
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetic and mechanistic study on gas phase reactions of ozone with a series of cis-3-hexenyl esters†
Qilei Zhang‡
ab,
Xiaoxiao Lin‡a,
Yanbo Gai *a,
Qiao Maab,
Weixiong Zhaoa,
Bo Fanga,
Bo Longc and
Weijun Zhang*ad
*a,
Qiao Maab,
Weixiong Zhaoa,
Bo Fanga,
Bo Longc and
Weijun Zhang*ad
aLaboratory of Atmospheric Physico-Chemistry, Anhui Institute of Optics and Fine Mechanics, Chinese Academy of Sciences, Hefei, 230031, Anhui, China. E-mail: gaiyanbo@aiofm.ac.cn
bUniversity of Science and Technology of China, Hefei 230026, China
cSchool of Materials Science and Engineering, Guizhou Minzu University, Guiyang 550025, China
dSchool of Environmental Science and Optoelectronic Technology, University of Science and Technology of China, Hefei, 230026, Anhui, China
First published on 23rd January 2018
Abstract
As an important group of green leaf volatiles (GLVs), C6 hexenyl esters, are found to be widely emitted into the atmosphere by plants and vegetation, especially when they suffer mechanical damage. It is indispensable to understand their atmospheric fate for environmental assessment and model simulation. In this paper, the rate constants for reactions of O3 with four cis-3-hexenyl esters have been measured using an absolute method in a flow tube reactor at 298 K and atmospheric pressure. The measured rate constants (in 10−17 cm3 per molecule per s) were 4.06 ± 0.66 for cis-3-hexenyl formate, 5.77 ± 0.70 for cis-3-hexenyl acetate, 7.62 ± 0.88 for cis-3-hexenyl propionate, and 12.34 ± 1.59 for cis-3-hexenyl butyrate, respectively. Theoretical calculations were also carried out for the title reactions to better understand their kinetics and mechanism using density functional theory (DFT) and transition state theory (TST). Geometry optimizations, energy and harmonic vibrational frequency calculations were performed for all of the stationary points at the BHandHLYP/6-311+G(d,p) level of theory. The calculated rate constants were in good agreement with the experimental values. The results showed that the reactivity of the studied compounds towards O3 was obviously dependent on their chemical structure, such as the nature of the substituent, and the relative positions of the double bond and the substituent. The results were also discussed in terms of their atmospheric importance in the degradation of these unsaturated esters by comparing their lifetimes with respect to their reactions with O3 and other main atmospheric oxidants.
Introduction
Large quantities of volatile organic compounds (VOCs) are emitted directly into the atmosphere from biogenic and anthropogenic sources, of which biogenic volatile organic compounds (BVOCs) account for about 90% estimated on a global basis.1,2 Because of their relatively high atmospheric reactivity, BVOCs are believed to play a central role in determining atmospheric composition and the oxidizing capacity of the atmosphere.3–5 Many studies have found great discrepancy between measured and simulated OH concentration, total OH reactivity and also the production of ozone.6–8 In addition to the uncertainty in the understanding of VOC emissions, the lack of systematic understanding of the reaction process of identified VOCs is also an important reason for this discrepancy. Thus, it is very important to thoroughly study the reaction processes, including atmospheric degradation mechanisms, and kinetic information, for all the VOCs emitted into the atmosphere.Green leaf volatiles (GLVs) are one of the most important groups of BVOCs, represented by a series of C5 and C6 oxygenated hydrocarbons. They are produced within plant cells through biochemical conversion of some fatty acid, and emitted into the atmosphere by vegetation during the developmental processes or in response to external stress.9,10 As an important class of components, C6 hexenyl esters have been extensively detected. For example, many studies have shown that cis-3-hexenyl acetate is one of the major components of vegetation emissions,11–14 and sometimes accounts for more than 40% of the total emissions.15,16 It has also been demonstrated as the predominant wound-inducible volatile signal that mediates indirect defense responses by directing tritrophic (plant–herbivore–natural enemy) interactions.17 Besides cis-3-hexenyl acetate, other C6 hexenyl esters have also been recognized to contribute a significant fraction to the total BVOC flux, such as cis-3-hexenyl formate, cis-3-hexenyl butyrate and cis-3-hexenyl isovalerate.18–20
Although reaction with OH may be the dominant degradation pathway for these esters in the atmosphere, reaction with O3 may also play an important role, especially in polluted areas.4 Because of the higher concentration of O3 than that of OH, when rate constants for reactions of these unsaturated esters with ozone reach 10−17 cm3 per molecule per s, oxidation of these esters by O3 would be competitive with OH reactions. In the literature, reaction of cis-3-hexenyl acetate with O3 has been studied by two groups and the rate constants reported were at (5.4 ± 1.4) × 10−17 cm3 per molecule per s and (5.9 ± 0.87) × 10−17 cm3 per molecule per s, respectively, which indicates that reaction with O3 cannot be ignored for the atmospheric degradation of cis-3-hexenyl acetate.21,22
In this work, we have studied the reaction kinetics of O3 with four cis-3-hexenyl esters (cis-3-hexenyl formate, cis-3-hexenyl acetate, cis-3-hexenyl propionate and cis-3-hexenyl butyrate) and isoprene using experimental methods in a flow tube reactor as well as using theoretical methods. As far as we know, there are still no kinetic data for the O3 reactions with cis-3-hexenyl formate, cis-3-hexenyl propionate and cis-3-hexenyl butyrate in the literature. Thus, this work provides the first kinetic study for these reactions. The results can help to evaluate the lifetime, main removal pathways and influence area of these esters in the atmosphere and can also be used as input data of atmospheric chemical models for more accurate air quality predictions.
Experimental and theoretical methods
Experimental
Experiments were carried out in a coaxial double-tube flow reactor. The outer tube (i.d. 36 mm, length 100 cm) was for the VOC flow. The inner (i.d. 4 mm, length 120 cm) was for the O3 flow, which was movable along a range of axial positions. The end of the inner tube was designed with a series of small emitting holes to allow the reagent flows from the inner and the outer tubes better mixed. The schematic diagram of the experimental setup and gas flow is shown in Fig. S1 in the (ESI†).Clean zero air was used as bath gas, which was generated from a zero air generator (AADCO, 737-15). O3 was generated via electrical discharge by an ozone generator (COM-AD-01, ANSEROS, Germany). A known amount of the liquid VOC reagent was injected into a heated three-necked flask with microsyringe and flushed into a Teflon bag by zero air to obtain certain concentration of VOCs. A sampling pump (GM-1.0A, Jinteng Experimental Equipment Co., Ltd.) operated at the central outlet of the reactor draws forward the gas flow through the reactor. The O3 flow and VOC flow were set at about 0.5 L min−1 and 1.6 L min−1, respectively, by mass flow controllers (MFC, D08-8C, Beijing Sevenstar Electronics). Thus, the initial concentration of the VOC in the flow reactor could be calculated from its original concentration in the Teflon bag and the flow rates of both the O3 and VOC flow in the reactor. An ozone analyzer (Model 49i, Thermo Scientific) was used to measure the concentration of O3 at the end of the reactor tube. The reaction time was varied in the range of 0–25 s by adjusting the position of the inner tube relatively to the sampling point (0–80 cm). In our experimental conditions, the Reynolds number calculated was ∼90 with the linear flow velocity at ∼3.44 cm s−1, corresponding to a laminar flow. Experiments were conducted at a total pressure of 760 Torr and room temperature (298 ± 2 K) with the relative humidity ≤5%.
The rate constants were measured under pseudo-first-order conditions with the range of [VOC]0/[O3]0 at about 100–500, which had been used in our previous works.23,24 The temporal profile of [O3] can be given as:
[O3]t = [O3]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−k′t) exp(−k′t)
| (I) |
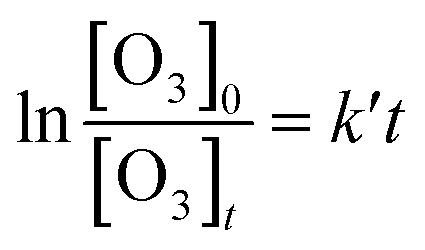 | (II) |
| k′ = ki[VOC]0 + kW | (III) |
The VOC reagents involved in this work included isoprene and a series of cis-3-hexenyl esters. The sources and their stated purity levels were as follows: cis-3-hexenyl formate (97%), cis-3-hexenyl acetate (98%), cis-3-hexenyl propionate (98%), cis-3-hexenyl butyrate (98%), Adamas Reagent Co., Ltd.; isoprene (99%), Alfa Asaer Co., Ltd. Cyclohexane (≥99.5%) was also used in some experiments as OH radical scavenger, which was from Sinopharm chemical reagent Co., Ltd.
Theoretical calculations
The reactions of cis-3-hexenyl formate, cis-3-hexenyl acetate, cis-3-hexenyl propionate and cis-3-hexenyl butyrate with O3 have been described at the density functional theory (DFT) level, employing the BHandHLYP functional and the 6-311+G(d,p) basis sets. Geometry optimizations, energies and the harmonic vibrational frequencies calculations were performed for all of the stationary points at the BHandHLYP/6-311+G(d,p) level of theory. Each local minimum has all positive frequencies and each transition state (TS) has one imaginary frequency. In addition, the connection between each minimum and the corresponding transition state (TS) was confirmed with intrinsic reaction path (IRC) method calculations.25 Quantum chemical calculations were performed with the Gaussian 09 program.26 The reaction rate constants were calculated using multiconformer transition state theory (MC-TST) as the reactions involve multiple conformers.27 For example, it includes two conformers (cis and trans) of the reactant and eight conformers of the transition state (TS1–TS8) in the reaction of cis-3-hexenyl formate with O3.Results and discussion
Absolute rate constant measurements
The initial concentrations used in the reactions were (0.74–2.58) × 1012 molecule per cm3 for O3 and (1.51–6.51) × 1014 molecule per cm3 for cis-3-hexenyl esters. In the absence of O3, the wall loss of the cis-3-hexenyl esters in the flow tube was first measured using a gas chromatography-flame ionization detector (GC-FID, 7820A, Agilent Technologies), which was showed to be negligible. Moreover, the GC-measured concentration of the studied esters in the reactor was very consistent with the concentration calculated based on its original concentration in the Teflon bag and the flow rates in the reactor. Thus, the calculated initial concentrations were used in the following experiments. It is well known that OH radicals could be produced in the ozone reactions with unsaturated VOCs,28–30 which would cause possible change in the VOC concentration. Thus, comparative experiments with and without cyclohexane as OH scavenger were conducted, and no significant difference was found in the rate constants. Fig. S2† gives one example of comparative experiments in the reaction of O3 with cis-3-hexenyl acetate. Actually, since the initial concentration of the esters was in large excess than O3, it would almost not change during the reaction. So that loss of the esters caused by reaction with OH radicals would have a negligible effect on the experiments.To validate the setup and the methods, rate constant for the reaction of O3 with isoprene was at first measured, which has been widely studied in the literatures.31,32 The initial concentrations of isoprene used were in the range of (5.25–10.5) × 1014 molecule per cm3. In all experiments, the ozone concentration with the inner tube at different positions was measured, corresponding to the ozone concentration at different reaction time. Experiments at each initial concentrations of isoprene were repeated at least twice. One typical set of pseudo-first-order plots for O3 reactions with different concentrations of isoprene is shown in Fig. S3.† The slope of each straight line gives the pseudo-first-order rate constants, k′. The absolute rate constant, ki, then was obtained by plotting all the data sets of k′ against their corresponding [isoprene]0 in Fig. 1. The determined rate constant was (1.23 ± 0.16) × 10−17 cm3 per molecule per s. It was in very good agreement with the IUPAC recommended value for kO3+isoprene which was 1.27 × 10−17 cm3 per molecule per s.32
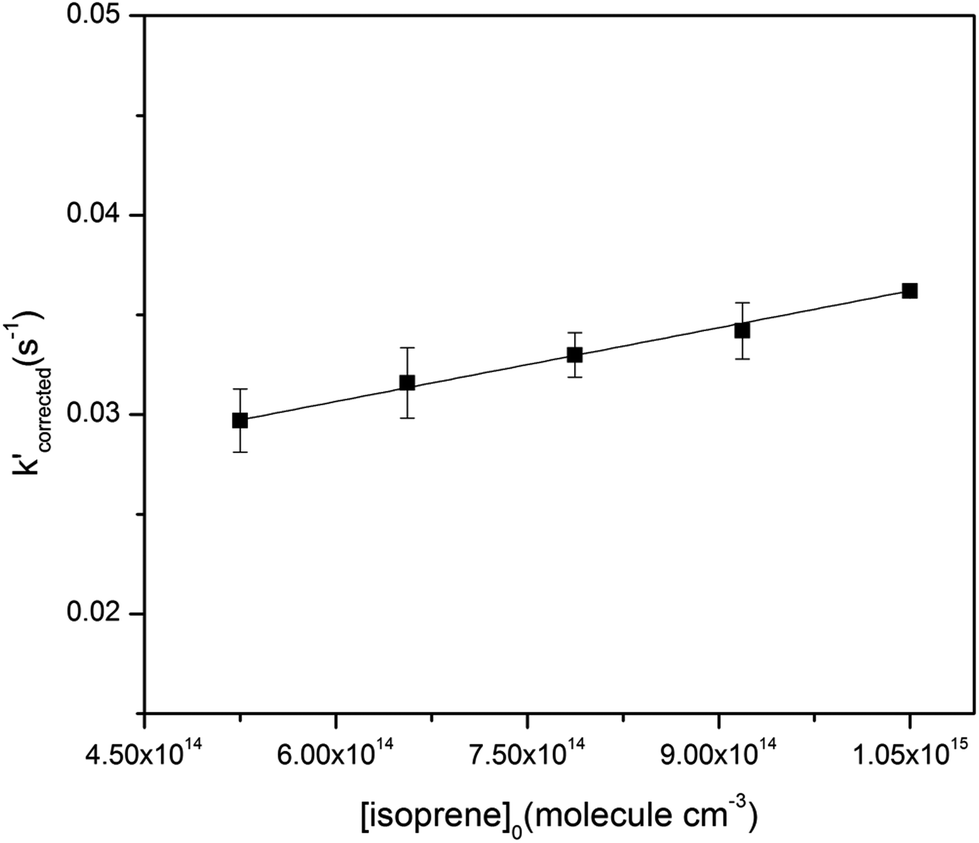 | ||
| Fig. 1 Plot of k′ (corrected) versus initial isoprene concentrations. | ||
Then, reactions of O3 with four cis-3-hexenyl esters, cis-3-hexenyl formate, cis-3-hexenyl acetate, cis-3-hexenyl propionate and cis-3-hexenyl butyrate, were studied. Fig. 2 shows one group of the typical pseudo-first-order plots for O3 reactions with different concentrations of cis-3-hexenyl acetate. Pseudo-first-order plots for other esters studied are given in Fig. S4–S6 in ESI.† In the flow reactor, the pseudo-first-order rate constants obtained may be subject to diffusion processes.33–35 So corrections were made to the experimentally obtained k′ values considering the axial and radial diffusion of O3 by:
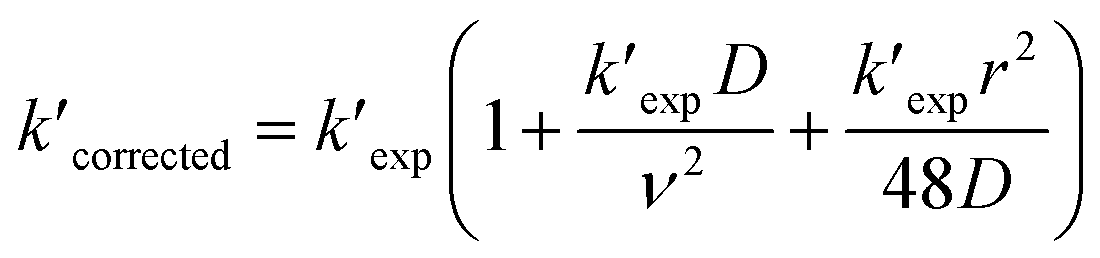 | (IV) |
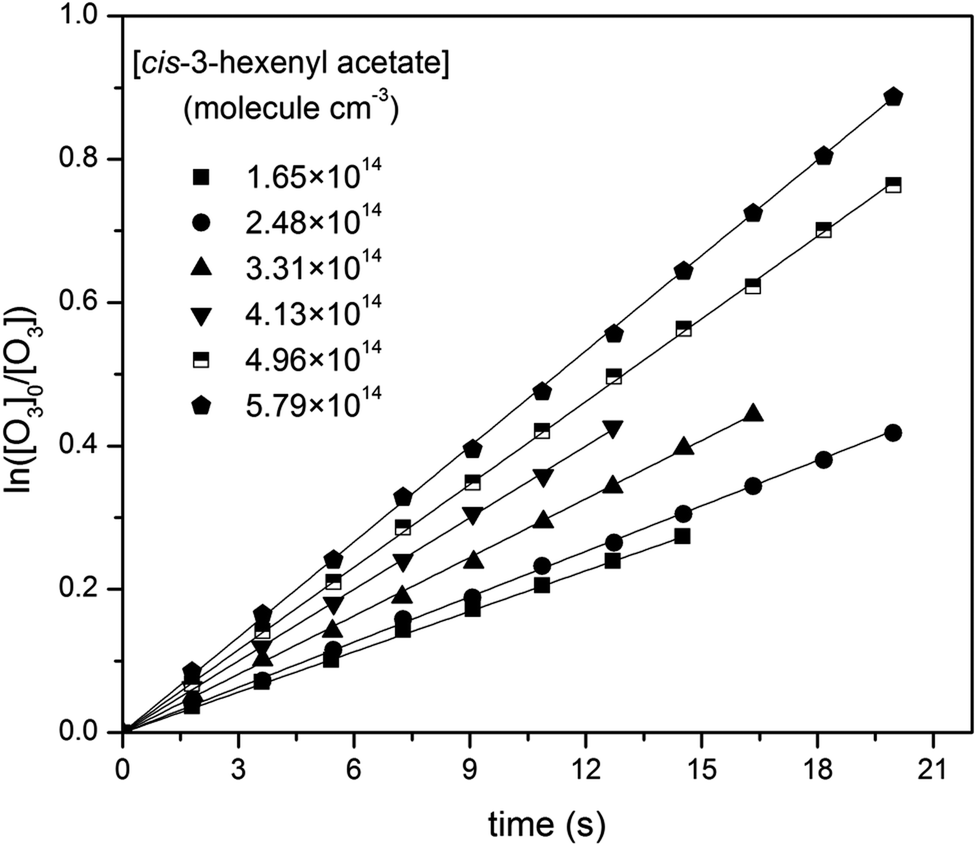 | ||
| Fig. 2 Pseudo-first-order plots for O3 reactions with different concentrations of cis-3-hexenyl acetate (in molecule per cm3). | ||
Plotting k′corrected against the initial ester concentrations yields the second order rate constant. Plots for four studied esters are shown in Fig. 3, all of which showed good linearity. As a result, the acquired rate constants for the studied reactions are as follows, with the errors including 2σ from the least-square fitting and a systematic error estimated ∼10% which was believed to include errors from the measurement of [O3], the initial concentration calculation of the esters, the flow measurement in the reactor, and some other possible errors.
| kcis-3-hexenyl formate = (4.06 ± 0.66) × 10−17 cm3 per molecule per s |
| kcis-3-hexenyl acetate = (5.77 ± 0.70) × 10−17 cm3 per molecule per s |
| kcis-3-hexenyl propionate = (7.62 ± 0.88) × 10−17 cm3 per molecule per s |
| kcis-3-hexenyl butyrate = (12.34 ± 1.59) × 10−17 cm3 per molecule per s |
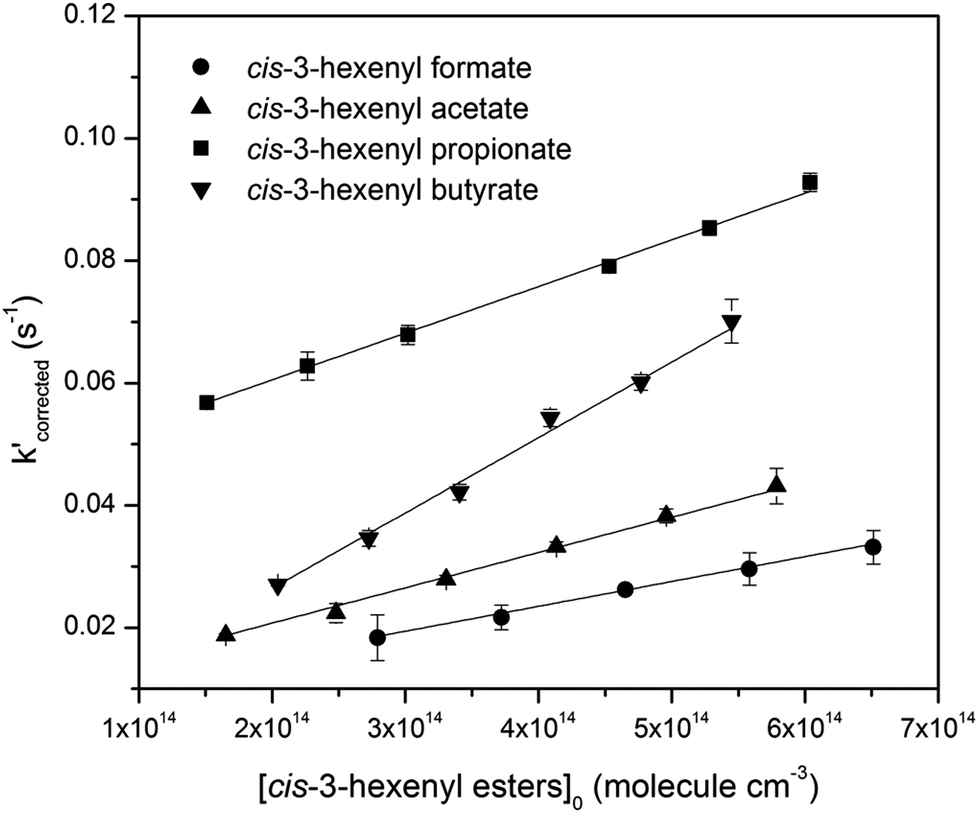 | ||
| Fig. 3 Plots of k′ (corrected) versus initial ester concentrations at room temperature. The lines represent linear least-squares fitting (in order to express more clearly, the plots of cis-3-hexenyl propionate was artificially shifted upward by 0.04). | ||
Of the four cis-3-hexenyl esters studied here, only reaction of cis-3-hexenyl acetate has been studied previously. Using cis-2-butene as reference compound, Atkinson et al. gave a relative rate constant of (5.4 ± 1.4) × 10−17 cm3 per molecule per s.21 Then Grosjean et al. reported an absolute rate constant for this reaction, which was (5.9 ± 0.87) × 10−17 cm3 per molecule per s.22 The value we measured in this work at (5.77 ± 0.70) × 10−17 cm3 per molecule per s agreed very well with both of these literature data within the stated uncertainties. For reactions of O3 with other three esters, to the best of our knowledge, no previously reported kinetic values are available, and thus, no direct comparisons with literature are made here.
Reaction mechanism and theoretical rate constants
The O3 reactions with unsaturated compounds are initiated by the 1,3-dipolar addition of O3 to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond through a transition state (TS), which leads to the production of a primary ozonide (POZ).24,37,38 Scheme 1 illustrates the mechanism of cis-3-hexenyl formate with O3. Structures of the reactants, the transition states (TS) and the primary ozonide for the reaction of O3 with cis-3-hexenyl formate are given in Fig. 4, and those for other esters are given in Fig. S7–S9.† There are two conformers (cis and trans) of the reactant for the cis-3-hexenyl formate, cis-3-hexenyl acetate, cis-3-hexenyl propionate and cis-3-hexenyl butyrate, respectively. Eight transition states, TS1–TS8, were identified associated with the production of the primary ozonides (POZ1–POZ8) from each unsaturated ester ozonolysis reactions. The potential energy surface for the reaction pathways are presented in Fig. 5 and S10–S12† at the BH&HLYP/6-311+G(d,p) level of theory. All of the ozone addition reactions are highly exothermic, with the reaction energies more than 270 kJ mol−1.
C bond through a transition state (TS), which leads to the production of a primary ozonide (POZ).24,37,38 Scheme 1 illustrates the mechanism of cis-3-hexenyl formate with O3. Structures of the reactants, the transition states (TS) and the primary ozonide for the reaction of O3 with cis-3-hexenyl formate are given in Fig. 4, and those for other esters are given in Fig. S7–S9.† There are two conformers (cis and trans) of the reactant for the cis-3-hexenyl formate, cis-3-hexenyl acetate, cis-3-hexenyl propionate and cis-3-hexenyl butyrate, respectively. Eight transition states, TS1–TS8, were identified associated with the production of the primary ozonides (POZ1–POZ8) from each unsaturated ester ozonolysis reactions. The potential energy surface for the reaction pathways are presented in Fig. 5 and S10–S12† at the BH&HLYP/6-311+G(d,p) level of theory. All of the ozone addition reactions are highly exothermic, with the reaction energies more than 270 kJ mol−1.
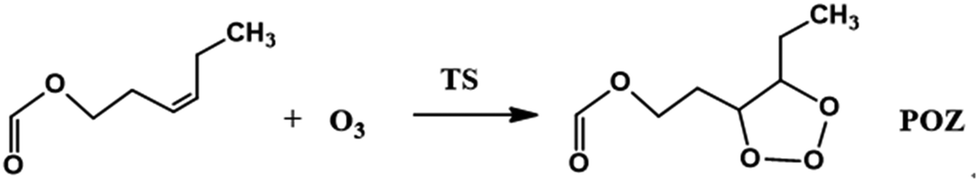 | ||
| Scheme 1 | ||
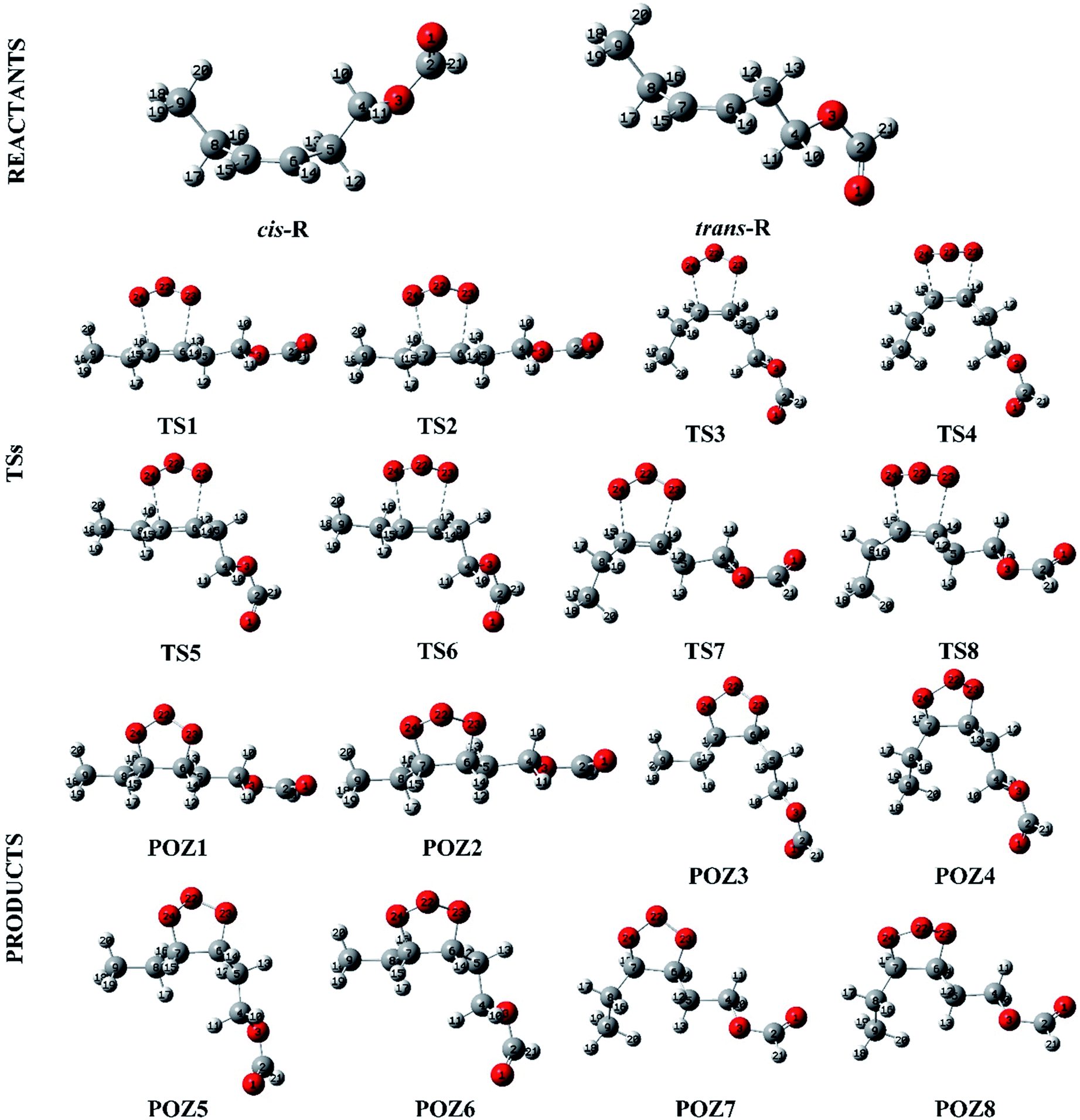 | ||
| Fig. 4 Structures for the reactants, transition states and products along the cis-3-hexenyl formate + O3 reaction profile. | ||
 | ||
| Fig. 5 BHandHLYP/6-311+G(d,p) energy profile (in kJ mol−1) for the cis-3-hexenyl formate ozonolysis. | ||
Rate constants including tunneling at 298 K for each unsaturated ester were calculated. There are several methods for tunneling corrections.39–41 In this work, Eckart tunneling42 was used, which has been performed on the TheRate program.43 The rate constant is expressed as
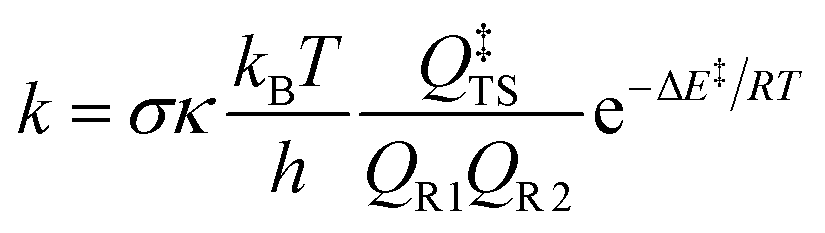
The calculated rate constants for the reactions of ozone with each unsaturated ester are 6.60 × 10−17 cm3 per molecule per s, 6.84 × 10−17 cm3 per molecule per s, 10.84 × 10−17 cm3 per molecule per s, and 17.89 × 10−17 cm3 per molecule per s, respectively.
Reactivity trends
As mentioned above, the reaction of ozone with unsaturated VOCs begins with the electrophilic addition of ozone to the carbon–carbon double bond. Thus, the factors that affect the electronic density on the double bond will also affect the reactivity of unsaturated VOCs toward ozone. Table 1 shows the rate constants for the reactions studied in this work and those for reactions of several compounds with similar structures in the literature.| Esters | Formula | kO3 |
|---|---|---|
| a This work. | ||
| Vinyl acetate | CHCH2OC(O)CH3 |
3.0 × 10−18 (ref. 48) |
| Vinyl propionate | CHCH2OC(O)CH2CH3 |
5.3 × 10−18 (ref. 49) |
| cis-2-Hexenyl acetate | CH3(CH2)2CHCHCH2OC(O)CH3 |
2.50 × 10−17 (ref. 50) |
| cis-3-Hexene | CH3CH2CHCHCH2CH3 |
14.4 × 10−17 (ref. 51) |
| cis-3-Hexen-1-ol | CH3CH2CHCHCH2CH2OH |
5.47 × 10−17 (ref. 24) |
| cis-3-Hexenal | CH3CH2CHCHCH2CHO |
3.5 × 10−17 (ref. 52) |
| cis-3-Hexenyl formate | CH3CH2CHCH(CH2)2OC(O)H |
4.06 × 10−17a |
| cis-3-Hexenyl acetate | CH3CH2CHCH(CH2)2OC(O)CH3 |
5.77 × 10−17a |
| cis-3-Hexenyl propionate | CH3CH2CHCH(CH2)2OC(O)CH2CH3 |
7.62 × 10−17a |
| cis-3-Hexenyl butyrate | CH3CH2CHCH(CH2)2OC(O)(CH2)2CH3 |
12.34 × 10−17a |
It can be seen that, the rate constants for the four studied cis-3-hexenyl esters are all smaller than that of cis-3-hexene, reflecting the electron-withdrawing effect of the –OC(O)R function group. And the rate constants toward O3 increase with the length of R group (R from H, CH3, CH2CH3 to CH2CH2CH3). The theoretical calculation results show similar reactivity trend, i.e., the reaction rate increases from cis-3-hexenyl formate to cis-3-hexenyl butyrate. When the ester group is separated from the CC double bond by CH2 group, comparing vinyl acetate with cis-3-hexenyl acetate, and vinyl propionate with cis-3-hexenyl propionate in Table 1, the electron-withdrawing influence of the ester group on the reactivity reduced, and the rate constants increased by more than 10 times, from 10−18 cm3 per molecule per s to 10−17 cm3 per molecule per s. Obviously, the farther the distance between the ester group and the CC bond, the smaller the influence of the ester group on the reactivity and the greater the reaction rate constants. For vinyl acetate, cis-2-hexenyl acetate (one CH2 group between the ester group and CC bond) and cis-3-hexenyl acetate (two CH2 groups between the ester group and CC bond), the rate constants increased from 3.0 × 10−18 cm3 per molecule per s to 2.50 × 10−17 and 5.77 × 10−17 cm3 per molecule per s, respectively. For the cis-3-hexenyl compounds, the reactivity toward O3 also varies with the nature of the substituent. Table 1 compares the reactivity of cis-3-hexene, cis-3-hexen-1-ol (replacing –CH3 by –CH2OH), cis-3-hexenyl formate (replacing –CH3 by –CH2OC(O)H) and cis-3-hexenal (replacing –CH3 by –C(O)H), with the rate constants at 14.4 × 10−17, 5.47 × 10−17, 4.06 × 10−17 and 3.5 × 10−17 cm3 per molecule per s, respectively, indicating the relative magnitude of electron-withdrawing effect of these substituents in the order –C(O)H > –CH2OC(O)H > –CH2OH. The rate constants of cis-3-hexenyl acetate, cis-3-hexenyl propionate and cis-3-hexenyl butyrate are all larger than that of cis-3-hexen-1-ol, which is the result that the alkyl group in ester group weakens the electron withdrawing effect of the ester group.
Atmospheric implications
The atmospheric implications of this work can be analyzed through comparison of the lifetimes of the cis-3-hexenyl esters with respect to reaction with the main atmospheric oxidants, such as OH, NO3, O3 and Cl. Lifetimes were calculated using the expression τ = 1/(k[X]), where [X] represents the concentration of the oxidant and k is the corresponding rate constant. Rate constants for these reactions are correlated with the highest occupied molecular orbital (HOMO) energies of the unsaturated VOCs.53,54 Thus, the rate constants without reported experimental values were estimated using their HOMO energies which were calculated at the HF/6-31G** level of theory using the Gaussian 09 program and listed in Table S1.† The obtained rate constants for these cis-3-hexenyl esters and the calculated lifetimes are summarized in Table 2.| Esters | k (cm3 per molecule per s) | τf (hours) | ||||||
|---|---|---|---|---|---|---|---|---|
| kOH × 1011 | kNO3 × 1014c | kO3 × 1017d | kCl × 1010 | τOH | τNO3 | τO3 | τCl | |
| a Rodriguez et al., 2015.55b Atkinson et al., 1995.21c King et al., 1999, ln(kOH) = 1.13EHOMO − 13.11; ln(kNO3) = 6.37EHOMO + 30.54.53d This work.e Timerghazin et al., 2001, ln(kCl) = 0.8EHOMO − 13.8.54f Atmospheric concentrations of the oxidants used were: [O3] = 7 × 1011 molecule per cm3,56 [OH] = 1.0 × 106 molecule per cm3,57 [NO3] = 5 × 108 molecule per cm3,58 [Cl] = 5 × 103 molecule per cm3.59 | ||||||||
| cis-3-Hexenyl formate | 4.61a | 3.67 | 4.06 | 2.45a | 6.0 | 15.1 | 9.8 | 226.8 |
| cis-3-Hexenyl acetate | 7.84b | 5.77 | 5.77 | 4.77e | 3.5 | 9.6 | 6.9 | 116.5 |
| cis-3-Hexenyl propionate | 4.07c | 6.09 | 7.62 | 4.80e | 6.8 | 9.1 | 5.2 | 115.7 |
| cis-3-Hexenyl butyrate | 4.08c | 6.25 | 12.34 | 4.82e | 6.8 | 8.9 | 3.2 | 115.3 |
It is clear from Table 2 that reactions with OH, NO3 radicals and O3 should all play important role in the atmospheric removal of the studied cis-3-hexenyl esters. Lifetimes estimated for these esters are all in the range of a few hours, which means that these esters are likely to be rapidly removed in the vicinity of their emission source. In the daytime, OH radical plays a major role; in the night, NO3 radical becomes the main oxidant for these esters; while ozone can compete with them either day or night. The contribution of Cl atoms seems small. However, due to their high reaction rate, it may be significant in areas with high chlorine atom concentrations, such as coastal areas and some mid-continental polluted areas.60–62 Other possible removal processes for these esters, such as photolysis, dry and wet depositions, could be considered negligible, since they do not absorb radiation in the actinic region, and are highly volatile and insoluble in water.63 It should be noted that the ozone concentration used in Table 2 is only the background concentration (28 ppb) in the remote region, where O3 reactions have already been comparable to the reaction with OH and NO3 radicals. In some urban areas, ozone concentrations frequently exceed 120 ppb and are even much higher.64,65 In this case, the reaction with ozone will become the most important degradation pathway of these unsaturated esters in the atmosphere.
Reactions of these unsaturated esters with ozone are expected to produce some carbonyl compounds (such as aldehydes, ketones or esters) and some organic nitrates, which could further be subject to reaction with OH or photolysis, contributing to the formation of secondary organic aerosol (SOA) or photochemical smog. Therefore, further studies should be conducted on the product distribution of these reactions and their contribution to the formation of SOA. In addition, for reactions with OH, NO3 radicals and Cl atoms, both theoretical and experimental studies are still necessary to obtain their accurate rate constants and to reevaluate the atmospheric implications of these reactions.
Conclusions
In summary, the rate constants of the reactions of O3 with four cis-3-hexenyl esters and isoprene were measured using absolute method in a laminar flow reactor. At 298 K and atmosphere pressure, the following values (in 10−17 cm3 per molecule per s) were obtained: 4.06 ± 0.66 for cis-3-hexenyl formate, 5.77 ± 0.70 for cis-3-hexenyl acetate, 7.62 ± 0.88 for cis-3-hexenyl propionate, 12.34 ± 1.59 for cis-3-hexenyl butyrate, and 1.23 ± 0.16 for isoprene. The results of isoprene and cis-3-hexenyl acetate showed very good agreement with the literature data, indicating the reliability of this reactor in the kinetic study. To the best of our knowledge, it was the first kinetic study for the O3 reactions with cis-3-hexenyl formate, cis-3-hexenyl propionate and cis-3-hexenyl butyrate. Theoretical calculations were also performed to better understand the kinetics and the mechanism of the title reactions. The results showed that reactions of these unsaturated esters with O3 are slower than their corresponding parent alkene because of the electron-withdrawing effect of the –OC(O)R function group, and the extent of this effect was obviously dependent on the structure of the R group, as well as the relative distance between the double bond and the ester group. It can be found from the calculation of atmospheric lifetimes that reaction with O3 is one of the most important removal pathways for these esters in the atmosphere. They are expected to have short atmospheric lifetimes due to their high reactivity with OH, NO3 radicals and O3. Therefore, further research on the reaction products will be necessary to assess the effect of these reactions on regional pollution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 91544228, 41575125, 41605102, 41775125), the National Key Research and Development Program of China (2016YFC0202205), and the Presidential Foundation of Hefei Institutes of Physical Science (No. YZJJ201508), CAS.Notes and references
- A. Guenther, C. N. Hewitt, D. Erickson, R. Fall, C. Geron, T. Graedel, P. Harley, L. Klinger, M. Lerdau, W. A. Mckay, T. Pierce, B. Scholes, R. Steinbrecher, R. Tallamraju, J. Taylor and P. Zimmerman, J. Geophys. Res., 1995, 100, 8873–8892 CrossRef CAS.
- A. H. Goldstein and I. E. Galbally, Environ. Sci. Technol., 2007, 41, 1514–1521 CrossRef CAS PubMed.
- J. D. Fuentes, M. Lerdau, R. Atkinson, D. Baldocchi, J. W. Bottenheim, P. Ciccioli, B. Lamb, C. Geron, L. Gu, A. Guenther, T. D. Sharkey and W. Stockwell, Bull. Am. Meteorol. Soc., 2000, 81, 1537–1575 CrossRef.
- R. Atkinson and J. Arey, Atmos. Environ., 2003, 37, S197–S219 CrossRef CAS.
- E. G. Colman, M. B. Blanco, I. Barnes and M. A. Teruel, RSC Adv., 2015, 5, 30500–30506 RSC.
- A. Hofzumahaus, F. Rohrer, K. D. Lu, B. Bohn, T. Brauers, C. C. Chang, H. Fuchs, F. Holland, K. Kita, Y. Kondo, X. Li, S. R. Lou, M. Shao, L. M. Zeng, A. Wahner and Y. H. Zhang, Science, 2009, 324, 1702–1704 CrossRef CAS PubMed.
- L. K. Whalley, D. Stone, B. Bandy, R. Dunmore, J. F. Hamilton, J. Hopkins, J. D. Lee, A. C. Lewis and D. E. Heard, Atmos. Chem. Phys., 2016, 16, 2109–2122 CrossRef CAS.
- X. R. Ren, D. van Duin, M. Cazorla, S. Chen, J. Q. Mao, L. Zhang, W. H. Brune, J. H. Flynn, N. Grossberg, B. L. Lefer, B. Rappengluck, K. W. Wong, C. Tsai, J. Stutz, J. E. Dibb, B. T. Jobson, W. T. Luke and P. Kelley, J. Geophys. Res.: Atmos., 2013, 118, 5770–5780 CAS.
- Lipids in Plant and Algae Development. Subcellular Biochemistry, ed. Y. Nakamura and Y. Li-Beisson, Springer, Cham, 2016, vol. 86, pp. 427–443 Search PubMed.
- M. M. Maja, A. Kasurinen, T. Holopainen, R. Julkunen-Tiitto and J. K. Holopainen, Sci. Total Environ., 2016, 547, 39–47 CrossRef CAS PubMed.
- J. Arey, A. M. Winer, R. Atkinson, S. M. Aschmann, W. D. Long and C. L. Morrison, Atmos. Environ., Part A, 1991, 25, 1063–1075 CrossRef.
- G. Konig, M. Brunda, H. Puxbaum, C. N. Hewitt, S. C. Duckham and J. Rudolph, Atmos. Environ., 1995, 29, 861–874 CrossRef.
- F. Brilli, T. M. Ruuskanen, R. Schnitzhofer, M. Müller, M. Breitenlechner, V. Bittner, G. Wohlfahrt, F. Loreto and A. Hansel, PLoS One, 2011, 6, e20419 CAS.
- K. Jardine, G. A. Barron-Gafford, J. P. Norman, L. Abrell, R. K. Monson, K. T. Meyers, M. Pavao-Zuckerman, K. Dontsova, E. Kleist, C. Werner and T. E. Huxman, Photosynth. Res., 2012, 113, 321–333 CrossRef CAS PubMed.
- W. Kirstine, I. Galbally, Y. R. Ye and M. Hooper, J. Geophys. Res., 1998, 103, 10605–10619 CrossRef CAS.
- F. Brilli, L. Hortnagl, I. Bamberger, R. Schnitzhofer, T. M. Ruuskanen, A. Hansel, F. Loreto and G. Wohlfahrt, Environ. Sci. Technol., 2012, 46, 3859–3865 CrossRef CAS PubMed.
- W. E. Chehab, R. Kaspi, T. A. Savchenko and K. Dehesh, Proceedings of ANAS, Biological Sciences, 2010, 65, 145–151 Search PubMed.
- D. Helmig, L. F. Klinger, A. Guenther, L. Vierling, C. Geron and P. Zimmerman, Chemosphere, 1999, 38, 2163–2187 CrossRef CAS PubMed.
- J. Ruther, J. Chromatogr. A, 2000, 890, 313–319 CrossRef CAS PubMed.
- M. M. Maja, A. Kasurinen, T. Holopainen, S. Kontunen-Soppela, E. Oksanen and J. K. Holopainen, Tree Physiol., 2015, 35, 975–986 CrossRef PubMed.
- R. Atkinson, J. Arey, S. M. Aschmann, S. B. Corchnoy and Y. H. Shu, Int. J. Chem. Kinet., 1995, 27, 941–955 CrossRef CAS.
- E. Grosjean and D. Grosjean, Int. J. Chem. Kinet., 1998, 30, 21–29 CrossRef CAS.
- Y. B. Gai, M. F. Ge and W. G. Wang, Atmos. Environ., 2011, 45, 53–59 CrossRef CAS.
- X. X. Lin, Q. Ma, C. Q. Yang, X. F. Tang, W. X. Zhao, C. J. Hu, X. J. Gu, B. Fang, Y. B. Gai and W. J. Zhang, RSC Adv., 2016, 6, 83573–83580 RSC.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- K. H. Møller, R. V. Otkjær, N. Hyttinen, T. Kurtén and H. G. Kjaergaard, J. Phys. Chem. A, 2016, 120, 10072–10087 CrossRef PubMed.
- D. Johnson and G. Marston, Chem. Soc. Rev., 2008, 37, 699–716 RSC.
- S. M. Aschmann, J. Arey and R. Atkinson, Atmos. Environ., 2002, 36, 4347–4355 CrossRef CAS.
- T. L. Malkin, A. Goddard, D. E. Heard and P. W. Seakins, Atmos. Chem. Phys., 2010, 10, 1441–1459 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Atmos. Chem. Phys., 2006, 6, 3625–4055 CrossRef CAS.
- IUPAC, Task Group on Atmospheric Chemical Kinetic Data Evaluation-Data sheets for gas phase organic reactions: Ox + VOC, http://iupac.pole-ether.fr/.
- F. Kaufman, J. Phys. Chem., 1984, 88, 4909–4917 CrossRef CAS.
- L. F. Keyser, J. Phys. Chem., 1984, 88, 4750–4758 CrossRef CAS.
- H. Chen, Y. Ren, M. Cazaunau, V. Daële, Y. Hu, J. Chen and A. Mellouki, Chem. Phys. Lett., 2015, 621, 71–77 CrossRef CAS.
- W. J. Massmana, Atmos. Environ., 1998, 32, 1111–1127 CrossRef.
- L. Vereecken and J. S. Francisco, Chem. Soc. Rev., 2012, 41, 6259–6293 RSC.
- E. Gaona-Colman, M. B. Blanco, I. Barnes, P. Wiesen and M. A. Teruel, RSC Adv., 2017, 7, 2733–2744 RSC.
- W. H. Miller, Y. Zhao, M. Ceotto and S. Yang, J. Chem. Phys., 2003, 119, 1329 CrossRef CAS.
- C. Aieta, F. Gabas and M. Ceotto, J. Phys. Chem. A, 2016, 120, 4853–4862 CrossRef CAS PubMed.
- C. Aieta and M. Ceotto, J. Chem. Phys., 2017, 146, 214115 CrossRef PubMed.
- C. Eckart, Phys. Rev., 1930, 35, 1303–1309 CrossRef CAS.
- W. T. Duncan, R. L. Bell and T. N. Truong, J. Comput. Chem., 1998, 19, 1039–1052 CrossRef CAS.
- B. Long, W. J. Zhang, X. F. Tan, Z. W. Long, Y. B. Wang and D. S. Ren, J. Phys. Chem. A, 2011, 115, 1350–1357 CrossRef CAS PubMed.
- B. Long, X. F. Tan, C. R. Chang, W. X. Zhao, Z. W. Long, D. S. Ren and W. J. Zhang, J. Phys. Chem. A, 2013, 117, 5106–5116 CrossRef CAS PubMed.
- J. R. Alvarez-Idaboy, N. Mora-Diez and A. Vivier-Bunge, J. Am. Chem. Soc., 2000, 122, 3715–3720 CrossRef CAS.
- J. R. Alvarez-Idaboy, N. Mora-Diez, R. J. Boyd and A. Vivier-Bunge, J. Am. Chem. Soc., 2001, 123, 2018–2024 CrossRef CAS PubMed.
- B. Picquet-Varrault, M. Scarfogliero and J. F. Doussin, Environ. Sci. Technol., 2010, 44, 4615–4621 CrossRef CAS PubMed.
- E. Gaona Colman, M. B. Blanco, I. Barnes and M. Teruel, Chem. Phys. Lett., 2013, 579, 11–15 CrossRef CAS.
- J. Li, Y. H. Sun, H. J. Cao, D. D. Han and M. X. He, Struct. Chem., 2014, 25, 71–83 CrossRef CAS.
- E. Grosjean and D. Grosjean, Int. J. Chem. Kinet., 1996, 28, 461–466 CrossRef CAS.
- J. H. Xing, M. Ono, A. Kuroda, K. Obi, K. Sato and T. Imamura, J. Phys. Chem. A, 2012, 116, 8523–8529 CrossRef CAS PubMed.
- M. D. King, C. E. Canosa-Mas and R. P. Wayne, Phys. Chem. Chem. Phys., 1999, 1, 2231–2238 RSC.
- Q. K. Timerghazin and P. A. Ariya, Phys. Chem. Chem. Phys., 2001, 3, 3981–3986 RSC.
- D. Rodriguez, A. Rodriguez, I. Bravo, A. Garzon, A. Aranda, Y. Diaz-de-Mera and A. Notario, Int. J. Environ. Sci. Technol., 2015, 12, 2881–2890 CrossRef CAS.
- J. A. Logan, J. Geophys. Res., 1985, 90, 10463–10482 CrossRef.
- W. J. Bloss, M. J. Evans, J. D. Lee, R. Sommariva, D. E. Heard and M. J. Pilling, Faraday Discuss., 2005, 130, 425–436 RSC.
- Y. Shu and R. Atkinson, J. Geophys. Res., 1995, 100, 7275–7281 CrossRef CAS.
- A. A. P. Pszenny, W. C. Keene, D. J. Jacob, S. Fan, J. R. Maben, M. P. Zetwo, M. Springer-Young and J. N. Galloway, Geophys. Res. Lett., 1993, 20, 699–702 CrossRef CAS.
- A. Rodriguez, I. Bravo, D. Rodriguez, M. Tajuelo, Y. Diaz-de-Mera and A. Aranda, RSC Adv., 2016, 6, 21833–21843 RSC.
- J. A. Thornton, J. P. Kercher, T. P. Riedel, N. L. Wagner, J. Cozic, J. S. Holloway, W. P. Dube, G. M. Wolfe, P. K. Quinn, A. M. Middlebrook, B. Alexander and S. S. Brown, Nature, 2010, 464, 271–274 CrossRef CAS PubMed.
- X. X. Liu, H. Qu, L. G. Huey, Y. H. Wang, S. Sjostedt, L. M. Zeng, K. D. Lu, Y. S. Wu, M. Hu, M. Shao, T. Zhu and Y. H. Zhang, Environ. Sci. Technol., 2017, 51, 9588–9595 CrossRef CAS PubMed.
- M. B. Blanco, I. Bejan, I. Barnes, P. Wielsen and M. A. Teruel, J. Phys. Chem. A, 2009, 113, 5958–5965 CrossRef CAS PubMed.
- Y. Wang, P. Konopka, Y. Liu, H. Chen, R. Muller, F. Ploger, M. Riese, Z. Cai and D. Lu, Atmos. Chem. Phys., 2012, 12, 8389–8399 CrossRef CAS.
- X. Pu, T. J. Wang, X. Huang, D. Melas, P. Zanis, D. K. Papanastasiou and A. Poupkou, Sci. Total Environ., 2017, 603–604, 807–816 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13369c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |