
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriple network hydrogels (TN gels) prepared by a one-pot, two-step method with high mechanical properties
Xiangong Wang,
Fang Zhao,
Bo Pang,
Xuping Qin* and
Shengyu Feng *
*
Key Laboratory of Special Functional Aggregated Materials, Key Laboratory of Colloid and Interface Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, P. R. China. E-mail: fsy@sdu.edu.cn; qinxvping@sdu.edu.cn
First published on 12th February 2018
Abstract
In this work, poly(vinyl alcohol) (PVA) was incorporated into the networks of polyacrylamide/polyacrylic acid (PAM/PAA) to prepare novel PAM/PAA/PVA Triple-network (TN) hydrogels by an in situ polymerization and repeated freezing–thawing (F–T) process. The TN hydrogels have not only high mechanical strength, but also a moderate swelling ability by varying the weight ratio of calcium chloride (CaCl2) and PVA and free shaping. The compressive stress of the as-prepared hydrogels could reach 11 MPa, and the highest stretching stress could reach 0.8 MPa. Upon mechanical loading, the coordination network between PAA and CaCl2 served as sacrificial bonds to efficiently dissipate energy. However, they can reform when the mechanical load is released, resulting from the fast coordination between PAA and Ca2+. Therefore, TN hydrogels have potential application in biomaterials.
Introduction
Hydrogels are hydrophilic three-dimensional polymers that have remarkable capacities to hold water and have achieved much progress in sensing,1,2 drug delivery,3 actuation,4 tissue engineering and so on.5–8 Both synthetic and natural polymers can be used for the production of hydrogels,9–12 achieved by methods such as physical crosslinking,13 chemical gelation or self-assembly.14–16The combination of covalent and physical bonds has been proven as an ideal method to produce a hydrogel with high strength, elongation, and excellent recovery properties.17 Cations can form coordination bonds with the appropriate ligands and, in most cases, dissociate and associate reversibly and rapidly.18–20 This has led to their use as physical crosslink points for preparing high strength hydrogels because of the ease of energy dissipation and recovery of mechanical properties in a short amount of time.21,22
Articular cartilage can reach 60% of compressibility strain, and possesses the excellent mechanical properties of quick stress relaxation, large energy dissipation, strain stiffening, and fast recovery of the mechanical properties after the release of load.23 The cartilage has a water content of 70–80% from its extracellular matrix composition.24,25 Inspired by the mechanical properties of articular cartilage, many tough hydrogels have been designed with double-network hydrogels (DN gels).26,27 For instance, Suo et al. prepared a new type of DN hydrogel through combining the networks of PAM and alginate (ionic), which is notch-insensitive, very tough and stretchable up to 20 times the initial length, but still soft, with a tensile strength of only ≈160 kPa.28 Yu et al. designed a stretchable and tough hydrogel cross-linked between Ca2+ and graphene oxide. But its tensile stress is also only about 150 kPa.
PVA is a water soluble synthetic polymer of great interest because of its many desirable characteristics, such as its viscoelastic nature, high water content, and biocompatibility. Therefore, it is advantageous to use it as a biomaterial for fabricating medical devices to be used as synthetic articular cartilage.29 PVA can be a semi-crystalline structure, formed through intramolecular hydrogen bonding, resulting from the possession of a large number of hydroxyl side groups. PVA can be made into hydrogels through chemical or physical cross-linking, such as glutaraldehyde acting as a functional cross-linking agents,30 or ionizing radiation. Physically cross-linked PVA-based hydrogels in water can be prepared by the F–T process, in which, the concentrated PVA aqueous solution induces crystallization, leading to the formation of a network structure.31 Alternatively, PVA physical hydrogels have drawn even greater research attention, in which, this method addresses toxicity issues because it does not result in the reagent residuals and chemical toxicity; consequently, no toxicity agents leaching from the gel. Furthermore, these physically cross-linked hydrogels, which are inter-connected by hydrogen bonding, exhibit more porous, spongy, rubbery and higher elastic properties than PVA hydrogels prepared by other methods.32 Currently, these gel matrices have been widely implemented in biotechnology fields. Crystallites serve as physical cross-links that are both reversible and stable, but, unfortunately, PVA hydrogels synthesized using this method are brittle.33
Most hydrogels have low stiffness (10 kPa), strength (100 kPa) and toughness (10 J m−2).34,35 Despite recent progress, it is imperative to develop effective methods of preparing novel hydrogels with excellent comprehensive performance of in terms of mechanical properties,36,37 the ability to control water-absorption, and recovery properties in the as-prepared state, as well as in the swelling equilibrium states.
In this work, we have prepared hydrogels with a low covalently cross-linked PAM plus PAA network, a physically cross-linked PVA network through crystallizing by the F–T process, and an ionically cross-linked network between PAA and Ca2+. In this gels, acrylamide (AM) and acrylic acid (AA) were made for the first network cross-linking, with micro-scale a chemical cross-linker, through an in situ polymerization. Meanwhile, the chains of PVA interpenetrated with network of PAM, PAA and the F–T process, resulting in crystallites, which acted as junction points in the hydrogel. At the same time, the ionic cross-linking between PAA and Ca2+ made another interpenetration with PVA, in which the third network was created (Scheme 1). All PAM, PAA chains and PVA chains form a continuous network, which closely correlates with the mechanical properties of the TN gels. The as-prepared hydrogels coexisted physical network and covalent network. Ionic bonds and crystallites have been used as the sacrificial network, which greatly improved the recovery of the mechanical stability. Based on the coexisting high density physical bonds, stress could be greatly dissipated by unzipping physical bonds during the deformation process.
 | ||
| Scheme 1 Preparation of TN gels by using one-pot, two step method. | ||
The Triple network (TN) has a synergetic effect, which can dramatically increase the mechanical properties. Because the robust hydrogels were synthesized via a one-pot, two-step procedure, the hydrogels can be prepared in different desired shapes controlled by the shapes of the mould.
Results and discussion
Mechanical properties
The typical stress–strain curves of the prepared hydrogels are shown in Fig. 1. From Fig. 1a, with a fixed weight ratio of AM/AA at 2 and the content of CaCl2 at 4.2% (w/v), the tensile stress and toughness gradually increase with the increasing content of PVA, while the elongation decreased. The tensile strength of the 2-P0-C4.17 TN gel is only ≈61 kPa at a strain of 1045%. In contrast, the stress of the 2-P8.3-4.17 TN gel can reach 0.804 MPa, which is 13 times higher than that of the 2-P0-C4.17 TN gels. The corresponding tensile strain is 365%, which is much less than that of the 2-P8.3-4.17 TN gels, and the toughness38 is 1.505 M J−3. The results indicate that the introduction of PVA can significantly improve the mechanical strength, excluding the tensile strain. This occurs from the physical cross-linked density of the as-prepared hydrogel, which is increased through the F–T process and the molecular entanglement between PAM, PAA and PVA, which was strengthened by introducing more PVA. Futheremore, the tensile strain decreased with increasing physical cross-linked density. | ||
| Fig. 1 Stress–strain curves (a), compressive stress–strain curves (b) and toughness (c) of the hydrogels at various content of PVA. | ||
The compressive stress–strain curves of the hydrogels are shown in Fig. 1b. The as-prepared hydrogels could be compressed to a strain of 90%, and the stress at this strain was used to evaluate the compressive strength of the as-prepared hydrogels. The curves of the as-prepared hydrogels, which went through the F–T process for a total of three cycle showed that the compressive stress decreased with increasing content of PVA. At this condition, the ionic coordination bonds of the hydrogels play an important role in maintaining the excellent compressive performance.
Another series of experiments' fixed the content of PVA and CaCl2 but with a changing weight ratio of AM/AA (Fig. 2). When the weight ratio of AM/AA increased from 0.2 to 1, the tensile strength increased considerably, from 64.3 kPa to 0.572 MPa, and the corresponding ruptured strain increased from 417% to 612% (Fig. 2a). For the different weight ratio of AM/AA, 0.572 MPa was the highest tensile strength. When the weight ratio of AM/AA increased from 1 to 5, the tensile strength decreased considerably from 0.572 MPa to 0.263 MPa and the corresponding ruptured strains decreased from 612% to 491%. The toughness increased dramatically, from near 0.6 M J m−3 to 1.7 M J m−3, with the decreasing weight ratio of AM/AA (Fig. 2c). The results indicated that the mechanical strength of the hydrogel has a strong dependence on the weight ratios of AM/AA. Mechanical strength can be changed by regulating the content of AA, influencing the coordination interaction.
 | ||
| Fig. 2 Stress–strain curves (a), compressive stress–strain curves (b) and toughness (c) of the hydrogels with changing the weight ratio of AM/AA. | ||
The compressive strain–stress curves of the as-prepared hydrogels are shown in Fig. 2b. The compressive stress increased with the decreasing weight ratio of AM/AA, in which the rules of change differ from those of the stretching stress. The results indicated that the compressive stress was enhanced with increasing AA content and excess Ca2+ cross-linking with PAA, resulting in increased ion coordination bonds, and improving the compressive strength resulting from the physical cross-linked density.
Another series of experiments were conducted, in which the content of PVA and ratio of AM/AA were fixed, with only the content of CaCl2 varied (Fig. 3). The breaking strain changed slightly with the increasing content of CaCl2, and the stretching stress of the 2-P4.17-C0.92 TN gels increased from 0.30 MPa to 0.35 MPa of the 2-P4.17-C4.17 TN gels. Therefore, the results suggest that the influence of CaCl2 on the stress and strain was getting slightly with the increasing content of CaCl2. Excess CaCl2 limited improvement of the stress and strain of the as-prepared hydrogels, such that superfluous Ca2+ diffused into the hydrogels, leading to enlargement of the ratio of mono-dentate/bi-edentates. The toughness (Fig. 3c) showed that different content of CaCl2 had different influences on the stress and strain. The results of the compressive strain–stress analysis of the as-prepared hydrogels are shown in Fig. 3b; compressive stress increased with the decreasing content of CaCl2.
 | ||
| Fig. 3 Stress–strain curves (a), compressive stress–strain curves (b) and toughness (c) of the hydrogels at various content of CaCl2. | ||
All samples could sustain knotting (Fig. 4a), torsion (Fig. 4b), elongation (Fig. 4c–d), compression (Fig. 4e–g). Moreover, they had the ability to recover to the initial state when the load was released. The knotting behaviour of the TN gels indicated that they were highly elastic and stretchable. The 2-P4.17-C0.21 TN gels with a diameter of 8 mm, could lift up a steel block of ≈1.5 kg (Fig. 4h and i) which directly indicates that the hydrogels have high mechanical strength. For the sake of comparison, the 0.2-P4.17-C4.17 TN gels went freezing–thawing for a single cycle and three cycles to reveal if the number of F–T cycle has an important influence on the compressive stress of the as-prepared hydrogels. The results (Fig. 5a) showed that the compressive stress could increase 10 times with three cycles of the F–T process, in comparison to a single cycle of the hydrogel. Therefore, the number of F–T cycles can improve the compressive stress of the hydrogels, increasing the physical cross-linked density with the increasing number of F–T cycles.
 | ||
| Fig. 4 Compressive and tensile properties of TN gels. (a) Knotting. (b) Torsion. (c, d) Elongation. (e–g) Compression. (h, i) The 2-P4.17-C0.21 TN gels with a diameter of 8 mm lift up steel block of ≈1.5 kg in weight. | ||
 | ||
| Fig. 5 (a) Compressive stress–strain curves of the 0.2-P4.17-C4.17 TN gels with one cycle or three cycles of the F–T process. (b) Five successive loading–unloading cycles of the as–prepared samples. (c) Compressive stress–strain curves at a strain of 80% for three successive loading-unloading cycles. | ||
The loading–unloading tests were measured to evaluate the energy dissipation of the TN gels. Successive loading–unloading cycles were applied to the 2-P4.17-C0.21 TN gels, with testing aimed to make the hydrogels experience enough fatigue. Five cycles of 275% stretching loading–unloading results are shown in Fig. 5b and 3 cycles of 80% compressing loading–unloading are shown in Fig. 5c. For the as-prepared hydrogels, the residual strain is almost 0%, and a large obvious hysteresis loop can be observed in the first loading–unloading. During the immediate second to fifth cycles of loading–unloading experiments, the residual strain is the same to the first loading–unloading test, with almost no hysteresis observed. This indicated that the TN structure was only changed by the first – loading process, owing to the hydrogel' robustness and ability to undergo multiple loading–unloading cyclic stretching experiments without damage. Meanwhile, the almost unchanged hysteresis loops also demonstrated that during the multiple loading-unloading cyclic stretching experiments, stress was well dissipated by the physical coactions of the Ca2+ and PAA chain unzipping process and the damage to PVA crystallites, while chemical cross-linked networks of the hydrogels remained intact. From Fig. 5c, the 2-P4.17-C0.21 TN gels expressed a large hysteresis loop in the first loading–unloading cycle, while in the second and third compression loading–unloading cycles, no hysteresis can be observed, with the last 2 cycles almost overlapping with the first unloading curve. The hydrogels can almost fully recover their mechanical properties within a few seconds. This finding indicates that the TN structure was changed at the first compression loading process and that the hydrogel only dissipated energy effectively at the first compression cycle. The pure PVA hydrogels produced by the F–T process are unrecoverable; the physical bonds result from the fast self-assembly speed and high self-assembly fidelity between PAA and Ca2+. The almost unchanged hysteresis loops indicated that, during the multiple loading–unloading cyclic compression experiments, stress was well dissipated by the physical coaction during the Ca2+ and PAA unzipping process and damage to the PVA crystallites.
Fig. 6a shows the loading–unloading curves at different strains for the 2-P4.17-C0.21 TN gels. Apparent hysteresis loops are observed on the loading–unloading curves, indicating that the 2-P4.17-C0.21 TN gels dissipate energy effectively. The data are in Fig. 6b, which clearly shows that the as-prepared hydrogel could dissipate energy as much as 0.086 M J m−3 at a strain of 325%. Furthermore, there was no hysteresis loop for the sample hydrogel at a strain of 100%, indicating that the hydrogel could not dissipate energy and that it exhibited typical rubber elastic behaviour.
 | ||
| Fig. 6 (a) Loading–unloading tests of the 2-P4.17-C0.21 TN gels under different strain (50%, 100%, 200%, 250%, 325%), (b) the calculated total toughness and the dissipated toughness of the 2-P4.17-C0.21 TN gels during the loading–unloading tests under different strains. | ||
The compressive stress–strain curves of the as-prepared hydrogel and hydrogel with 91.1% water content are shown in Fig. 7a. The results of compressive experiments show that the hydrogel with 91.1% water content began its destruction at a stress of 4.5 MPa, without reaching a strain of 90%. Alternatively, the as–prepared hydrogel could reach a stress of 5 MPa at a strain of 90% without destruction. However, the compressive stress of the hydrogel with 91.1% water content was higher than the as-prepared hydrogel at the same strain (before an 80% strain). Therefore, the results indicate that hydrogels with high water content have excellent performance during the compressing process before reaching an 80% strain, and the highest compressive stress of the as-prepared hydrogel reached 11 MPa (Fig. 7b).
 | ||
| Fig. 7 (a) Compressive stress–strain curves of the 2-P4.17-C4.17 TN gels with different content of water. (b) Compressive stress–strain curves of the 0.2-P4.17-C4.17 TN gel with a total of three cycles of the F –T process. | ||
SEM results
Fig. 8 depicts the SEM image of cross-section morphologies of the sample. According to Fig. 8a and b, the PAM hydrogels exhibited a closely packed honeycomb-like structure. The cross-section morphology of the 2-P4.17-C0.21 TN gels (freeze-dried) is shown in Fig. 8c and d. From Fig. 8c, numerous spines and a few pores were present. And the rough structure indicates the molecular entanglement between the chains of PAM/PAA and PVA. This morphological change explains the swelling capacity of the gels. With the introduction of PVA, the physical crosslinking density of the hydrogel was increased by the F–T process cycles, and the molecular entanglement between PAM, PAA and PVA was strengthened, leading to a decreased hydrogel water content but increased tensile strength. In the freeze-drying process (hydrogels dehydrated by lyophilization), the PVA of the hydrogel was frozen into a crystal area and the pores of the PAM, PAA produced by lyophilization were eventually filled with the chains or the crystal area of PVA. | ||
| Fig. 8 (a, b) SEM images of PAM hydrogels. (c, d) SEM images of the 2-P4.17-C0.21 TN gels. | ||
Fourier transform infrared (FTIR) spectroscopy
The FTIR results of the gels are shown in Fig. 9a. A broad band appearing at 3430 cm−1 in the PVA gel spectra corresponds to stretching vibrations of the hydroxyl groups. And a band appearing in the spectra of 1095 cm−1 is related to C–O stretching. The PAM gel showed bands at 3435 cm−1 and 3200 cm−1, assigned to a stretching vibration of N–H. The bands at 1658 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1622 cm−1 (N–H deformation for primary amine), 1450 cm−1 (CH2 in-plane scissoring), 1418 cm−1 (C–N stretching for primary amide), 1317 cm−1 (C–H deformation), and 1120 cm−1 (NH2 inplane rocking) were also detected. The FTIR spectra of the TN gel was shown in Fig. 9a. The characteristic peaks of the PAM and PVA gel can be found compared with them.
O stretching), 1622 cm−1 (N–H deformation for primary amine), 1450 cm−1 (CH2 in-plane scissoring), 1418 cm−1 (C–N stretching for primary amide), 1317 cm−1 (C–H deformation), and 1120 cm−1 (NH2 inplane rocking) were also detected. The FTIR spectra of the TN gel was shown in Fig. 9a. The characteristic peaks of the PAM and PVA gel can be found compared with them.
 | ||
| Fig. 9 (a) FTIR spectra of pure PVA, PAM and TN gel. (b) XRD patterns of PVA, PAM, 2-P6.25-C4.17 TN and 2-P4.17-C4.17 TN gels. | ||
X-ray diffraction (XRD)
The XRD results (Fig. 9b) showed a amorphous diffraction for the PAM gels with a broad peak at 2θ = 41 degree. And the PVA gel exhibited a typical peak that appeared at 2θ = 20 degree, corresponding to the mixture of (101) and (200) plane of PVA semi-crystalline in pure PVA gel.39,40 PVA crystallinity decreased with decreasing PVA content in the TN gels. And PVA semi-crystalline can be formed in TN gels. The molecular entanglement between PAM, PAA and PVA could account for the decreasing of PVA crystallinity.Swelling behaviour
Fig. 10a displays the equilibrium swelling ratio (ESR) of the hydrogels in deionized water (DI). The swelling ability of the hydrogels increased with declining PVA content. The 2-P0-C4.17 TN gels showed a maximum swelling ratio of 1015.6%, compared to 587.0% for the 2-P8.33-C4.17 TN gels. Less cross-linked hydrogels tended to show higher water absorption ability, because the highly cross-linked structure could not sustain a large amount of water within the gel structure, reducing the space and ability to take up water during the F–T process. For the TN gels, when the PVA content decreased, the physical cross-linked density of the hydrogel network decreased, and molecular entanglement between PAM/PAA and PVA was weakened, which led to improvement in the hydrogel swelling ability. At the same time, the swelling capability was strongly influenced by the hydrophilicity of the hydrogels. The swelling ability of the gels, with a changing AM/AA ratio, is shown in Fig. 10b. By enhancing the weight ratio of AM/AA, the cross-linking between PAA and Ca2+ decreased, and the hydrophilic groups of the hydrogel networks increased, and consequently, the hydrogel absorbed more water, demonstrating improved swelling ability. Fig. 10c shows the ESR of the hydrogels in DI with changing CaCl2; the weight ratio of AM/AA and PVA were unchanged. Furthermore, the ESR of the hydrogels gradually increased with the decreasing CaCl2 content, excess CaCl2 was cross-linked with PAA with the increasing AA content. Therefore, the hydrophilic groups were decreased, leading to a decreased swelling ability. The swelling ability of the prepared hydrogels can be improved by controlling the ratio of AA/AM, AM/PVA and AA/CaCl2, according to the swelling experiments.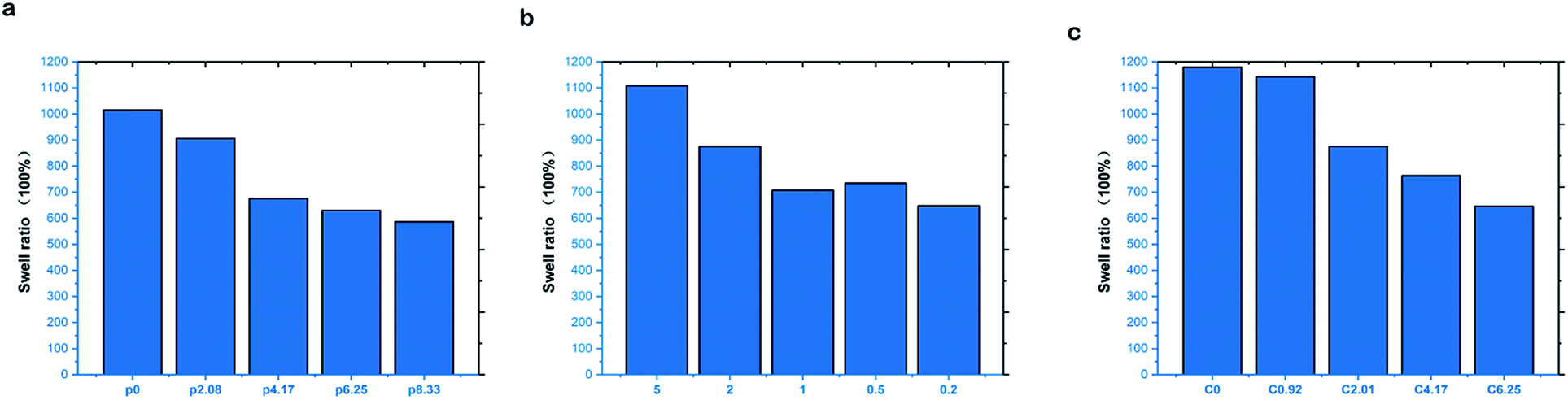 | ||
| Fig. 10 (a) The swelling ability of the hydrogels at various weight ratio of AM/AC. (b) The swelling ability of the prepared hydrogels at various content of PVA. (c) The swelling ability of the prepared hydrogels at different content of CaCl2. | ||
Experimental
Materials
AM, AA, and CaCl2 were purchased from Kermel. PVA (average Mw = 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was purchased from Sinopharm Chemical Co, Ltd. Ammonium persulfate (APS), N,N,N′,N′-tetramethylethylenediamine (TEMED) and N,N′-methylenebisacrylamide (BIS) were purchased from Energy Chemical. The concentrations of the stock solution of TEMED (liquid), APS, and BIS were fixed at 5% v/v, 1% w/v and 1% w/v, respectively. All materials were used as received.
000 g mol−1) was purchased from Sinopharm Chemical Co, Ltd. Ammonium persulfate (APS), N,N,N′,N′-tetramethylethylenediamine (TEMED) and N,N′-methylenebisacrylamide (BIS) were purchased from Energy Chemical. The concentrations of the stock solution of TEMED (liquid), APS, and BIS were fixed at 5% v/v, 1% w/v and 1% w/v, respectively. All materials were used as received.
Preparation of TN gels
The hydrogels were prepared using a two-step method. First, the hydrogels were synthesized through an in situ radical polymerization of the monomers (AM and AA) in an aqueous solution of PVA with a small amount of BIS, which acted as a cross-linker. The TN gels were then obtained by an F–T process. The initial concentration of AM plus AA was kept at 25% w/v, whereas the concentration of PVA was varied between 0 and 8.33% w/v; the concentrations of TEMED, APS, and BIS were 0.208% v/v, 0.083% w/v, and 0.025% w/v, respectively. First, PVA was added to DI and stirred continuously at 90 °C for about 2 h to form a homogeneous, transparent solution with the designated concentration. AM, AA, CaCl2, BIS and PVA solutions were then added into a flask, and the solution was stirred vigorously with a magnetic stirrer at room temperature for 2 h. Argon gas was bubbled through the solution for 30 min, and the APS (1 ml) and TEMED (1 ml) were introduced. The mixture solution was transferred into the mould, and the radical polymerization was carried out in a water bath at 65 °C for 8 h. The hydrogels were removed from the mould after polymerization, frozen at −20 °C for 12 h and thawed at 25 °C for 8 h, for three consecutive cycles. Finally, the samples were stored in the as-prepared state for mechanical property testings.In this article, the as prepared TN gels are denoted as x–Py–Cz, where P and C indicate PVA and CaCl2, respectively, x represents the weight ratio of AM/AA, and y, z represent the corresponding concentrations in the form PVA/DI (w/v) and CaCl2/DI (w/v), respectively. For example, 2-P2.1-C0.92 TN gels represent a gel containing AM/AA with a ratio of 2, PVA with a PVA/DI ratio of 2.1% w/v and CaCl2 with a CaCl2/DI ratio of 0.92% w/v. FT represents the Freezing–thawing process. S and W represent the cycles of loading–unloading experiments and water content, respectively.
Measurements of mechanical properties
Tensile and compressive tests were performed on the as-prepared hydrogel samples with a Universal Testing Machine (WDW-10) at room temperature. The hydrogel samples for tensile testing were rod-like shapes 50 mm in length, while the diameter of the as-prepared hydrogel samples was 7 mm, with a crosshead speed of 100 mm min−1. The tensile strain was taken as the length change related to the initial length, and the tensile stress was evaluated on the cross-section of the initial sample. The toughness was calculated from the area of the stress–strain curves. For compression testing, cylindrical samples of the as-prepared hydrogel were (30 mm diameter and 30 mm height). The crosshead speed was 5 mm min−1, and the compression strength was obtained using the stress at deformation of 90%. The hysteresis of the as-prepared gels samples was measured at a deformation of 80%.Swelling experiments
The swelling properties of the hydrogel were characterized by a gravimetric method.41 The dried hydrogels were immersed in excess DI to reach the state of swelling equilibrium at room temperature. The equilibrium swelling ratio was calculated as:where Ws is the weight of the swollen gel and Wd is the weight of the dry gel.
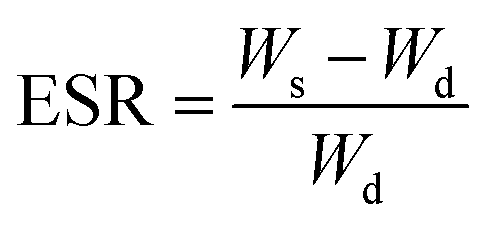
Scanning electron microscopy (SEM)
The surface morphology of the hydrogels after being freeze dried was investigated by scanning electron microscope (JSM-6700F, Japan) using 3.0 kV voltage. Prior to observation, the freeze dried fracture sections were coated with a thin layer of platinum to enhance conductivity.X-ray diffraction (XRD)
A D8 Advance X-ray diffractometer (Bruker, Germany, 40 kV, 40 mA, Cu (Kα) radiation, λ = 1.54184 Å) was used to measure the crystallinity of the TN gels. The XRD results were obtained from 5° to 80°, 2θ at a rate of 10° min−1.Fourier transform infrared (FTIR) spectroscopy
The hydrogels were freeze-dried by a lyophilizer for 24 h at −50 °C to eliminate water molecules from the gels. FTIR spectra were recorded between 400 and 4000 cm−1 on a FTIR Spectrometer from Bruker Optics Inc.Conclusions
In this work, we synthesized and characterized TN gels. A PAM, PAA network and PVA network can be simultaneously mixed and interspersed together in a unique one-pot, two-step reaction, with the resulting hydrogels having anti-fatigue and super-tough properties. The PAM, PAA network provided a rigid scaffold for holding up the shape of the TN gels, whereas the PVA polymers in the PAM/PAA networks play a role in energy absorption. Some PVA chains entangled with the PAM, PAA chains, and the PAA and Ca2+ made another network by ionic cross-linking. This simple two-step method is particularly interesting when a chemical, physical and ionic network is mixed into the hydrogels to enhance the mechanical strength. Even hydrogels with high water content had the excellent compressive performance. The mechanical properties and absorbing ability of the TN gels can be adjusted by regulating the concentration of PVA and the number of F–T cycles; the compressive stress can be regulated by controlling the AM/AA weight ratio. The as-prepared hydrogels have more potential applications as materials for articular cartilage and artificial muscles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21774070 and 21274080) and Fund for Shandong province major scientific and technological innovation projects (No. 2017CXGC1112).Notes and references
- C. Zhang, G. G. Cano and P. V. Braun, Adv. Mater., 2014, 26, 5678–5683 CrossRef CAS PubMed.
- Y. Wang, L. Wang, T. Yang, X. Li, X. Zang, M. Zhu, K. Wang, D. Wu and H. Zhu, Adv. Funct. Mater., 2014, 24, 4666–4670 CrossRef CAS.
- J. Lu, C. Cheng, Y. S. He, C. Lyu, Y. Wang, J. Yu, L. Qiu, D. Zou and D. Li, Adv. Mater., 2016, 28, 4025–4031 CrossRef CAS PubMed.
- M. K. Shin, G. M. Spinks, S. R. Shin, S. I. Kim and S. J. Kim, Adv. Mater., 2009, 21, 1712–1715 CrossRef CAS.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed.
- N. Annabi, S. R. Shin, A. Tamayol, M. Miscuglio, M. A. Bakooshli, A. Assmann, P. Mostafalu, J. Y. Sun, S. Mithieux, L. Cheung, X. S. Tang, A. S. Weiss and A. Khademhosseini, Adv. Mater., 2016, 28, 40–49 CrossRef CAS PubMed.
- M. A. Haque, T. Kurokawa and J. P. Gong, Polymer, 2012, 53, 1805–1822 CrossRef CAS.
- K. Yasuda, N. Kitamura, J. P. Gong, K. Arakaki, H. J. Kwon, S. Onodera, Y. M. Chen, T. Kurokawa, F. Kanaya, Y. Ohmiya and Y. Osada, Macromol. Biosci., 2009, 9, 307–316 CrossRef CAS PubMed.
- K. Haraguchi and T. Takada, Macromolecules, 2010, 43, 4294–4299 CrossRef CAS.
- E. Ozturk, O. Arlov, S. Aksel, L. Li, D. M. Ornitz, G. Skjak-Braek and M. Zenobi-Wong, Adv. Funct. Mater., 2016, 26, 3649–3662 CrossRef CAS PubMed.
- J. Liu, D. Su, J. Yao, Y. Huang, Z. Shao and X. Chen, J. Mater. Chem. A, 2017, 5, 4163–4171 CAS.
- E. Tous, J. L. Ifkovits, K. J. Koomalsingh, T. Shuto, T. Soeda, N. Kondo, J. H. Gorman III, R. C. Gorman and J. A. Burdick, Biomacromolecules, 2011, 12, 4127–4135 CrossRef CAS PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- S. Hong, D. Sycks, H. F. Chan, S. Lin, G. P. Lopez, F. Guilak, K. W. Leong and X. Zhao, Adv. Mater., 2015, 27, 4035–4040 CrossRef CAS PubMed.
- W. Sun, B. Xue, Y. Li, M. Qin, J. Wu, K. Lu, J. Wu, Y. Cao, Q. Jiang and W. Wang, Adv. Funct. Mater., 2016, 26, 9044–9052 CrossRef CAS.
- X. Qin, F. Zhao, Y. Liu, H. Wang and S. Feng, Colloid Polym. Sci., 2009, 287, 621–625 CAS.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- P. Lin, S. Ma, X. Wang and F. Zhou, Adv. Mater., 2015, 27, 2054–2059 CrossRef CAS PubMed.
- M. X. Wang, C. H. Yang, Z. Q. Liu, J. Zhou, F. Xu, Z. Suo, J. H. Yang and Y. M. Chen, Macromol. Rapid Commun., 2015, 36, 465–471 CrossRef CAS PubMed.
- L. Zhang, N. R. Brostowitz, K. A. Cavicchi and R. A. Weiss, Macromol. React. Eng., 2014, 8, 81–99 CrossRef CAS.
- Z. Wei, J. He, T. Liang, H. Oh, J. Athas, Z. Tong, C. Wang and Z. Nie, Polym. Chem., 2013, 4, 4601 RSC.
- Z. Li, Z. Hou, H. Fan and H. Li, Adv. Funct. Mater., 2017, 27, 1604379 CrossRef.
- J. Hou, X. Ren, S. Guan, L. Duan, G. H. Gao, Y. Kuai and H. Zhang, Soft Matter, 2017, 13, 1357–1363 RSC.
- Q. Chen, L. Zhu, C. Zhao, Q. Wang and J. Zheng, Adv. Mater., 2013, 25, 4171–4176 CrossRef CAS PubMed.
- H. M. Nizam El-Din, S. G. Abd Alla and A. W. M. El-Naggar, J. Macromol. Sci., Part A: Pure Appl. Chem., 2007, 44, 47–54 CrossRef CAS.
- J. P. Gong, Soft Matter, 2010, 6, 2583 RSC.
- R. E. Webber, C. Creton, H. Brown and J. P. Gong, Macromolecules, 2007, 40, 2919–2927 CrossRef CAS.
- J. Y. Sun, X. Zhao, W. R. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- H. Li, D. Hao, J. Fan, S. Song, X. Guo, W. Song, M. Liu and L. Jiang, J. Mater. Chem. B, 2016, 4, 4662–4666 RSC.
- H. Bodugoz-Senturk, C. E. Macias, J. H. Kung and O. K. Muratoglu, Biomaterials, 2009, 30, 589–596 CrossRef CAS PubMed.
- Q. Wei, Y. Wang, Y. Che, M. Yang, X. Li and Y. Zhang, J. Mech. Behav. Biomed. Mater., 2017, 65, 565–573 CrossRef CAS PubMed.
- L. Zhang, J. Zhao, J. Zhu, C. He and H. Wang, Soft Matter, 2012, 8, 10439 RSC.
- M. Fukasawa, T. Sakai, U.-i. Chung and K. Haraguchi, Macromolecules, 2010, 43, 4370–4378 CrossRef CAS.
- L. W. Xia, R. Xie, X. J. Ju, W. Wang, Q. Chen and L. Y. Chu, Nat. Commun., 2013, 4, 2226 Search PubMed.
- X. Zhao, Soft Matter, 2014, 10, 672–687 RSC.
- J. Wang, L. Lin, Q. Cheng and L. Jiang, Angew. Chem., Int. Ed., 2012, 51, 4676–4680 CrossRef CAS PubMed.
- Z. Hu and G. Chen, Adv. Mater., 2014, 26, 5950–5956 CrossRef CAS PubMed.
- S. S. Kulkarni, A. A. Kittur, M. I. Aralaguppi and M. Y. Kariduraganavar, J. Appl. Polym. Sci., 2004, 94, 1304–1315 CrossRef CAS.
- L. Lu, F. Peng, Z. Jiang and J. Wang, J. Appl. Polym. Sci., 2006, 101, 167–173 CrossRef CAS.
- F. Zhao, X. Qin and S. Feng, RSC Adv., 2016, 6, 100511–100518 RSC.
| This journal is © The Royal Society of Chemistry 2018 |