
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solution growth and thermal treatment of crystals lead to two new forms of 2-((2,6-dimethylphenyl)amino)benzoic acid†
Rong Hu‡
a,
Yunping Zhoujin‡a,
Meng Liua,
Mingtao Zhangb,
Sean Parkind,
Panpan Zhou c,
Jianzhi Wanga,
Faquan Yu*a and
Sihui Long*a
c,
Jianzhi Wanga,
Faquan Yu*a and
Sihui Long*a
aKey Laboratory for Green Chemical Process of Ministry of Education, School of Chemical Engineering and Pharmacy, Wuhan Institute of Technology, 693 Xiongchu Road, Wuhan, Hubei 430073, China. E-mail: fyuwucn@gmail.com; sihuilong@wit.edu.cn; longsihui@yahoo.com; Tel: +86 02787194980 Tel: +86 15549487318
bComputational Center for Molecular Science, College of Chemistry, Nankai University, Tianjin, China
cDepartment of Chemistry, Lanzhou University, Lanzhou, Gansu, China
dDepartment of Chemistry, University of Kentucky, Lexington, Kentucky, USA
First published on 24th April 2018
Abstract
We report the discovery of two new forms (II and III) of a potential non-steroidal anti-inflammatory and thyroid drug, 2-((2,6-dimethylphenyl)amino)benzoic acid (HDMPA) through solution growth and thermal treatment of crystals. Form II has been discovered through crystal growth in a variety of solvents, and characterized by single-crystal X-ray diffraction, powder X-ray diffraction (PXRD), FT-IR, and Raman spectroscopy. Form II converts into form III upon thermal treatment, as indicated by the phase behavior study of form II with differential scanning calorimetry (DSC). Form III has been characterized by IR, Raman and PXRD. Conformational flexibility of the molecule seems to lead to the polymorphism of the system. A conformational scan shows the conformational minima correspond to the conformers in the polymorphs. Lattice energy calculations show energies of −48.14 and −50.31 kcal mol−1 for forms I and II, providing information on the relative stability for each form. Hirshfeld analysis revealed that intermolecular interactions such as C⋯C, H⋯H, C⋯H, and H⋯O contribute to the stability of the crystal forms.
1. Introduction
N-Anthranilic acids, the nitrogen isosteres of salicylic acid, constitute an important class of non-steroidal anti-inflammatory drugs (NSAIDs).1 Classic anthranilic acid NSAIDs include mefenamic acid, meclofenamic acid, chlofenamic acid, and other structurally related compounds.2 Their medical application is not limited to NSAIDs, and they can also be used as analgesics and antirheumatics.1 Recently, this class of compounds have been investigated as therapeutics for neurodegenerative and amyloid diseases as well as cancer, and some have shown great promise.3–5 At the same time, these compounds can be used as ligands in inorganic complexes in the search for anticancer drugs.6 In addition, fenamic acids are also synthetic precursors to acridones and acridines, which present bioactive properties such as anti HIV, anticancer, antibacterial, antifungal, and antimalarial activities.7Likely due to their intrinsic conformational flexibility (as diarylamines), N-anthranilic acids tend to be polymorphic, i.e., existing in more than one crystal form. Several important N-anthranilic acid NSAIDs have multiple crystal forms. For example: two, five, nine, two, and four forms have been found for mefenamic acid,8 tolfenamic acid,9 flufenamic acid,10 niflumic acid,11 and clonixin,12 respectively. The existence of nine forms of flufenamic acid is reported to be the current world record for most forms having been structurally characterized by single-crystal X-ray diffraction. Recently, clonixin was found to form a solvate with N,N-dimethylformamide.13 Polymorphism is important both theoretically (CSP)14,15 and practically.16–19 Polymorphism is of particular significance in pharmaceuticals because different forms of the same API (Active Pharmaceutical Ingredient) may have different kinetic, thermodynamic, surface, mechanical, and packaging properties, which can affect clinical formulation and eventual bioavailability.20,21
HDMPA is an N-anthranilic acid similar to other NSAIDs with potent anti-inflammatory properties.22 In addition, it has ramifications in the treatment of disorders caused by thyroid hormone excess by inhibiting triiodothyronine uptake by the hepatocytes.23
In 2004, in an attempt to synthesize phenylorganotin complexes as anti-tuberculosis drugs, Dokorou et al. discovered the first crystal structure of HDMPA (designated form I). The asymmetric unit of form I consists of two crystallographically independent molecules (Z′ = 2) and the two molecules are distinct in geometry.24
Our lab has been interested in the polymorphism of diarylamines for the past decade. We have systematically investigated a series of compounds using a combination of both experimental and theoretical approaches.25–29 Recently we also studied the polymorphism of an important N-anthranilic acid, 4-chloro-phenylanthranilic acid and highlighted the role played by both the conformational flexibility and sp2 C–H⋯Cl hydrogen bond.30
In this study, we report the discovery and characterization of a 2nd form of HDMPA, as well as the structural comparison of forms I and II, and the generation and characterization of a third form (III) of the compound, and the corresponding theoretical investigation to shed light on the polymorphism of this system.
2. Experimental section
2.1. General
All solvents and reagents were purchased from commercial sources and used as received: 2-chlorobenzoic acid was from J&K Chemical (Beijing, China), 2,6-dimethylaniline was from Energy Chemical (Shanghai, China), and Cu2O was from Aladdin (Shanghai, China); Cu, K2CO3, 2-ethoxyethanol, and the solvents used for crystal growth were from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). The IR and Raman spectra were recorded using an FT-IR Perkin-Elmer LX10-8873 and a Thermo Electron DXR Laser Confocal Microscopy Raman Spectrometer, respectively. NMR spectra were obtained on a Varian INOVA spectrometer at an observation frequency of 400 MHz. Thermal analyses were performed on an SII DSC 6220 (SEIKO, Japan) apparatus, using a heating rate of 10 °C min−1.2.2. Synthesis and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, Rf = 0.50) as eluent. The product was obtained as colorless solid (230 mg, yield%: 11) (Scheme 1).
:1, Rf = 0.50) as eluent. The product was obtained as colorless solid (230 mg, yield%: 11) (Scheme 1).
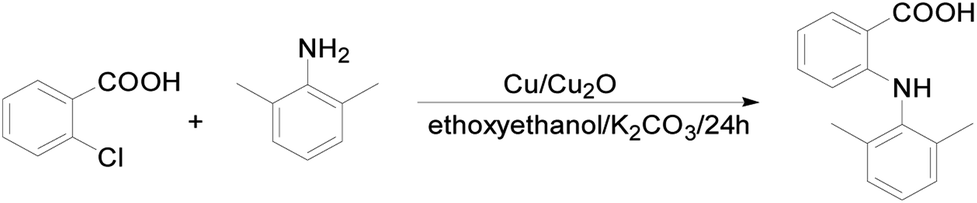 | ||
| Scheme 1 Synthesis of HDMPA. | ||
1H-NMR (400 MHz, CDCl3): δ ppm 8.90 (s, 1H), 8.05 (d, 1H), 7.26 (t, 1H), 7.16 (s, 3H), 6.67 (t, 1H), 6.29 (d, 1H), 2.23 (s, 6H); 13C-NMR (100 MHz, CDCl3): δ ppm 174.2, 150.7, 136.9, 135.6, 132.5, 129.1, 128.0, 127.3, 128.3, 115.8, 112.7, 108.7, 18.2; IR (KBr, cm−1) 3346 (s), 3018–2539 (m), 1651 (s), 1578 (s), 1498(s), 1417 (s), 1250 (s), 1170 (s), 923 (m), 785 (s), 741 (s); MS (ESI): 242.1 (M + 1); mp: 208 °C.
2.3. Crystal growth
Slow evaporation was applied for polymorph screening of HDMPA.32 The experiment is as follows: pure HDMPA was dissolved in eighteen different solvents (the compound is insoluble in water), forming saturated solutions at ambient temperature (∼22 °C) (Table 1). The solutions were set to evaporate in a vibration-free environment until single crystals appeared or no solvent remained. For example, 50 mg of HDMPA was suspended in 5 mL HPLC grade acetonitrile. The mixture was agitated overnight and the remaining solid was pipette filtered. A vial containing the saturated solution was covered with perforated parafilm. Slow evaporation led to single crystals in about a week. Quench cooling was used to obtain form I, as reported in the literature. For quench cooling, a supersaturated solution of HDMPA in a mixture of THF and acetone (1:1 ratio) at 50 °C was rapidly cooled in a freezer of −20 °C. All crystallization experiments were conducted in an unmodified atmosphere. Each experiment was repeated multiple times. All experiments led to crystal form II. The crystals were identified by single-crystal X-ray diffraction when high-quality single crystals were available, and by powder X-ray diffraction for tiny crystals.
| Solvents | Method | Form |
|---|---|---|
| Acetone | Slow evaporation | II |
| Chloroform | Slow evaporation | II |
| Ethyl acetate | Slow evaporation | II |
| Methanol | Slow evaporation | II |
| Dichloromethane | Slow evaporation | II |
| Ethanol | Slow evaporation | II |
| Hexane | Slow evaporation | II |
| Pet ether | Slow evaporation | II |
| Acetonitrile | Slow evaporation | II |
| Ether | Slow evaporation | II |
| iso-Propanol | Slow evaporation | II |
| Dimethyl sulfoxide | Slow evaporation | II |
| Tetrahydrofuran | Slow evaporation | II |
| Dimethylformamide | Slow evaporation | II |
| Toluene | Slow evaporation | II |
| Benzene | Slow evaporation | II |
| Acetic acid | Slow evaporation | II |
| Tetrahydrofuran & acetone (1:1) |
Slow evaporation | II |
| Tetrahydrofuran & acetone (various ratios) | Quench cooling | II |
2.4. Crystal structure determination
The crystal structures of form II HDMPA were determined by single-crystal X-ray diffraction at both 90 K (low temperature, LT) and 296 K (room temperature, RT).Data collection was carried out at 90 K on a Nonius kappaCCD diffractometer with MoKα radiation (λ = 0.71073 Å),33 and at 296 K on a Bruker SMART APEX II diffractometer.
Cell refinement and data reduction were done using SCALEPACK and DENZO-SMN for the low temperature structure,34 and SADABS and Bruker SMART for the room temperature structure. Structure solution and refinement were carried out using the SHELXS and SHELXL2016 programs, respectively.35,36
Powder X-ray diffraction (PXRD) data for each sample were collected on a Rigaku X-ray diffractometer with CuKα radiation (40 kV, 40 mA, λ = 1.5406 Å) between 5.0–50.0° (2θ) at ambient temperatures. The finely ground sample was placed on a quartz plate in an aluminum holder.
2.5. Thermal analyses
Phase behavior for the solid forms were studied by differential scanning calorimetry (DSC), vide supra.2.6. Spectroscopic studies
For IR spectrum recording, samples were dispersed in KBr pellets. For Raman measurement, samples were compressed in a gold-coated sample holder.2.7. Computational details
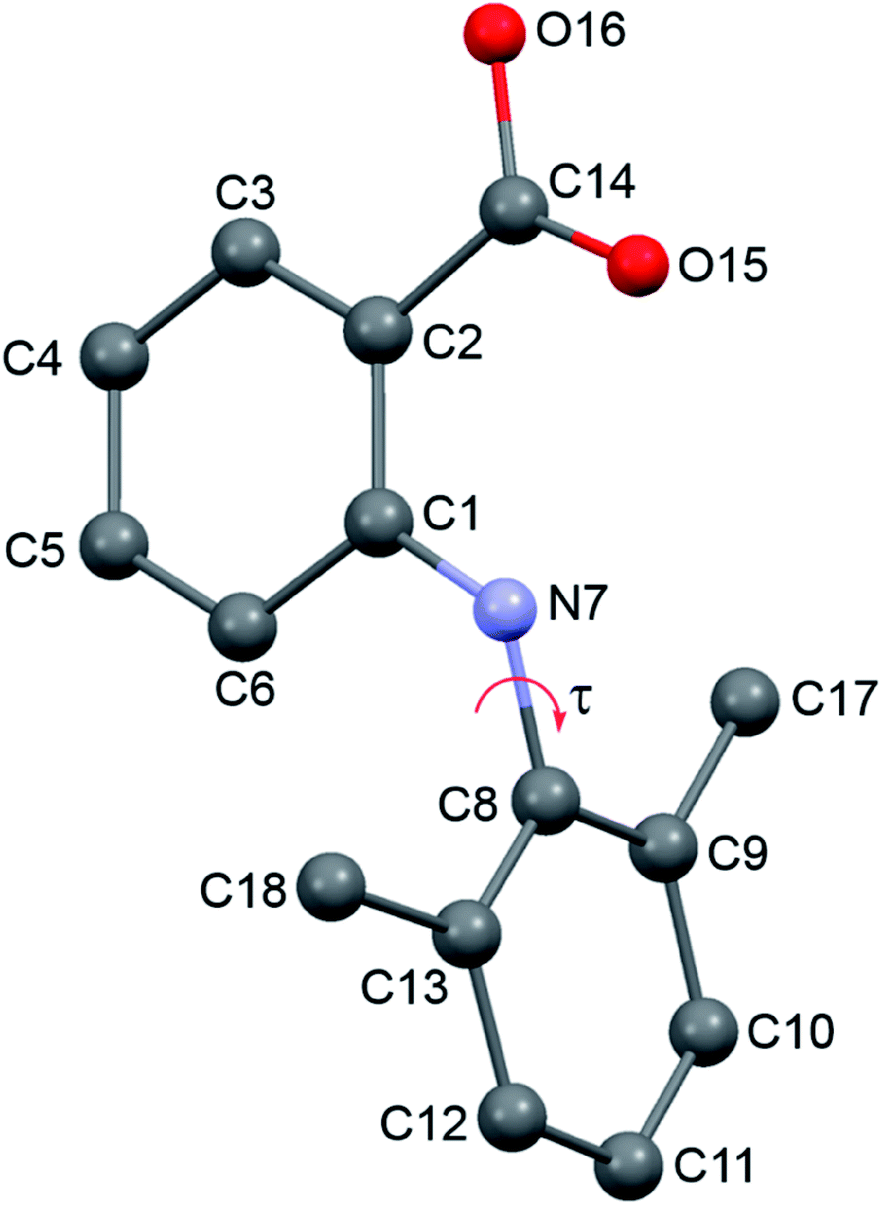 | ||
| Fig. 1 Labeling of HDMPA. | ||
| Conformer | Bond length (Å) | |
|---|---|---|
| C1–N7 | N7–C8 | |
| I-A | 1.380 | 1.432 |
| I-B | 1.363 | 1.465 |
| II (RT) | 1.375 | 1.441 |
| II (LT) | 1.378 | 1.441 |
3. Results and discussion
3.1. Crystal structures
Two new polymorphs (II and III) of HDMPA have been discovered in this study (Fig. 2). Form II crystals were grown as colorless blocks from (but not limited to) acetone, and form III was generated upon thermal treatment of form II samples. Form I crystals were obtained as colorless plates from a mixture of THF and acetone.24 Structure determination by single-crystal X-ray diffraction found form II to be triclinic, space group P (Z = 2), and form I was reported to be triclinic, space group P (Z = 4). No single crystal structure was determined for form III. Crystallographic data for form I and two structures of form II are given in Table 3; for complete CIF files, see the ESI.† The crystallographically independent conformations in each of the asymmetric units are slightly different, as suggested by the dihedral angle between the two aromatic rings (88.25(38)° for I-A and 86.69(34)° for I-B in form I, 88.91(4)° for II in form II). Conformational variability is also shown by a superposition of all three experimental conformations (Fig. 3). | ||
| Fig. 2 SEM micrographs of crystals of HDMPA form II from different solvents and form III. | ||
| I | II (LT) | II (RT) | |
|---|---|---|---|
| Formula | C15H15NO2 | C15H15NO2 | C15H15NO2 |
| Formula weight | 241.28 | 241.28 | 241.28 |
| Crystal size (mm) | 0.50 × 0.50 × 0.15 | 0.30 × 0.20 × 0.20 | |
| Crystal system | Triclinic | Triclinic | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
| a/Å | 15.8375(16) | 7.4127(1) | 7.524(2) |
| b/Å | 7.5311(7) | 8.0298(1) | 8.103(2) |
| c/Å | 11.1845(12) | 10.9583(2) | 11.191(3) |
| α/° | 83.728(9) | 73.3544(8) | 72.582(4) |
| β/° | 104.806(9) | 83.3366(8) | 83.740(4) |
| γ/° | 79.038(8) | 73.6078(8) | 73.854(2) |
| Z, Z′ | 4, 2 | 2, 1 | 2, 1 |
| V/Å3 | 1248.560 | 599.091(16) | 625.1(3) |
| Dcal/g cm−3 | 1.284 | 1.338 | 1.276 |
| T/K | 296 | 90(2) | 296(2) |
| Abs. coeff. (mm−1) | 0.089 | 0.085 | |
| F(000) | 256 | 254 | |
| Range (deg) | 1.94–27.49 | 1.91–25.00 | |
| Limiting indices | −9 ≤ h ≤ 9 | −6 ≤ h ≤ 8 | |
| −10 ≤ k ≤ 10 | −9 ≤ k ≤ 9 | ||
| −14 ≤ l ≤ 14 | −13 ≤ l ≤ 12 | ||
| Completeness to 2θ | 99.5% | 98.5% | |
| Unique reflections | 2737 | 2170 | |
| R1[I > 2σ(I)] | 0.0553 | 0.0434 | 0.0490 |
| wR2 (all data) | 0.0553 | 0.1190 | 0.1552 |
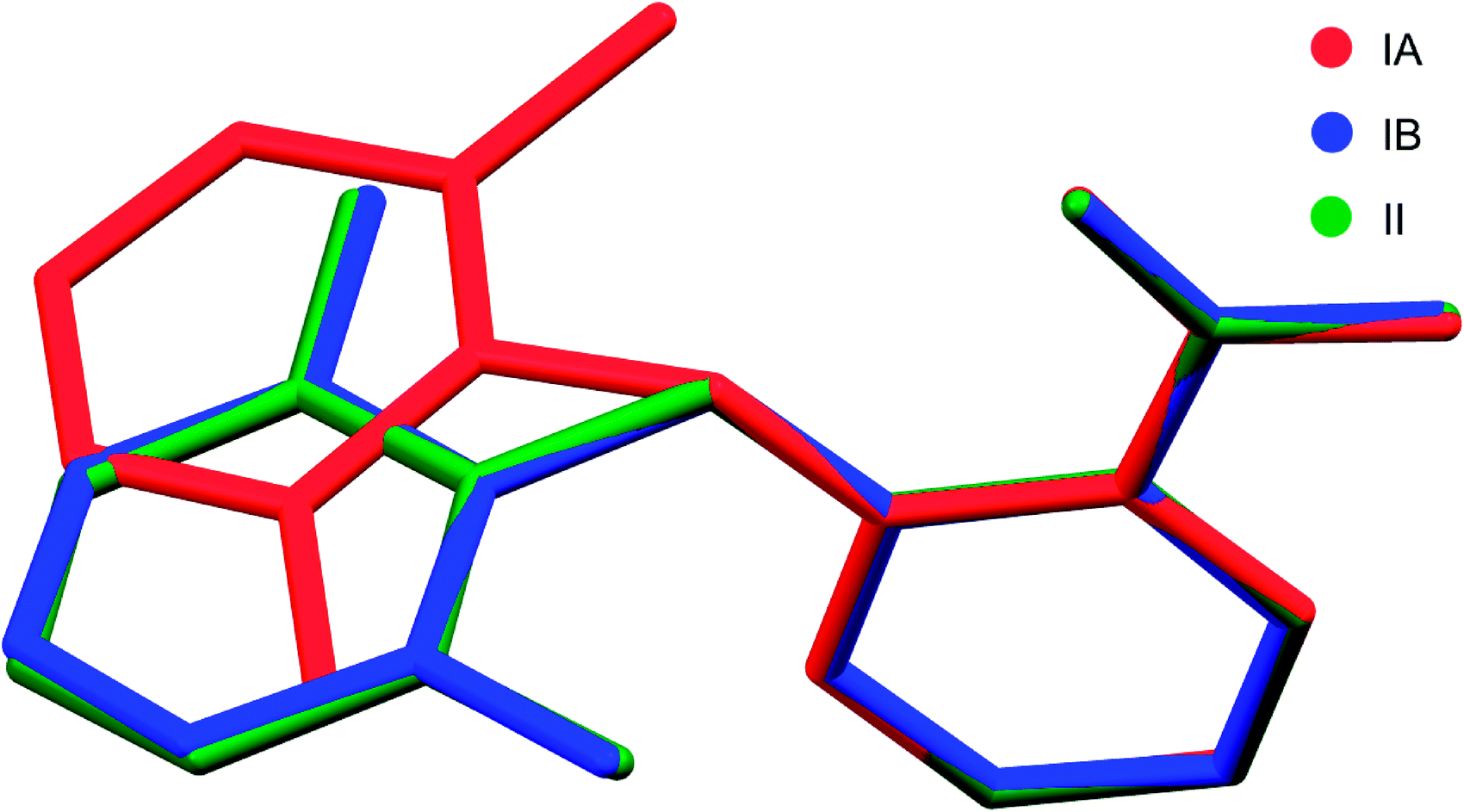 | ||
| Fig. 3 Superposition of all three molecular conformations in the asymmetric units of forms I and II of HDMPA. | ||
In form I, the asymmetric unit consists of two molecules (Z′ = 2), each having a twisted conformation. The two molecules form an acid–acid dimer hydrogen-bonding motif (R22(8)).44–46 The intermolecular hydrogen bond has a bond distance and bond angle of 1.888 Å and 138.28° for O16–H16⋯O15 for molecule A, and 1.830 Å and 140.31° for O16–H16⋯O15 for molecule B. In addition to the intermolecular hydrogen bonds, an intramolecular hydrogen bond exists between the carboxylic acid carbonyl O and the NH bridging the two aromatic rings, with a bond distance of 2.140 Å and bond angle 119.94° for molecule A and 2.071 Å and 120.79° for molecule B (Fig. 4).
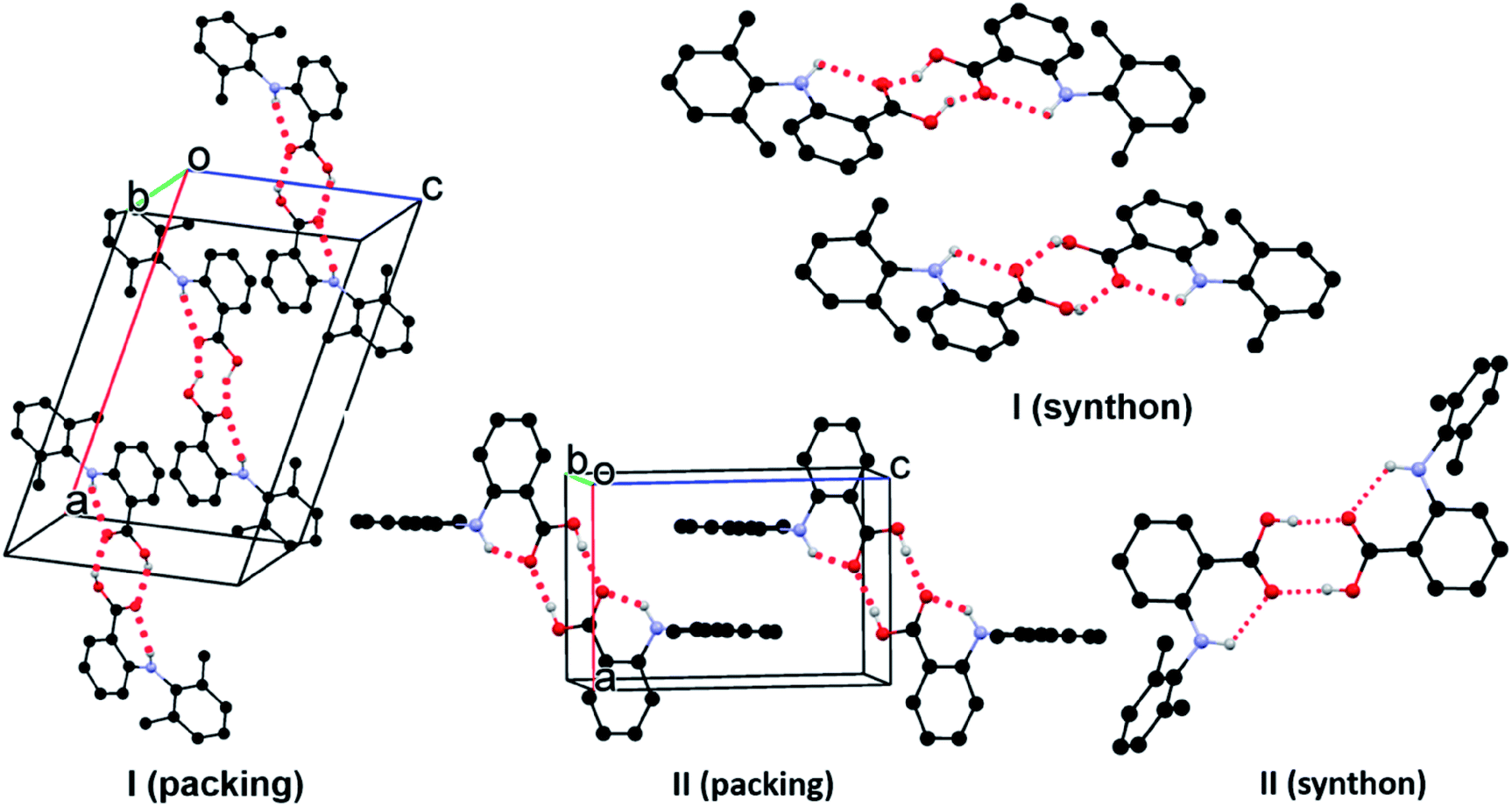 | ||
| Fig. 4 Crystal packing of I and II. For clarity, only hydrogens involved in hydrogen bonds (dotted line) are shown. | ||
Form II has one molecule in the asymmetric unit (Z′ = 1), and the molecule shows a twisted conformation, with the aromatic rings being perpendicular, as suggested by a dihedral angle of 88.91(4)°. The molecules form centrosymmetric acid–acid dimers. The intermolecular hydrogen bond has a bond distance and bond angle of 1.684 Å and 174.34° for O16–H16⋯O15. Both the bond distance and bond angle are significantly different from those in form I. The intramolecular hydrogen bond between the carbonyl O of the carboxylic acid and the anilino NH has a bond distance of 2.020 Å and bond angle of 135.16°. The intramolecular hydrogen bond parameters are also different from those in form I.
An exhaustive polymorph screening was performed to obtain the literature form I, yet only the new form II was generated. Questions were raised on the authenticity of the literature form. Simulated PXRD patterns are almost identical between forms I and II. But disappearing polymorphism could be the reason of the absence of form I.47
3.2. Thermal properties
DSC was conducted to investigate the thermal properties of form II, shown in Fig. 5. Form II has two endothermic DSC peaks. The first, with an onset temperature of 199.2 °C, appears to be a phase transition to a new form that melts at approximately 201.6 °C. Based on the DSC thermograms, we can infer that form II converts into a third form when heated. As mentioned before, form I has Z′ = 2 and form II has Z′ = 1 and most of the time, the Z′ = 1 form is more stable than the high Z′ forms,48,49 so it is unlikely that form II transforms into form I, suggesting that the new form (labelled III above) could be a third form. | ||
| Fig. 5 DSC thermograms of HDMPA form II before and after thermal treatment. | ||
We also investigated the reversibility of the phase transition. When the sample was heated to a temperature just below the melting (200 °C) and then cooled down to room temperature, the DSC of the new sample showed only one peak, which corresponds to the melting of the sample. PXRD was measured for form III. The patterns of form III is different from that of form II as shown in Fig. 6.
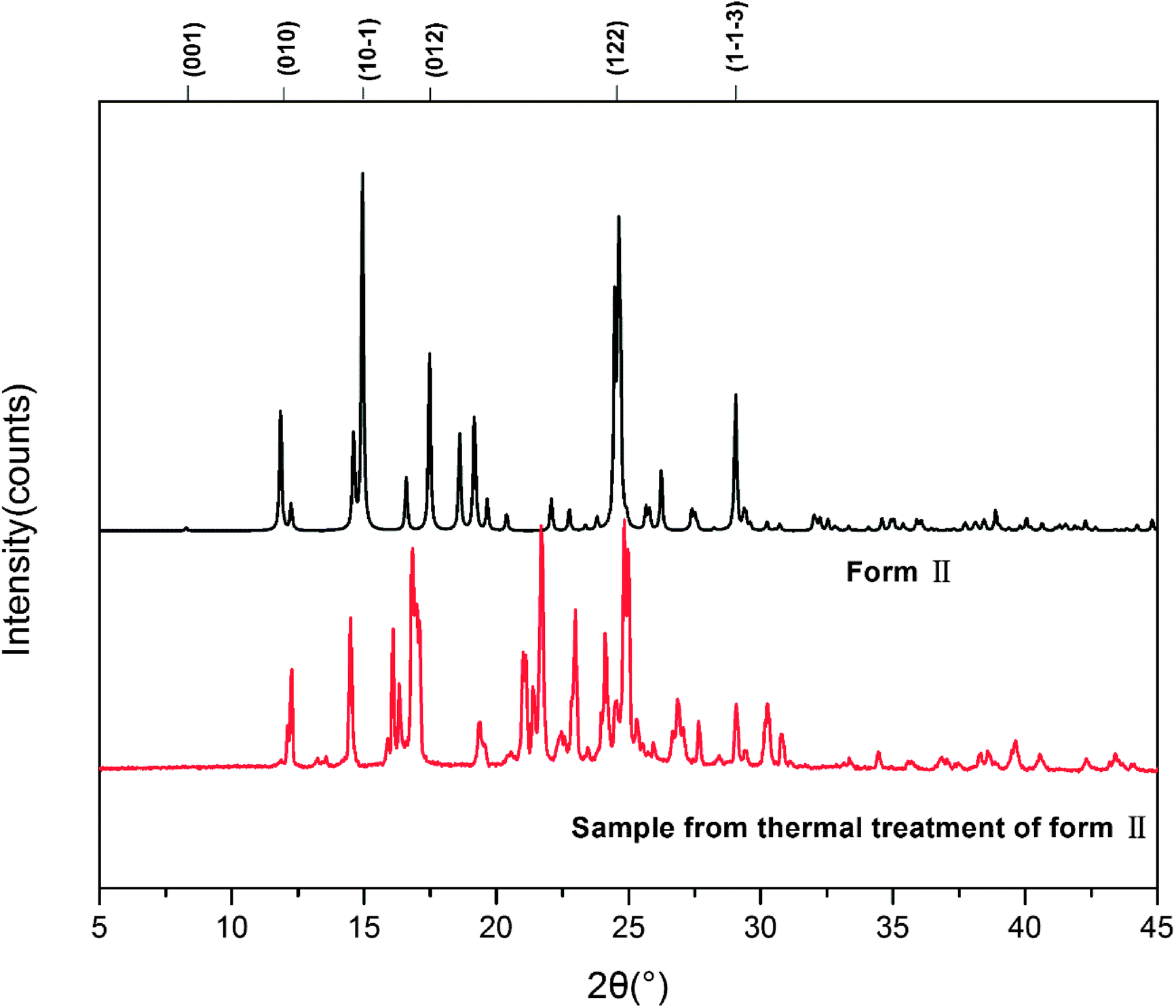 | ||
| Fig. 6 PXRD patterns of HDMPA form II before and after thermal treatment. | ||
Fig. 7 shows the calculated powder X-ray diffraction patterns of forms I and II collected at room temperature (a), along with the matching of PXRD patterns calculated from the single-crystal structures determined at both 90 K and 296 K and the experimental PXRD patterns. The calculated PXRD patterns of the literature form and the new form are nearly identical, and the experimental and calculated PXRD patterns for form II match extremely well.
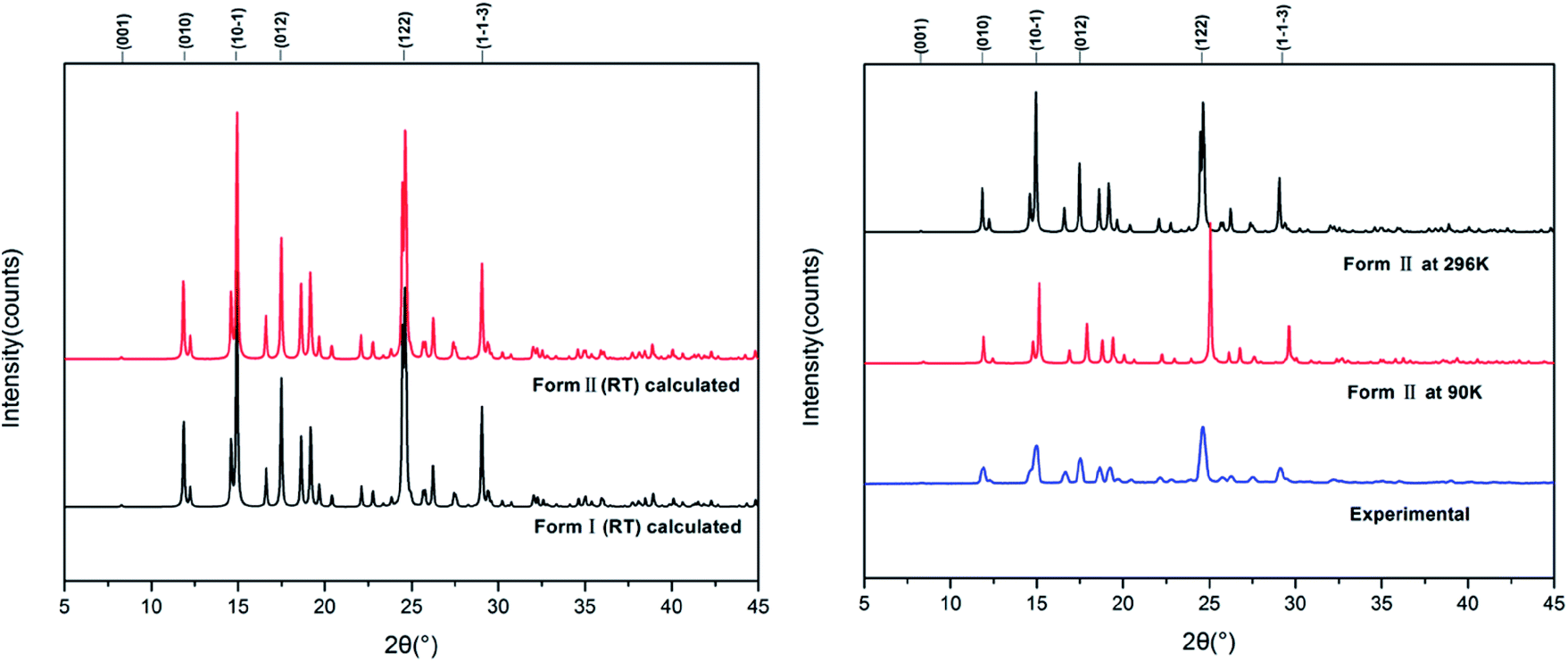 | ||
| Fig. 7 Experimental and calculated PXRD patterns of HDMPA polymorphs. | ||
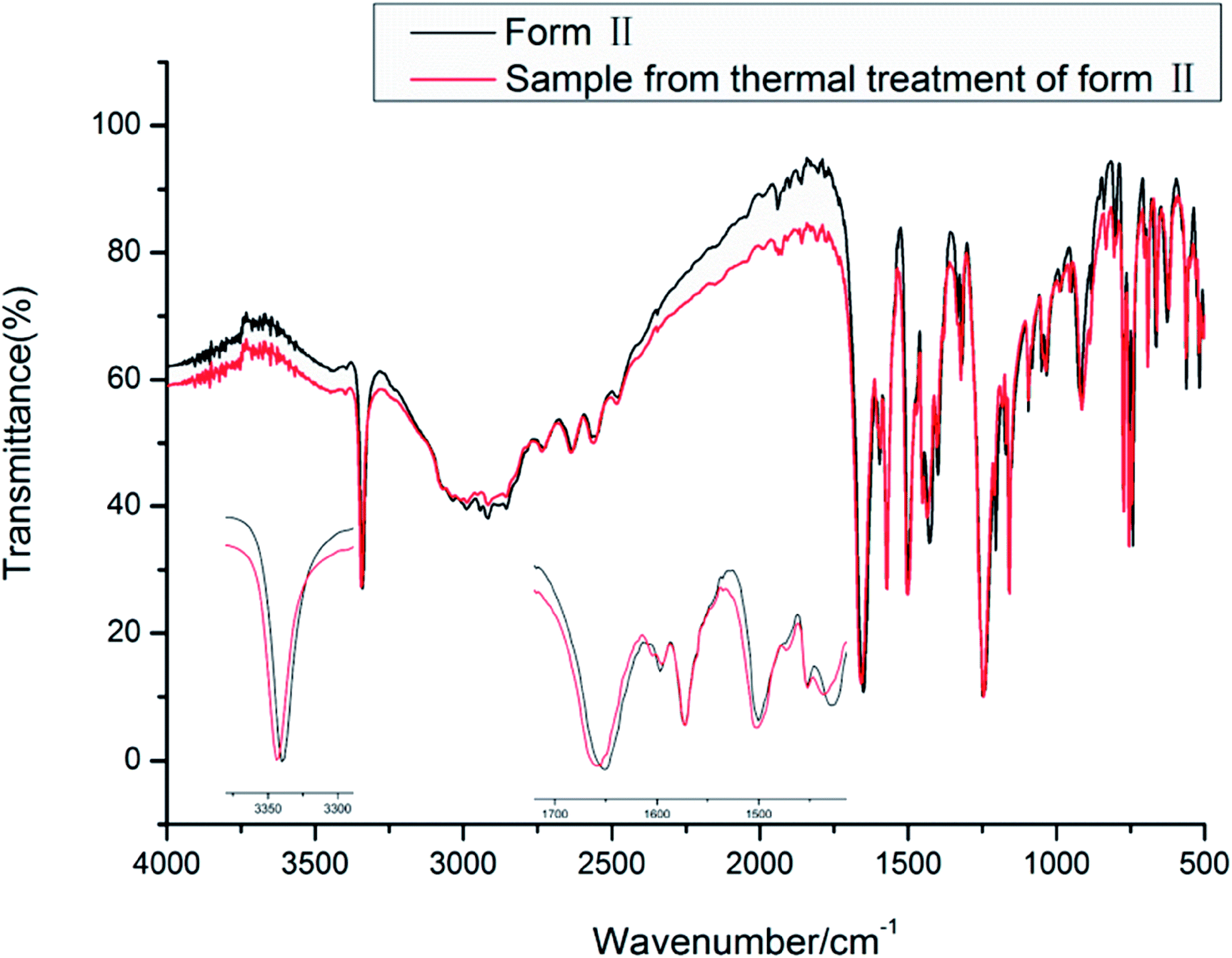 | ||
| Fig. 8 IR spectra of the new forms of HDMPA. | ||
| N–H stretch | Carboxyl O–H stretch and aromatic/aliphatic C–H stretch | Carboxyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch O stretch |
Aromatic CC stretch |
|
|---|---|---|---|---|
| Form II | 3339.5 | 3031.7–2565.3 | 1650.8 | 1596.6 |
| 1572.9 | ||||
| 1500.1 | ||||
| Form III | 3343.3 | 3077.6–2561.0 | 1658.4 | 1595.0 |
| 1572.5 | ||||
| 1501.8 |
The vibrational spectra show the effect of the intramolecular hydrogen bond between NH and OC. The NH stretch is at a lower frequency of around 3339.5 and 3343.3 cm−1 for forms II and III, respectively. The carboxyl CO stretch is at 1650.8 and 1658.4 cm−1 for forms II and III. The carboxyl O–H and aromatic/aliphatic C–H as well as aromatic CC stretches show subtle differences between forms II and III as indicated in Table 4.
The Raman spectra of the two new forms of HDMPA were measured for comparision (Fig. 9). Subtle yet distinguishable differences are observed (Table 5).
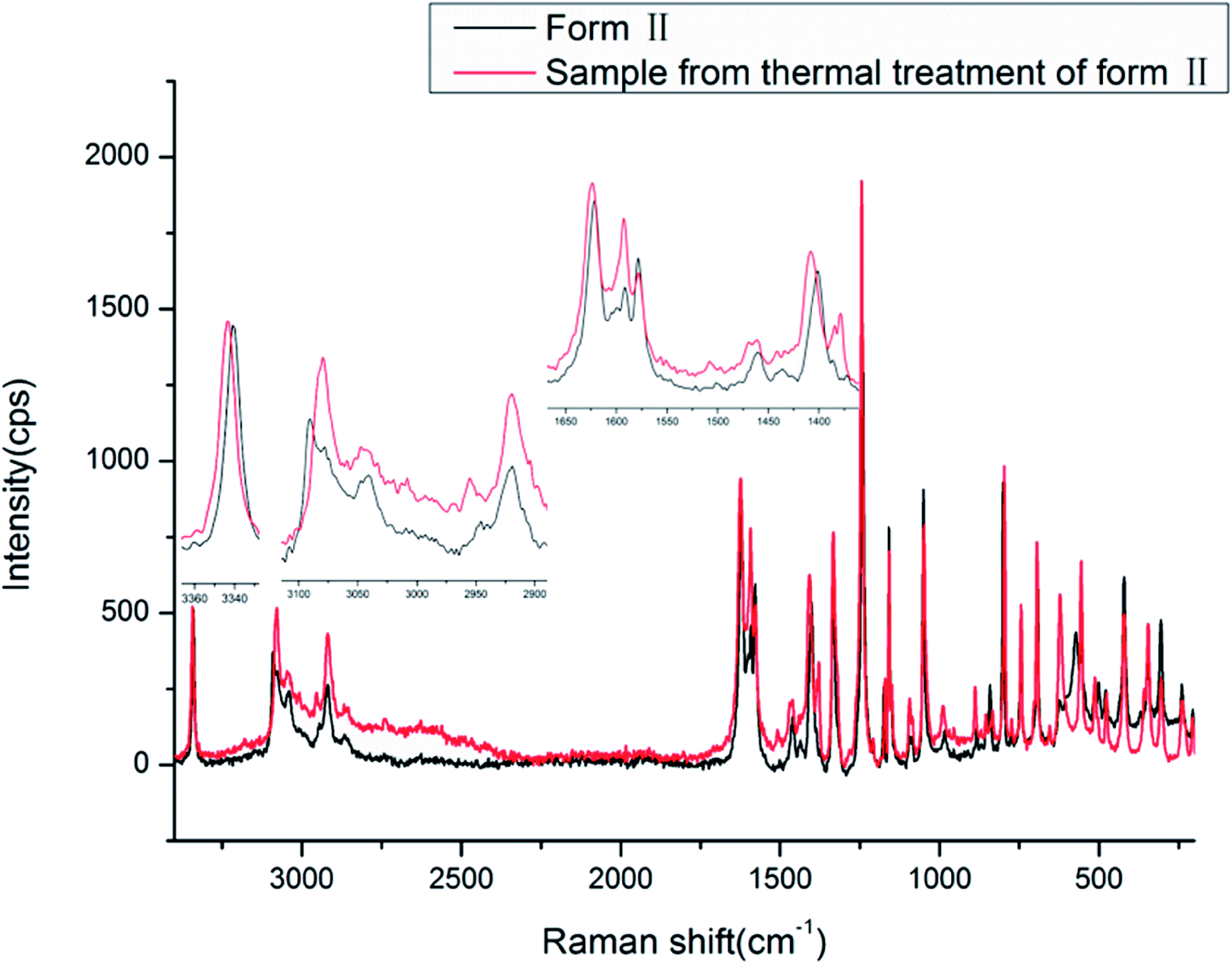 | ||
| Fig. 9 Raman spectra of HDMPA polymorphs. | ||
| N–H stretch | Carboxyl O–H stretch and aromatic/aliphatic C–H stretch | Carboxyl CO stretch |
Aromatic CC stretch |
|
|---|---|---|---|---|
| Form II | 3340.6 | 3090.5–2919.0 | 1621.5 | 1591.4 |
| 1578.3 | ||||
| 1401.2 | ||||
| Form III | 3343.4 | 3079.4–2919.4 | 1623.7 | 1592.5 |
| 1578.0 | ||||
| 1408.2 |
The Raman spectra also show the effect of the intramolecular hydrogen bond between NH and OC. The NH stretch is at a lower frequency of around 3340.6 and 3343.4 cm−1, and the carboxyl CO stretch is at 1621.5 and 1623.7 cm−1 for forms II and III, respectively. The carboxyl O–H and aromatic/aliphatic C–H as well as aromatic CC stretches also show subtle differences between forms II and III as indicated in Table 5.
3.3. Computational results
For forms I and II, calculated lattice energies based on the empirically augmented DFT method are −48.14 and −50.31 kcal mol−1. The energy difference between forms I and II is 9.07 kJ mol−1, slightly higher than 6 kJ mol−1, which is in agreement with the observation that conformational polymorphs have higher lattice energy differences.50 Two forms have similar calculated densities at 296 K (I: 1.284 g cm−3 and II: 1.276 g cm−3). While form I is slightly denser than form II, its lattice energy is higher than that of form II, which might be due to the hydrogen bond difference between the two forms, since form II has a shorter and more linear hydrogen bond than form I. Due to the uncertainty inherent to the computational methods employed (which may range from a few kJ mol−1 to a few kcal mol−1, or even higher), the small energy difference between forms I and II should not be regarded as evidence of which is more stable. Also it should be pointed out that discrepancy could arise since the computation implicitly assumed a temperature of 0 K, whereas the densities are based on the structures measured at 296 K, and the DSC experiments were performed at elevated temperatures.Although steric hindrance can limit the flexibility of HDMPA, polymorphism could arise from its intrinsic conformational flexibility. The conformational scan conducted over τ for a single HDMPA molecule is illustrated in Fig. 10. The global minima of τ are identified at ±78.5° and ±98.0° due to the symmetry of the benzene ring. The conformers of both forms are located near the minima, but in two different energy valleys. Thus it appears that the polymorphic system is one of conformational polymorphism, not merely due to conformational adjustment.51 The torsion angle of all three conformations is listed in Table 6.
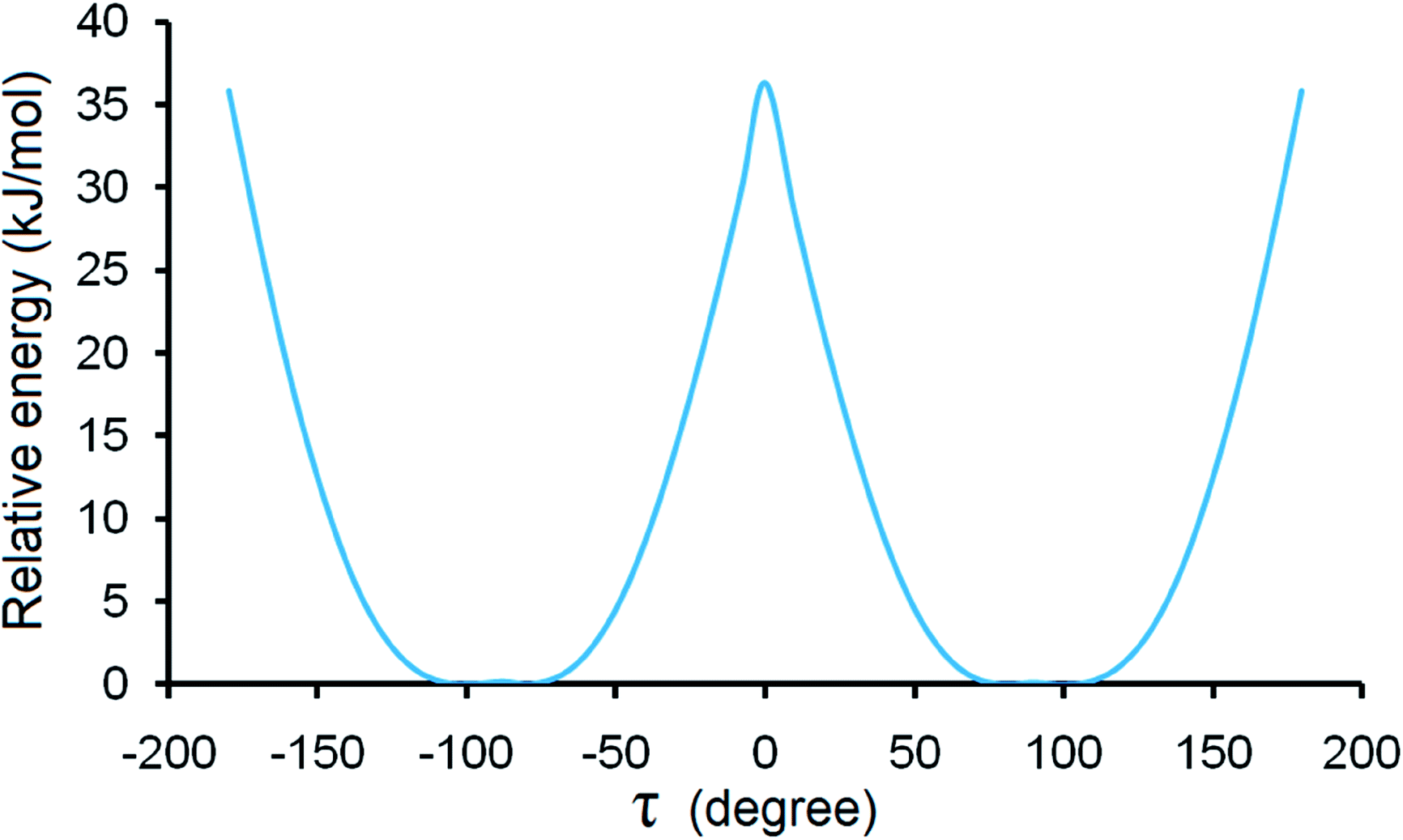 | ||
| Fig. 10 Conformation scan of single HDMPA molecule. | ||
| Conformer | Torsion angle (C6–N7–C8–C9/C13) (°) |
|---|---|
| I-A | −102.32/79.77 |
| I-B | 94.88/−79.51 |
| II | 79.63/−99.50 |
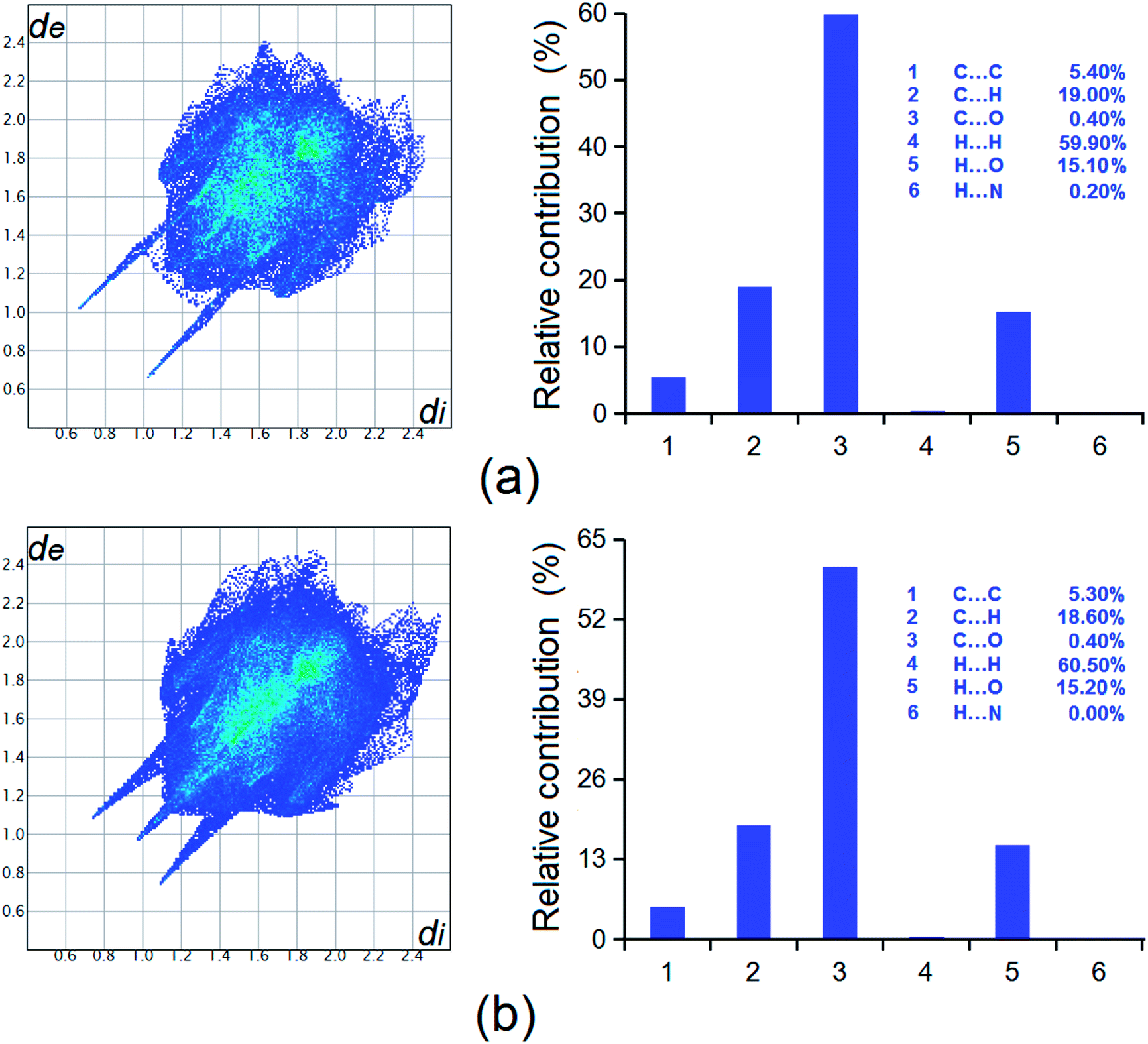 | ||
| Fig. 11 Hirshfeld surface analysis of the two forms of HDMPA, (a) form II; and (b) form I. | ||
4. Conclusions
Two new forms have been discovered for HDMPA, a fenamic acid with potential as an NSAID and in treatment of thyroid hormone disorders. Form II was characterized by structure determination by single-crystal X-ray diffraction, PXRD and spectroscopic methods such as FT-IR and Raman. The polymorphism appears to result from the conformational flexibility of the molecule, as suggested by the conformers in the two modifications and the conformational scan. The phase behaviors of the new forms were studied by DSC. The metastable form II appears to convert into form III on the application of thermal energy. Form III was characterized by PXRD, FT-IR and Raman spectroscopy. Lattice energies were calculated to be −48.14 and −50.31 kcal mol−1 for forms I and II suggesting the relative stabilities of the polymorphs, and Hirshfeld analysis indicated intermolecular interactions such as H⋯H, C⋯H, and H⋯O contribute significantly to the overall stability of the forms. This study clearly indicates that thermal treatment of crystalline samples could be an efficient approach for new polymorph generation. We are currently attempting to obtain single crystals for form III.Conflicts of interest
There are no conflicts to declare.Acknowledgements
RH and SL thanks Natural Science Foundation of Hubei Province (2014CFB787) and the President's Fund of Wuhan Institute of Technology (CX2016075) for financial support. PPZ acknowledges the financial support by the National Natural Science Foundation of China (Grant No. 21403097) and the Fundamental Research Funds for the Central Universities (lzujbky-2016-45).References
- S. Bajaj, S. S. Sambi and A. K. Madan, Bioorg. Med. Chem., 2004, 12, 3695 CrossRef CAS PubMed.
- J. S. Kaltenbronn, R. A. Scherrer, F. W. Short, E. M. Jones, H. R. Beatty, M. M. Saka, C. V. Winder, J. Wax and W. R. Williamson, Arzneim. Forsch., 1983, 33, 621 CAS.
- L. Fábián, N. Hamill, K. S. Eccles, H. A. Moynihan, A. R. Maguire, L. McCausland and S. E. Lawrence, Cryst. Growth Des., 2011, 11, 3522 Search PubMed.
- C. M. Ulrich, J. Bigler and J. D. Potter, Nat. Rev. Cancer, 2006, 6, 130 CrossRef CAS PubMed.
- C. Wolf, S. Liu, X. Mei, A. T. August and M. D. Casimir, J. Org. Chem., 2006, 71, 3270 CrossRef CAS PubMed.
- V. Dokorou, A. Primikiri and D. Kovala-Demertzi, J. Inorg. Biochem., 2011, 105, 195 CrossRef CAS PubMed.
- A. Martín, R. F. Pellón, M. Mesa, M. L. Docampo and V. Gómez, J. Chem. Res., 2006, 9, 561 Search PubMed.
- F. Kato, M. Otsuka and Y. Matsuda, Int. J. Pharm., 2006, 321, 18 CrossRef CAS PubMed.
- V. López-Mejías and A. J. Matzger, Cryst. Growth Des., 2015, 15, 3955 Search PubMed.
- V. Lópezmejías, J. W. Kampf and A. J. Matzger, J. Am. Chem. Soc., 2012, 134, 9872 CrossRef PubMed.
- P. P. Bag and C. M. Reddy, Cryst. Growth Des., 2012, 12, 2740 CAS.
- S. S. Kumar and A. Nangia, Cryst. Growth Des., 2014, 14, 1865 CAS.
- S. Long, T. Mao, P. Chen, M. Liu, S. Parkin, M. Zhang, T. Li, P. Zhou and F. Yu, ChemistrySelect, 2017, 1, 1 Search PubMed.
- G. R. Desiraju, Science, 1997, 278, 404 CrossRef CAS.
- J. Thun, M. Schoeffel and B. Josef, Mol. Simul., 2008, 34, 1359 CrossRef CAS.
- Y. Xu, Z. Xie, H. Zhang, F. Shen and Y. Ma, CrystEngComm, 2016, 18, 6824 RSC.
- J. Bauer, S. Spanton, R. Henry, J. Quick, W. D. Porter and K. Morris, Pharm. Res., 2001, 18, 859 CrossRef CAS.
- J.-R. Wang, B. Zhu, Z. Zhang, J. Bao, G. Deng, Q. Ding and X. Mei, Cryst. Growth Des., 2017, 17, 3482 CAS.
- D. Gur, M. Pierantoni, N. E. Dov, A. Hirsh, Y. Feldman, S. Weiner and L. Addadi, Cryst. Growth Des., 2016, 16, 4975 CAS.
- Polymorphism in Pharmaceutical Solids, ed. H. G. Brittain, Marcel Dekker, New York, 1999 Search PubMed.
- S. R. Byrn, R. R. Pfeiffer and J. G. Stowell, Solid State Chemistry of Drugs, SSCI, Inc., West Lafayette, IN, 2nd edn, 1999 Search PubMed.
- D. K. Chalmers, G. H. Scholz, D. J. Topliss, E. Koliniatis, S. L. Munro, D. J. Craik, M. Iskander and J. R. Stockigt, J. Med. Chem., 1993, 36, 1272 CrossRef CAS PubMed.
- J. S. Kaltenbrom, R. A. Scherrer, F. W. Short, H. R. Beatty, M. M. Saka, C. V. Winder, J. Wax and W. R. Williamson, Arzneim.-Forsch., 1983, 33, 621 Search PubMed.
- V. Dokorou, D. Kovala-Demertzi, J. P. Jasinskib, A. Galania and M. A. Demertzisa, Helv. Chim. Acta, 2004, 87, 1940 CrossRef CAS.
- S. Long and T. Li, Cryst. Growth Des., 2010, 10, 2465 CAS.
- S. Long, P. Zhou, S. Parkin and T. Li, Cryst. Growth Des., 2014, 14, 27 CAS.
- S. Long, S. Parkin, A. Siegler, C. Brock, A. Cammers and T. Li, Cryst. Growth Des., 2008, 8, 9 Search PubMed.
- S. Long and T. Li, Cryst. Growth Des., 2009, 9, 4993 CAS.
- S. Long, S. Parkin, M. A. Siegler, A. Cammers and T. Li, Cryst. Growth Des., 2008, 8, 4006 CAS.
- M. Liu, C. Yin, P. Chen, M. Zhang, S. Parkin, P. Zhou, T. Li, F. Yu and S. Long, CrystEngComm, 2017, 19, 4345 RSC.
- X. Mei, A. T. August and C. Wolf, J. Org. Chem., 2006, 71, 142 CrossRef CAS PubMed.
- P. Chen, Y. Jin, P. Zhou, S. Parkin, Z. Zhang and S. Long, J. Chin. Chem. Soc., 2017, 64, 531 CrossRef CAS.
- B. V. Nonius, Nonius, Collect, Delft, The Netherlands, 2002 Search PubMed.
- Z. Otwinowski and W. Minor, in Methods in Enzymology: Macromolecular Crystallography, Part A, ed. C. W. Carter Jr and R. M. Sweet, Academic Press, New York, 1997, vol. 276, p. 307 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed.
- R. Dovesi, R. Orlando, A. Erba, C. M. Zicovich-Wilson, B. Civalleri, S. Casassa, L. Maschio, M. Ferrabone, M. De La Pierre, P. D'Arco, Y. Noel, M. Causa, M. Rerat and B. Kirtman, Int. J. Quantum Chem., 2014, 1287 CrossRef CAS.
- T. Li and S. Feng, Pharm. Res., 2006, 23, 2326 CrossRef CAS PubMed.
- S. Feng and T. Li, J. Chem. Theory Comput., 2006, 2, 149 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19 RSC.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer (Version 3.1), University of Western Australia, 2012 Search PubMed.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120 CrossRef CAS.
- M. C. Etter, J. C. Macdonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256 CrossRef.
- J. Bernstein, R. E. Davis, L. Shimoni and N. L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS.
- J. D. Dunitz and J. Bernstein, Acc. Chem. Res., 1995, 28, 193 CrossRef CAS.
- G. R. Desiraju, CrystEngComm, 2007, 9, 91 RSC.
- A. J. Cruz-Cabeza, S. M. Reutzel and J. Bernstein, Chem. Soc. Rev., 2015, 44, 8619 RSC.
- A. Bauer-Brandl, E. Marti, A. Geoffroy, A. Poso, J. Suurkuusk, E. Wappler and K. H. Bauer, J. Therm. Anal. Calorim., 1999, 57, 7 CrossRef CAS.
- A. J. Cruz-Cabeza and J. Bernstein, Chem. Rev., 2014, 114, 2170 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: CCDC 1590160 and 1590161. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra13353g |
| ‡ Rong Hu and Yunping Zhoujin contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2018 |