
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Penicilazaphilones D and E: two new azaphilones from a sponge-derived strain of the fungus Penicillium sclerotiorum†
Chao-Yi Wangab,
Jun-Di Haoab,
Xing-Yan Ningab,
Jing-Shuai Wuab,
Dong-Lin Zhaoab,
Chui-Jian Kongab,
Chang-Lun Shao*ab and
Chang-Yun Wang *abc
*abc
aKey Laboratory of Marine Drugs, The Ministry of Education of China, School of Medicine and Pharmacy, Ocean University of China, Qingdao 266003, People's Republic of China. E-mail: changyun@ouc.edu.cn; shaochanglun@ouc.edu.cn; Tel: +86-532-8203-1536 Tel: +86-532-8203-1381
bLaboratory for Marine Drugs and Bioproducts, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266003, People's Republic of China
cInstitute of Evolution & Marine Biodiversity, Ocean University of China, Qingdao 266003, People's Republic of China
First published on 24th January 2018
Abstract
Two new azaphilones, penicilazaphilones D (1) and E (2), along with four known analogs (3–6), were obtained from the sponge-derived fungus Penicillium sclerotiorum. Their structures were elucidated on the basis of comprehensive spectroscopic analyses, ECD calculation, single-crystal X-ray diffraction and comparison to the biosynthetically related compound 5. Compound 1 represents the second reported azaphilone with a C-4 aliphatic side chain, and 2 is the first azaphilone with a tetrahydrofuran ring at C-3. Compound 3 displayed strong antiviral activity against HSV (IC50 = 19.5 μM) and EV71 (IC50 = 132 μM), 16 and 3 fold more potent than the positive control ribavirin, respectively.
Introduction
Marine-derived fungi represent a huge potential for new natural products, which often show pharmaceutically relevant bioactivities.1,2 The genus Penicillium, which received great attention among all marine-derived fungi, is becoming a promising reservoir of marine natural products with novel structures and potential bioactivities.2,3 It was reported that sponges are the main hosts of marine symbiotic Penicillium sp. fungi for producing new marine natural products.3 Specifically, a growing number of azaphilone derivatives have been reported from the marine-derived fungi including Penicillium with various biologic activities, such as cytotoxic, antibacterial, antifungal, and enzyme inhibitory activities.4–8In the course of our ongoing investigation on new bioactive secondary metabolites from the genus Penicillium in the South China Sea,9–14 a sponge-derived fungal strain, P. sclerotiorum GDST-2013-0415, attracted our attention, because the extract of the fungal culture showed antibacterial activity. Chemical investigation of the EtOAc crude extract led to the isolation of two new azaphilone derivatives, penicilazaphilones D and E (1 and 2), together with four known compounds (+)-sclerotiorin (3),15 geumsanol C (4),16 WB (CAS no. 1701443-52-8, 6H-2-benzopyran-6-one, 5-chloro-3-[(1E,3R,4R,5S)-3,4-dihydroxy-3,5-dimethyl-1-hepten-1-yl]-1,7,8,8a-tetrahydro-7,8-dihydroxy-7-methyl-(7R,8R,8aS)) (5)17,18 and geumsanol G (6).16 The structures of compounds 1–6 were established on the basis of spectroscopic analysis, and the absolute configurations of 1 and 6 were determined by single-crystal X-ray diffraction analysis. Herein we report the isolation, structure elucidation, and biological activities of these compounds.
Results and discussion
The sponge-derived fungal strain, Penicillium sclertiorum GDST-2013-0415, was cultured with rice medium in fifty Erlenmeyer flasks at room temperature for 40 days. The fermented rice medium was extracted three times with EtOAc. The combined EtOAc layers were evaporated under reduced pressure to give the EtOAc extract (65 g). By repeated column chromatography (CC) over silica gel, octadecylsilyl (ODS), Sephadex LH-20, and semi-preparative HPLC, six compounds, 1–6, were obtained (Fig. 1).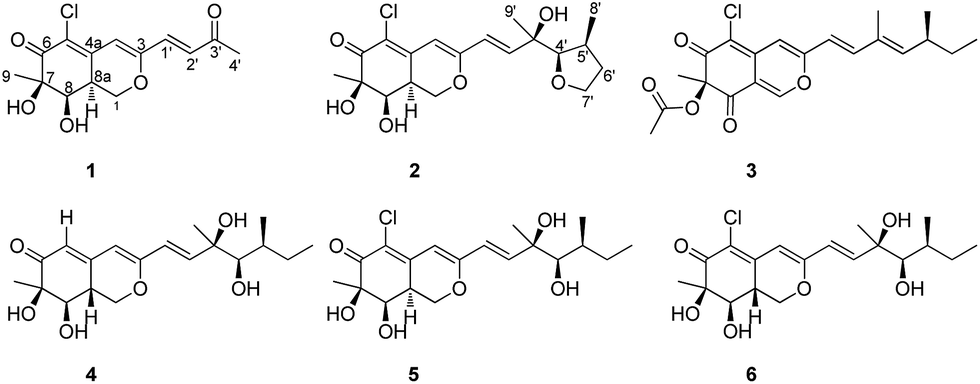 | ||
| Fig. 1 The structures of compounds 1–6. | ||
Penicilazaphilone D (1) was isolated as a yellow gum with the molecular formula of C14H15ClO5 based on HRESIMS, for which seven degrees of unsaturation were required. The IR spectrum of 1 exhibited the presence of hydroxyl (3633 cm−1), and conjugated carbonyl (1701 cm−1) groups. The 1H NMR spectroscopic data (Table 1) exhibited signals for three olefinic protons at δH 7.20 (d, J = 15.9 Hz), 6.61 (d, J = 15.9 Hz) and 6.50 (s), two methyl groups at δH 2.33 (s) and 1.35 (s), two methine protons at δH 4.18 (d, J = 3.1 Hz) and 3.29 (m), and one methylene group at δH 4.66 (dd, J = 11.0, 4.9 Hz) and 4.21 (dd, J = 13.0, 11.0 Hz). The 13C NMR spectroscopic data (Table 1) showed eight sp2-hybridized carbons in 1, including two carbonyl carbons at δC 197.9 and 194.5, six olefinic carbons at δC 159.5, 143.7, 136.1, 131.5, 120.5 and 108.7, and six sp3-hybridized carbons including two methyl groups at δC 27.7 and 23.4, two methine groups at δC 75.1 and 37.9, one methylene group at δC 69.5, and one quaternary carbon at δC 79.0. These spectroscopic features suggested that 1 shares the same bicyclic core of azaphilone as WB (CAS no. 1701443-52-8) (5), an azaphilone derivative isolated from a soil fungus and determined only the plane structure,17 then from an entomopathogenic fungus Hypocrella sp. reported the absolute configuration.18 The main differences were the presence of a 3-buten-2-one group [–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHC(O)CH3] at C-3 in 1 instead of the [–CHCHCOH(CH3)CH(OH)CH(CH3)CH2CH3] fragment in 5, which was confirmed by the COSY cross-peaks and HMBC correlations (Fig. 2). In the 1H–1H COSY spectrum, the cross peak of H-1′/H-2′, together with HMBC correlations from H-1′ to C-4 and C-3′, from H-2′ to C-3, and from H-4′ to C-2′ established a 3-buten-2-one unit linked with C-3. The relative configuration of 1 was determined by 1H–1H coupling constants and NOESY correlations. The cis-relationship of H-8/H-8a was deduced from the eq/ax coupling constant (J8,8a = 3.1 Hz). In the NOESY spectrum (Fig. 3), NOESY correlations were observed between H3-9 and H-8, indicating the cofacial orientation relative to these protons. On the basis of these results, the complete planar assignment for compound 1 was established.
CHC(O)CH3] at C-3 in 1 instead of the [–CHCHCOH(CH3)CH(OH)CH(CH3)CH2CH3] fragment in 5, which was confirmed by the COSY cross-peaks and HMBC correlations (Fig. 2). In the 1H–1H COSY spectrum, the cross peak of H-1′/H-2′, together with HMBC correlations from H-1′ to C-4 and C-3′, from H-2′ to C-3, and from H-4′ to C-2′ established a 3-buten-2-one unit linked with C-3. The relative configuration of 1 was determined by 1H–1H coupling constants and NOESY correlations. The cis-relationship of H-8/H-8a was deduced from the eq/ax coupling constant (J8,8a = 3.1 Hz). In the NOESY spectrum (Fig. 3), NOESY correlations were observed between H3-9 and H-8, indicating the cofacial orientation relative to these protons. On the basis of these results, the complete planar assignment for compound 1 was established.
| No. | 1 | 2 | ||
|---|---|---|---|---|
| δCa, type | δHa (J in Hz) | δCa, type | δHa (J in Hz) | |
| a In acetone-d6.b In CDCl3. | ||||
| 1 | 69.5, CH2 | 4.21, dd (13.0, 11.0) | 69.3, CH2 | 4.25, dd (13.2, 11.0)b |
| 4.66, dd (11.0, 4.9) | 4.60, dd (11.0, 4.9) | |||
| 3 | 159.5, C | 162.5, C | ||
| 4 | 108.7, CH | 6.50, s | 102.1, CH | 6.06, s |
| 4a | 143.7, C | 145.6, C | ||
| 5 | 120.5, C | 117.1, C | ||
| 6 | 194.5, C | 193.9, C | ||
| 7 | 79.0, C | 78.6, C | ||
| 8 | 75.1, CH | 4.18, d (3.1) | 75.1, CH | 4.12, d (3.0)b |
| 8a | 37.9, CH | 3.29, m | 38.0, CH | 3.22, m |
| 9 | 23.4, CH3 | 1.35, s | 26.5, CH3 | 1.340, s |
| 1′ | 136.1, CH | 7.20, d (15.9) | 122.2, CH | 6.36, d (15.5) |
| 2′ | 131.5, CH | 6.61, d (15.9) | 145.6, CH | 6.77, d (15.5) |
| 3′ | 197.9, C | 76.2, C | ||
| 4′ | 27.7, CH3 | 2.33, s | 80.5, CH | 3.52, d (2.0) |
| 5′ | 31.2, CH | 2.10, m | ||
| 6′ | 40.0, CH2 | 1.66, m | ||
| 1.53, m | ||||
| 7′ | 60.1, CH2 | 3.60, m | ||
| 8′ | 14.2, CH3 | 0.91, d (6.9) | ||
| 9′ | 23.7, CH3 | 1.336, s | ||
 | ||
| Fig. 2 1H–1H COSY and key HMBC correlations of compounds 1 and 2. | ||
 | ||
| Fig. 3 Selected NOESY correlations of 1 and 2. | ||
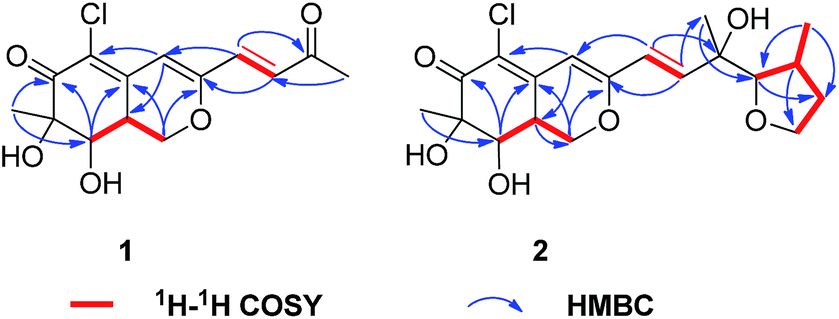
The absolute configuration of 1 was determined by comparing the theoretical calculation ECD with the experimental ECD. Conformational searches were carried out using Merck Molecular Force Field (MMFF) by means of the Spartan's 10 software to acquire meaningful conformers for 7R,8R,8aS-1, followed by DFT optimizations at the B3LYP/6-31+G(d,p) level in MeOH by CPCM polarizable conductor calculation model. The ECD spectrum was calculated by the TDDFT methodology with a larger basis set at the B3LYP/6-311+G(d,p) level and generated using SpecDis 1.6 according to Boltzmann distributions. The theoretical ECD spectrum for 7S,8S,8aR-1 was obtained by directly reversing the spectrum of 7R,8R,8aS-1. For compound 1 the experimental positive Cotton effect at 250 nm (Δε +14.32), and negative Cotton effects at 218 nm (Δε −6.14) and 328 nm (Δε −3.83) compared well with the calculated ECD curve for 7R,8R,8aS-1, which showed three corresponding Cotton effects around 257 nm (Δε +15.21), 223 nm (Δε −17.65), and 325 nm (Δε −2.90), respectively (Fig. 4). Therefore, qualitative analysis of the results allowed the assignment of the absolute configuration of 1 as 7R,8R,8aS.
 | ||
| Fig. 4 Experimental and calculated ECD spectra for 7R,8R,8aS-1. | ||
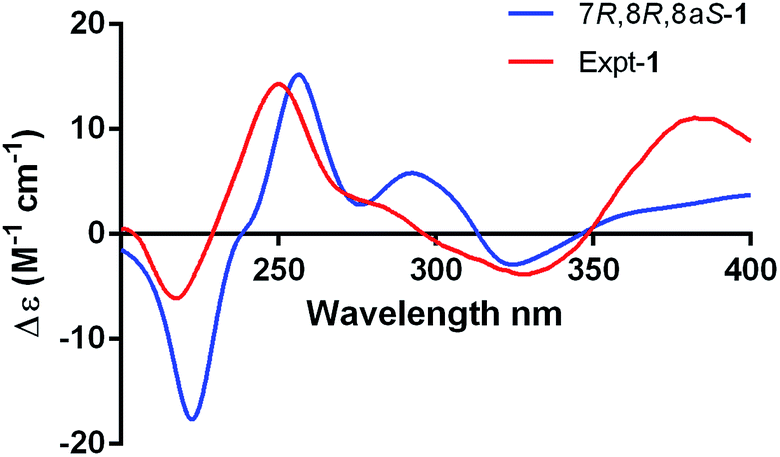
To confirm the absolute configuration, we undertook a single X-ray diffraction study. Ultimately, by slow crystallization from the mixture of CH2Cl2 and CH3OH, single crystals of 1 suitable for X-ray diffraction were obtained. The Mo Kα radiation used for the X-ray diffraction allowed the assignment of the absolute configuration of all of the stereogenic centers in 1, which contains heavy atom chlorine, as 7R, 8R and 8aS through refinement of Flack's parameter −0.07(3) (Fig. 5).
 | ||
| Fig. 5 Single-crystal X-ray structures of compounds 1 and 6. | ||
Penicilazaphilone E (2) was also obtained as a yellow gum, possessing a molecular formula of C19H25ClO6 with seven degrees of unsaturation. The IR spectrum indicated the presence of hydroxyl (3752 cm−1) and conjugated carbonyl (1703 cm−1) groups. The 13C NMR spectroscopic data (Table 1) displayed the presence of 19 carbon signals including one carbonyl carbon (δC 193.9) and six olefinic carbons, three methyl groups, three methylene groups, four methine groups, and two quaternary carbons. In the 1H NMR spectrum, three olefinic protons at δH 6.77 (d, J = 15.5 Hz), 6.36 (d, J = 15.5 Hz) and 6.06 (s), three methyl groups, three methylene groups, and four methine groups were observed. Detailed analysis of the 1H and 13C NMR spectroscopic data (Table 1) revealed that 2 is an azaphilone derivative, also closely related to WB (CAS no. 1701443-52-8) (5).17,18 Comparision of the 1H and 13C NMR data revealed that 2 differed from 5 only at the side chains. The terminal methyl of the side chain at δH 0.91, δC 11.8 in 5 were absent in 2. Instead, the signals for a methylene at δH 3.60, δC 60.1 were observed in 2. Furthermore, the above structural moiety accounts for six unsaturation degrees (one carbonyl, six olefinic carbons, and the bicyclic core of azaphilone), indicated that 2 must contain another ring. The contiguous sequence of correlations from H-4′ to H-7′ in the COSY spectrum and the HMBC correlations from H-5′ to C-7′and H-4′ to C-6′ indicated the presence of a tetrahydrofuran ring (Fig. 2).
The relative configuration of 2 was determined also by 1H–1H coupling constants and NOESY data revealing a bicyclic core with the same relative configurations as 1 and 5 (Fig. 3). Specifically, the cis-relationship of H-4′/H-5′ in the side chain was deduced from the coupling constant (J4′, 5′ = 2.0 Hz). The NOESY correlations of H3-9′/H-4′ indicated that these protons located on the same side of the moiety. Thus, the relative configurations of 2 were determined. The absolute configuration of 2 could not be assigned only by ECD method, while the absolute configuration of the side chain in 2 could not be determined by Mosher's method as 5 (ref. 18) because of the presence of tetrahydrofuran ring in the side chain. Unfortunately, the cultivation of single crystal of 2 was failed, even cultivated by the 4-bromobenzoate derivative of 2. And due to the limitation of compound amount, the absolute configuration of 2 was tentatively determined by biosynthetic pathways. Compound 2 was proposed to be a cyclized form of 5 between 7′-methyl and 4′-OH, that is, 5 was the precursor of 2. The plausible biosynthetic pathways from 5 to 2 were proposed. As depicted in Scheme 1, 5 is initially oxidized at side-chain by a cytochrome P450 hydroxylase to form a C-terminal hydroxylated product (a). Then the intermediate (a) undergoes a cyclic reaction between 7′-methyl and 4′-OH. It appears conceivable that a have two different cyclization pathways. Path 1: the dehydration of 7′-OH leads to the nucleophilic attack from 4′-O− to 7′-C+, which directly gives 2. Path 2: the dehydration of 4′-OH result in the nucleophilic attack from 7′-O− to 4′-C+, which could yield two possible products, 2 and b, on account of the rotation of the side chain. Owing to the fact that the relative configuration of 2 is identical with that of 5, the absolute configuration of 2 was proposed to be identical to that of the precursor 5 tentatively.
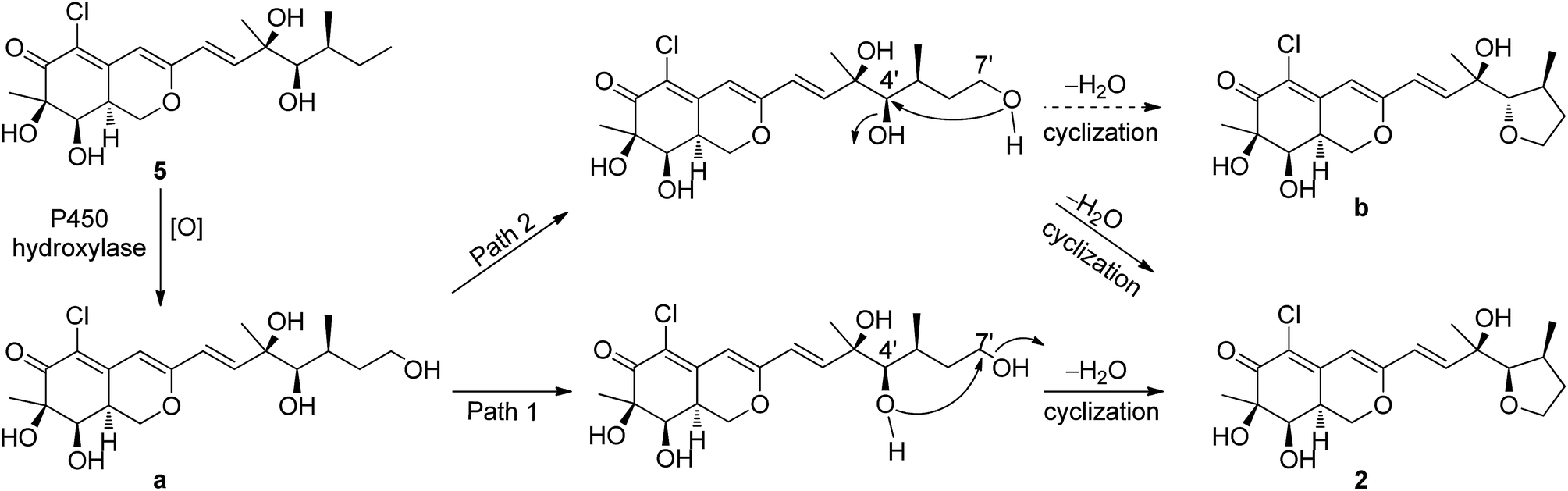 | ||
| Scheme 1 Proposed biosynthetic pathway of 2 from 5. | ||
Azaphilones are a class of structurally variable fungal polyketide metabolites isolated mainly from the ascomyceteous fungi, such as genera Penicillium, Aspergillus, Chaetomium, Talaromyces and Xylariaceae.8,19 The azaphilone derivatives could be defined as polyketides possessing a highly oxygenated pyranoquinone bicyclic core and a quaternary carbon center.8 In the present study, the new compounds 1 and 2 belong to the substructure type of sclerotiorin (3)-like azaphilones, which usually possess a γ-lactone, a conjugated ketone, a chlorine atom at C-5, and a branched C7 side chain at C-3.8 Their differences compared with sclerotiorin (3) lay on the imparity of the side chains at C-3 and substituent groups at C-7. Compound 1 represents the second reported azaphilone with a C4 aliphatic side chain,20 a 3-buten-2-one moiety. Besides, compound 2 is a firstly reported azaphilone with a tetrahydrofuran ring at C-3.
The known compounds 3–6 belong to sclerotiorin structural skeleton with a branched C7 side chain. As reported in the literature, the absolute configurations of the side chains [–CHCHCOH(CH3)CH(OH)CH(CH3)CH2CH3] in such azaphilones were determined by Mosher's method.16,18 Fortunately, by slow crystallization from the mixture of EtOAc and hexane, single crystals of 6 suitable for X-ray diffraction were obtained. By detailed analysis of the crystal structure, the absolute configuration of 6 was established unambiguously as 7R,8R,8aR,3′R,4′R,5′S (Fig. 5) through refinement of Flack's parameter 0.07(3). It was the first time to confirm the absolute configuration of 6 by single-crystal X-ray diffraction.
Compounds 1–6 were evaluated for their antiviral activities against four viral strains, including herpes simplex virus (HSV), human enterovirus 71 (EV71), respiratory syncytial virus 1 (RSV1), and coxsackievirus B3 (CoxB3). Among them, compound 3 exhibited promising antiviral activities against HSV with an IC50 values of 19.5 μM, 16 folds more potent than the positive control ribavirin (IC50 = 313 μM) (Table 2). Besides, 3 also displayed antiviral activity against EV71 (IC50 = 132 μM), 3 times stronger than the positive control ribavirin (IC50 = 418 μM). Comparison of their structures and antiviral activities revealed that the introduction of ester group in 3 may improve the antiviral activity.
Compounds 1–6 were also evaluated for their antibacterial activities towards six pathogenic bacteria. Only 3 showed moderate to weak antibacterial activities against Micrococcus lysodeikticus, Bacillus subtilis, Bacillus cereus, Micrococcus luteus, Bacillus megaterium, Shigella dysenteriae with the MIC values of 6.75, 6.75, 12.5, 12.5, 12.5, and 12.5 μM, respectively (Table S2†). Compounds 3–6 were tested for their cytotoxic activities against four tumor cell lines. However, none of them showed any cytotoxicities.
Experimental section
General experimental procedure
Optical rotations were measured on a JASCO P-1020 digital polarimeter. UV spectra were recorded on a Beckman DU 640 spectrophotometer. ECD spectra were recorded on a Jasco J-815-150S circular dichroism spectrometer. IR spectra were recorded on a Nicolet-Nexus-470 spectrometer using KBr pellets. NMR spectra were recorded on an Agilent DD2 500 MHz NMR spectrometer (500 MHz for 1H and 125 MHz for 13C), using TMS as an internal standard. The HRESIMS and ESIMS spectra were obtained from a Micromass Q-TOF spectrometer. The crystallographic data were collected on a Bruker SMART APEX-II CCD diffractometer equipped with graphite monochromatized Mo Kα radiation (λ = 0.71073 Å). Semi-preparative HPLC was performed on a Hitachi L-2000 HPLC system coupled with a Hitachi L-2455 photodiode array detector. A Kromasil C18 semi-preparative HPLC column (250 × 10 mm, 5 μm) was used. Silica gel (Qing Dao Hai Yang Chemical Group Co.; 200–300 mesh), Sephadex LH-20 (Amersham Biosciences) and octadecylsilyl silica gel (Unicorn; 45–60 μm) were used for column chromatography. Precoated silica gel GF254 plates (Yantai Zifu Chemical Group Co., Yantai, People's Republic of China) were used for analytical TLC.Fungal material
The fungal strain P. sclerotiorum GDST-2013-0415 was isolated from the inner part of an unidentified sponge GDST-2013-04 collected from the coral reef at a depth of 10 m in the sea area of Shantou, Guangdong Province of China, in May 2013. The fungal identification was performed by analysis of its ITS region of the rDNA as described previously.21 The sequence data was submitted to the Genbank, with accession number MF467908. The strain was deposited in the Key Laboratory of Marine Drugs, the Ministry of Education of China, School of Medicine and Pharmacy, Ocean University of China, Qingdao, PR China.Extraction and isolation
Fifty Erlenmeyer flasks of the fungal strain were cultivated in rice medium (3.6 g of natural sea salt from Yangkou saltern, China; 70 g of rice; 100 mL of H2O) for 40 days at room temperature. The fermented rice substrate was extracted repeatedly with EtOAc (400 mL for each flask), and the solvent was combined and concentrated in vacuo to afford a residue. The EtOAc residue (65 g) was subjected to silica gel CC using a step gradient elution with EtOAc–petroleum ether (0–100%) and then with MeOH–EtOAc (0–100%) to afford seven fractions (Fr.1–Fr.7). Fr.4 was first subjected to Sephadex LH-20 CC eluting with a mixture of CHCl3–MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) and then was further purified by using semi-preparative HPLC eluting with 50% MeOH–H2O (2 mL min−1) to give compounds 3 (27.0 mg) and 4 (4.1 mg). Fr.5 was separated on silica gel CC by using gradient elution of EtOAc–petroleum ether (0–100%) and then further purified on an ODS column eluting with 25% CH3CN–H2O to produce 1 (5.5 mg) and two subfractions, Fr.5-1 and Fr.5-2. Fr.5-1 was further purified by HPLC (20% CH3CN–H2O) to afford 5 (33.9 mg) and 6 (18.2 mg). Fr.6 was isolated by silica gel CC eluting with CHCl3–MeOH (30:1, v/v), then was further purified by using semi-preparative HPLC eluting with 80% MeOH–H2O to give compound 2 (1.3 mg).
ε): 208 (3.67), 338 (3.23) nm; IR (KBr) νmax 3633, 2922, 2362, 1701, 1540, 1383, 1187 cm−1; 1H NMR (acetone-d6, 500 MHz) and 13C NMR (acetone-d6, 125 MHz) see Table 1; ESIMS m/z 299 [M + H]+; HRESIMS m/z 299.0675 [M + H]+ (calcd for C14H16ClO5, 299.0681).ε): 206 (3.53), 256 (3.06), 332 (2.88) nm; IR (KBr) νmax 3753, 2928, 2377, 1703, 1565, 1371, 1150 cm−1; 1H NMR (acetone-d6, 500 MHz) and 13C NMR (acetone-d6, 125 MHz) see Table 1; ESIMS m/z 385 [M + H]+; HRESIMS m/z 385.1412 [M + H]+ (calcd for C19H26ClO6, 385.1412).557, reflections unique 6467 (Rint = 0.0993, Rsigma = 0.0566), final R indices for I > 2σ(I), R1 = 0.0495, wR2 = 0.1039, R indices for all data R1 = 0.0890, wR2 = 0.1209, Flack parameter = −0.07(3). Crystallographic data for 1 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 1445327.699, reflections unique 11415 (Rint = 0.0849, Rsigma = 0.0810), final R indices for I > 2σ(I), R1 = 0.0566, wR2 = 0.1465, R indices for all data R1 = 0.1298, wR2 = 0.1661, Flack parameter = 0.07(3). Crystallographic data for 6 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 1445322.
:1, v/v) and then was further purified by using semi-preparative HPLC eluting with 50% MeOH–H2O (2 mL min−1) to give compounds 3 (27.0 mg) and 4 (4.1 mg). Fr.5 was separated on silica gel CC by using gradient elution of EtOAc–petroleum ether (0–100%) and then further purified on an ODS column eluting with 25% CH3CN–H2O to produce 1 (5.5 mg) and two subfractions, Fr.5-1 and Fr.5-2. Fr.5-1 was further purified by HPLC (20% CH3CN–H2O) to afford 5 (33.9 mg) and 6 (18.2 mg). Fr.6 was isolated by silica gel CC eluting with CHCl3–MeOH (30:1, v/v), then was further purified by using semi-preparative HPLC eluting with 80% MeOH–H2O to give compound 2 (1.3 mg).
ε): 208 (3.67), 338 (3.23) nm; IR (KBr) νmax 3633, 2922, 2362, 1701, 1540, 1383, 1187 cm−1; 1H NMR (acetone-d6, 500 MHz) and 13C NMR (acetone-d6, 125 MHz) see Table 1; ESIMS m/z 299 [M + H]+; HRESIMS m/z 299.0675 [M + H]+ (calcd for C14H16ClO5, 299.0681).ε): 206 (3.53), 256 (3.06), 332 (2.88) nm; IR (KBr) νmax 3753, 2928, 2377, 1703, 1565, 1371, 1150 cm−1; 1H NMR (acetone-d6, 500 MHz) and 13C NMR (acetone-d6, 125 MHz) see Table 1; ESIMS m/z 385 [M + H]+; HRESIMS m/z 385.1412 [M + H]+ (calcd for C19H26ClO6, 385.1412).557, reflections unique 6467 (Rint = 0.0993, Rsigma = 0.0566), final R indices for I > 2σ(I), R1 = 0.0495, wR2 = 0.1039, R indices for all data R1 = 0.0890, wR2 = 0.1209, Flack parameter = −0.07(3). Crystallographic data for 1 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 1445327.699, reflections unique 11415 (Rint = 0.0849, Rsigma = 0.0810), final R indices for I > 2σ(I), R1 = 0.0566, wR2 = 0.1465, R indices for all data R1 = 0.1298, wR2 = 0.1661, Flack parameter = 0.07(3). Crystallographic data for 6 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 1445322.Antiviral activity assay
The antiviral activities against HSV, EV71, RSV1 and CoxB3 were determined by the CPE inhibition assay according to established procedures.22 Ribavirin was used as a positive control.Antibacterial activity assay
The antibacterial activity was evaluated by the conventional broth dilution assay.23 Six pathogenic bacterial strains, M. lysodeikticus, B. subtilis, B. cereus, M. luteus, B. megaterium, and S. dysenteriae, were used, and ciprofloxacin was used as a positive control.Cytotoxicity assay
The cytotoxicity against human erythroleukemia K562 cell lines was evaluated using MTT method.24 The cytotoxicity against human cervical carcinoma HeLa, human cancer of colon HCT-116, and human lung carcinoma A549 cell lines were evaluated using SRB method.25 The four human cancer cell lines above were purchased from Shanghai Institute of Biochemistry and Cell Biology (SIBCB), Chinese Academy of Sciences (CAS). Adriamycin was used as a positive control.Conclusion
In conclusion, two new azaphilones, penicilazaphilones D and E (1 and 2), along with four known analogs (3–6) were obtained from the sponge-derived fungus P. sclerotiorum. The absolute configurations of 1 and 2 were determined by single-crystal X-ray diffraction analysis and comparison to the biosynthetically related known compound 5, respectively. It was the first time to confirm the absolute configurations of known compound, geumsanol G (6) by single-crystal X-ray diffraction. Compound 1 represents the second reported azaphilone with a C-4 aliphatic side chain, while compound 2 is the first reported azaphilone with a tetrahydrofuran ring at C-3 side chain. Compound 3 showed potent antiviral activity against HSV and EV71 with the IC50 values of 19.5 and 132 μM, respectively, more stronger than the positive control ribavirin.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. U1606403; 81673350); the Fundamental Research Funds for the Central Universities (No. 201762017); the Open Research Fund Program of State Key Laboratory of Microbial Metabolism (Shanghai Jiao Tong University) (MMLKF1609), China; the Scientific and Technological Innovation Project Financially Supported by Qingdao National Laboratory for Marine Science and Technology (No. 2015ASKJ02); and the Taishan Scholars Program, China.Notes and references
- J. F. Imhoff, Mar. Drugs, 2016, 14, 19 CrossRef PubMed.
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2012, 29, 144–222 RSC.
- H. G. Ma, Q. Liu, G. L. Zhu, H. S. Liu and W. M. Zhu, J. Asian Nat. Prod. Res., 2016, 18, 92–115 CrossRef CAS PubMed.
- S. L. Zhou, M. Wang, H. G. Zhao, Y. H. Huang, Y. Y. Lin, G. H. Tan and S. L. Chen, Arch. Pharmacal Res., 2016, 39, 1621–1627 CrossRef CAS PubMed.
- M. Chen, N. X. Shen, Z. Q. Chen, F. M. Zhang and Y. Chen, J. Nat. Prod., 2017, 80, 1081–1086 CrossRef CAS PubMed.
- F. Cao, J. K. Yang, Y. F. Liu, H. J. Zhu and C. Y. Wang, Nat. Prod. Rep., 2016, 30, 2448–2452 CrossRef CAS PubMed.
- D. N. Chen, S. S. Ma, L. He, P. B. Yuan, Z. G. She and Y. J. Lu, Tuberculosis, 2017, 103, 37–43 CrossRef CAS PubMed.
- J. M. Gao, S. X. Yang and J. C. Qin, Chem. Rev., 2013, 113, 4755–4811 CrossRef CAS PubMed.
- Y. F. Liu, R. Zhong, F. Cao, C. Wang and C. Y. Wang, Chem. Nat. Compd., 2016, 52, 548–551 CrossRef CAS.
- X. M. Hou, C. Y. Wang, Y. C. Gu and C. L. Shao, Nat. Prod. Res., 2016, 30, 2274–2277 CrossRef CAS PubMed.
- D. L. Zhao, C. L. Shao, Q. Zhang, K. L. Wang, F. F. Guan, T. Shi and C. Y. Wang, J. Nat. Prod., 2015, 78, 2310–2314 CrossRef CAS PubMed.
- J. Qi, C. L. Shao, M. Liu, X. Qi and C. Y. Wang, Chem. Nat. Compd., 2014, 50, 568–570 CrossRef CAS.
- J. Qi, C. L. Shao, Z. Y. Li, L. S. Gan, X. M. Fu, W. T. Bian, H. Y. Zhao and C. Y. Wang, J. Nat. Prod., 2013, 76, 571–579 CrossRef CAS PubMed.
- C. L. Shao, C. Y. Wang, Y. C. Gu, M. Y. Wei, J. H. Pan, D. S. Deng, Z. G. She and Y. C. Lin, Bioorg. Med. Chem. Lett., 2010, 20, 3284–3286 CrossRef CAS PubMed.
- L. Pairet, S. K. Wrigley, I. Chetland, E. E. Reynolds, M. A. Hayes, J. Holloway, A. M. Ainsworth, W. Katzer, X. M. Cheng, D. J. Hupe, P. Charlton and A. M. Doherty, J. Antibiot., 2010, 48, 913–923 CrossRef.
- S. Son, S. K. Ko, J. W. Kim, J. K. Lee, M. Jang, I. J. Ryoo, G. J. Hwang, M. C. Kwon, K. S. Shin, Y. Futamura, Y. S. Hong, H. Oh, B. Y. Kim, M. Ueki, S. Takahashi, H. Osada, J. H. Jang and J. S. Ahn, Phytochemistry, 2016, 122, 154–164 CrossRef CAS PubMed.
- J. Yang, X. L. Qi, G. Shao, Y. H. Pei, X. S. Yao and S. Kitanaka, Chin. Chem. Lett., 1998, 9, 833–834 CAS.
- Q. F. Guo, L. L. Dong, X. Y. Zang, Z. J. Gu, X. Y. He, L. D. Yao, L. P. Cao, J. Z. Qiu and X. Guan, Nat. Prod. Res., 2015, 29, 2000–2006 CrossRef CAS PubMed.
- N. Osmanova, W. Schultze and N. Ayoub, Phytochem. Rev., 2010, 9, 315–342 CrossRef CAS.
- J. Ren, S. S. Ding, A. Zhu, F. Cao and H. J. Zhu, J. Nat. Prod., 2017, 80, 2199–2203 CrossRef CAS PubMed.
- X. Y. Qin, K. L. Yang, J. Li, C. Y. Wang and C. L. Shao, Mar. Biotechnol., 2015, 17, 99–109 CrossRef CAS PubMed.
- A. Grassauer, R. Weinmuellner, C. Meier, A. Pretsch, E. Prieschl-Grassauer and H. Unger, Virol. J., 2008, 5, 107 CrossRef PubMed.
- G. Appendino, S. Gibbons, A. Giana, A. Pagani, G. Grassi, M. Stavri, E. Smith and M. M. Rahman, J. Nat. Prod., 2008, 71, 1427–1430 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESIMS, HRESIMS, 1D, 2D NMR data of compounds 1 and 2; ECD calculation details of 1; 1H and 13C NMR data for 5; antibacterial activities of 3 against bacterial strains. CCDC 1445322 and 1445327. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra13327h |
| This journal is © The Royal Society of Chemistry 2018 |