 CrossRef; (c) G. Young, K. Sullivan, M. R. Zachariah and K. Yu, Combust. Flame, 2009, 2, 322–333 CrossRef.
- (a) D. Guan and N. Wang, Chin. J. Explos. Propellants, 1998, 2, 52–54 Search PubMed; (b) M. A. Trunov, V. K. Hoffmann, M. Schoenitz and E. L. Dreizin, Journal of Propulsion Powder, 2012, 2, 184–191 Search PubMed.
- (a) J. C. Trowbridge and J. D. Breazeale, US pat. 4877649, 1989; (b) I. M. Shyu and T. K. Liu, Combust. Flame, 1995, 4, 634–644 CrossRef.
- H. Li, X. Wan, X. Gao and J. Li, China Ceram., 2008, 8, 10–12 Search PubMed.
- S. He and B. Liu, Mater. Res. Appl., 2016, 2, 71–74 Search PubMed.
- (a) S. Thoru, I. Masahiro, N. Toshihiko and T. Naoe, EP328906 A2, 1989; (b) R. Wolfgang and S. Guenter. US pat. 5332767, 1994.
- J. Wang, G. Xu, X. Ji and R. Gong, Chin. J. Inorg. Chem., 2003, 9, 967–970 Search PubMed.
- W. Ma and A. Dong, Electron. Compon. Mater., 2006, 5, 62–64 Search PubMed.
- G. Yuan and S. Su, J. Dispersion Sci. Technol., 2010, 31, 327–331 CrossRef CAS.
- Z. Gao, N. I. Walton, A. Malugin, H. Ghandehari and I. Zharov, J. Mater. Chem., 2012, 22, 877–882 RSC.
- R. J. P. Lima, C. Dubois, O. Mader, R. Stowe and S. Ringuete, Int. J. Energ. Mater. Chem. Propul., 2010, 5, 437–446 Search PubMed.
- H. Xue, Y. Gao, B. Shreeve and J. M. Shreeve, Inorg. Chem., 2005, 14, 5068–5072 CrossRef PubMed.
-
(a) R. Lu, B. Qian, H. Deng and H. Yang, Chin. J. Chem., 1997, 6, 541–547 Search PubMed;
(b) M. Lesar, M.
![[Z with combining breve]](https://www.rsc.org/images/entities/char_005a_0306.gif) igon and T. Malavašič, J. Appl. Polym. Sci., 1993, 5, 805–814 CrossRef;
(c) H. Han, S. Li, X. Zhu, X. Jiang and X. Kong, RSC Adv., 2014, 4, 33520–33529 RSC.
igon and T. Malavašič, J. Appl. Polym. Sci., 1993, 5, 805–814 CrossRef;
(c) H. Han, S. Li, X. Zhu, X. Jiang and X. Kong, RSC Adv., 2014, 4, 33520–33529 RSC. - (a) T. Hirata, L. Twanmoh, H. Wood Jr, A. Goldin and J. Driscoll, J. Heterocycl. Chem., 1972, 1, 99–106 CrossRef; (b) G. Chipen, R. Bokaldere and V. Grinshtein, Chem. Heterocycl. Compd., 1971, 4, 546–547 CrossRef; (c) J. Reiter, L. Pongó and P. Dvortsák, J. Heterocycl. Chem., 1987, 6, 1685–1695 CrossRef; (d) T. Winkler and H. Kristinsson, Helv. Chim. Acta, 1983, 2, 694–700 CrossRef; (e) Y. L. Duc, E. Licsandru, D. Vullo, M. Barboiu and C. T. Supuran, Bioorg. Med. Chem., 2017, 5, 1681–1686 CrossRef PubMed.
- (a) C. Ronning, D. Schwen, S. Eyhusen, U. Vetter and H. Hofsass, Surf. Coat. Technol., 2002, 158, 382–387 CrossRef; (b) A. Jain, S. Anthonysamy, K. Ananthasivan, R. Ranganathan, V. Mittal, S. V. Narasimhan and P. R. Vasudeva Rao, Mater. Charact., 2008, 7, 890–900 CrossRef.
- (a) W. Moddeman, A. Burke, W. Bowling and D. Foose, Surf. Interface Anal., 1989, 14, 224–232 CrossRef CAS; (b) M. Ennaceur and B. Terreault, J. Nucl. Mater., 2000, 280, 33–38 CrossRef CAS; (c) T. Xu, J. Zheng, N. Wu, A. Nicholls, J. Roth, D. Dikin and R. Ruoff, Nano Lett., 2004, 5, 963–968 CrossRef; (d) W. Foo, J. Ozcomert and M. Trenary, Surf. Sci., 1991, 255, 245–258 CrossRef CAS.
- (a) D. Hendrickson, J. Hollander and W. Jolly, Inorg. Chem., 1970, 3, 612–615 CrossRef; (b) J. Schreifels, P. Maybury and W. Swartz, J. Catal., 1980, 65, 195–206 CrossRef CAS.
- (a) M. TrewhellaIain, J. Poplett and A. Grin, Fuel, 1986, 4, 541–546 CrossRef; (b) G. A. Nagana Gowda, Magn. Reson. Chem., 2001, 10, 581–585 CrossRef; (c) S. Barbuceanu, G. Saramet, G. Almajan, C. Draghicic, F. Barbuceanu and G. Bancescu, Eur. J. Med. Chem., 2012, 49, 417–423 CrossRef CAS PubMed.
- (a) H. Chen and Q. Shao, J. Instrum. Anal., 1995, 6, 21–25 Search PubMed; (b) F. J. Dinan, W. T. Schwartz, R. A. Wolfe, D. S. Hojnicki, T. St. Clair and J. Richard Pratt, J. Polym. Sci., Part A-1: Polym. Chem., 1992, 1, 111–118 CrossRef; (c) R. Foster, J. Iball, S. N. Scrimgeour and B. C. Williams, J. Chem. Soc., Perkin Trans. 2, 1976, 6, 682–685 RSC.
- (a) R. Manjari, V. C. Joseph, L. P. Pandureng and T. Sriram, J. Appl. Polym. Sci., 1993, 2, 271–278 CrossRef; (b) P. P. Vadhe, R. B. Pawar, R. K. Sinha, S. N. Asthana and A. Subhananda Rao, Combust., Expl., Shock Waves, 2008, vol. 4, pp. 461–477 Search PubMed.
- R. Rugunanan and M. Brown, J. Therm. Anal. Calorim., 1991, 37, 1193–1211 CrossRef CAS.
- (a) V. Bhingarkar and H. Singh, Def. Sci. J., 2006, 3, 345–351 CrossRef; (b) K. Krishnan, R. Ammal, B. Hariharanath, A. Rajendran and C. Kartha, Def. Sci. J., 2006, 3, 329–338 CrossRef; (c) Y. Ye, L. Shu and R. Shen, Chin. J. Energ. Mater., 2007, 1, 33–35 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |