
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComplement activation by gold nanoparticles passivated with polyelectrolyte ligands†
Quang Huy Quacha and
James Chen Kah *ab
*ab
aDepartment of Biomedical Engineering, Faculty of Engineering, National University of Singapore, Singapore 117583, Singapore
bNUS Graduate School for Integrative Science and Engineering, National University of Singapore, Singapore. E-mail: biekahj@nus.edu.sg
First published on 9th February 2018
Abstract
Gold nanoparticles passivated by polyelectrolyte ligands are widely used to confer stability and biofunctionality. While nanoparticles and polyelectrolytes have been reported as activators, their ability to activate the complement system as hybrid polyelectrolyte-coated nanoparticles is poorly characterized. Here, we found that gold nanoparticles passivated by common polyelectrolytes activated the system differently. The surface area of AuNPs appeared to be a major determinant of complement activation level as it determined the amount of adsorbed polyelectrolytes. Although a moderate negative correlation between AuNP surface hydrophilicity and their activation level was observed, the surface charge and functional group of polyelectrolyte ligands also influenced the final complement activation level.
Nanomaterials may elicit various biological responses upon contact with blood, of which activation of the complement system in innate immunity is one of the earliest.1–3 The complement system is a collection of over 40 soluble and membrane-bound proteins, which acts in any of three distinct enzymatic cascades: classical, lectin, and alternative leading to the formation of a C3 convertase complex, and its accompanying range of biological responses, including inflammation, opsonization and cytolysis.2,4
While these responses could lead to undesirable physiological responses from over-activation and rapid clearance of nanomaterials from circulation, the intricate link between complement activation and adaptive immunity5–7 also presents opportunities for exploitation in immune-related applications such as vaccines development. Therefore, complement activation by nanomaterials in biomedical applications has attracted great attention recently.3,8–10
We previously reported complement activation by gold nanoparticles (AuNPs) of different shapes in their as-synthesized citrate and CTAB coatings.11 Polyelectrolyte ligands are also widely used in the preparation of AuNPs not only to stabilize AuNPs against aggregation, but to also enhance their solubility and confer additional surface functionalities.12–14 These polyelectrolytes with different functional groups have been reported as complement activators in both their particulate and planar surface-immobilized forms.15–21 However, the ability of polymer-passivated AuNPs to activate the complement system and the underlying mechanism remain poorly understood, although our previous study demonstrated that passivating the surface of AuNPs by poly(ethylene glycol) (PEG) modulated the activation level.11
Herein, we stabilized different shapes of AuNPs with different polyelectrolytes and examined their levels of complement activation and underlying mechanism. This would provide rational guidelines on the use of polyelectrolytes to modulate complement activation. All polyelectrolytes used in this study are hydrophilic and widely used as delivery platforms5,22 and surface passivating ligands of nanomaterials23 (Scheme 1).
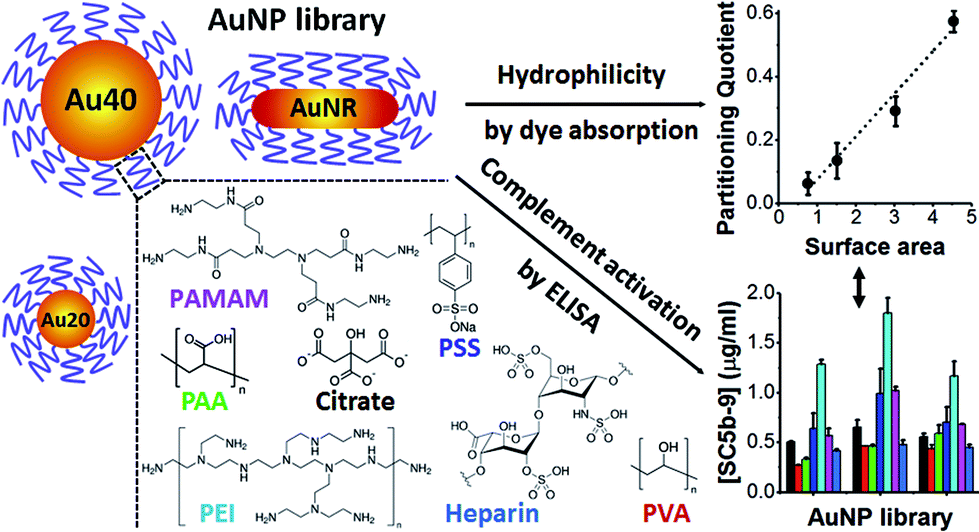 | ||
| Scheme 1 Schematic illustration of gold nanoparticles (AuNPs) core, polyelectrolytes and methods used in this study. AuNPs with spherical shape of 20 nm (Au20) and 40 nm (Au40) as well as rod-shape (AuNR) were used as AuNP cores. These AuNPs were passivated with various polyelectrolyte ligands, examined for their hydrophilicities and levels of complement activation. | ||
Spherical AuNPs of two diameters, 20 nm (Au20) and 40 nm (Au40), were synthesized by well-known citrate reduction method.24 AuNPs of rod-like shape (AuNR) were synthesized by hexadecyltrimethylammonium bromide (CTAB)-mediated method25 with the CTAB ligands subsequently replaced by citrate using previously established protocol.26 All AuNPs were then passivated by different polyelectrolytes following previously established layer-by-layer protocols (see Experimental section, ESI†). With this library of AuNPs, we sought to examine the effects of size and shape of AuNP core as well as polyelectrolyte shell on the level of complement activation.
The TEM images showed that Au20 and Au40 were spherical and homogenous while AuNR was rod-shaped with dimensions of approximately 40 × 10 nm (Fig. 1a–c). Hydrodynamic diameters, Dh, (Fig. 1d) were consistent with sizes of 20.2, 41.8, and 10.6 × 40.2 nm obtained from TEM images for Au20, Au40, and AuNR respectively (Fig. S1b–e, ESI†). The UV-Vis absorption spectra showed surface plasmon resonance (SPR) peaks of 523 nm and 530 nm for Au20 and Au40, respectively while AuNR had transverse peak of 509 nm and longitudinal peak of 800 nm (Fig. S1a, ESI†).27,28 All citrate-capped Au20, Au40 and AuNRs had similar zeta potentials of −30 mV, which became nearly neutral after passivated with poly(vinyl alcohol) (ζAuNP-PVA ≈ −5 mV) and polyamidoamine (ζAuNP-PAMAM ≈ −10 mV) (Fig. 1e). In contrast, surface passivation with poly(acrylic acid) (AuNP-PAA), poly(styrenesulfonate) (AuNP-PSS), and heparin (AuNP-Heparin) conferred AuNPs a more negative zeta potential of ≈−40 mV (Fig. 1e), while coating with poly(ethyleneimine) (AuNP-PEI) conferred AuNPs a positive surface charge of ≈+40 mV. The presence of polyelectrolytes on AuNPs was further confirmed by the increase in Dh of polyelectrolyte-passivated AuNPs (Fig. 1d).
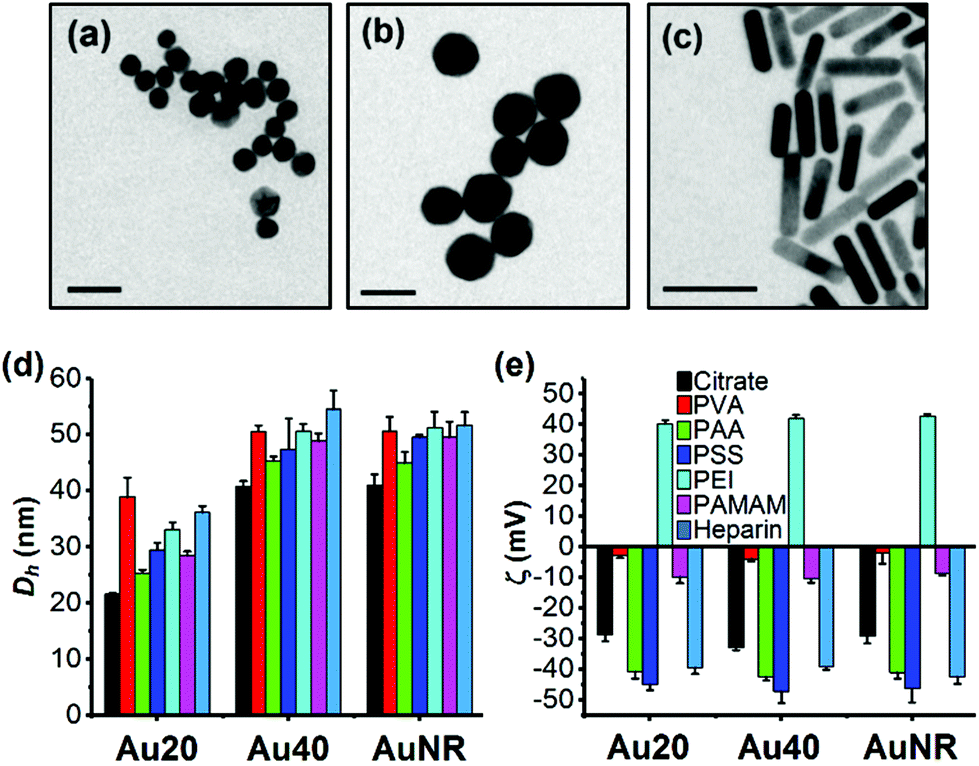 | ||
| Fig. 1 Physical properties of library of AuNPs. TEM images of (a) citrate-capped Au20, (b) Au40, and (c) AuNR. (d) Hydrodynamic diameter, Dh, and (e) zeta potential, ζ, of citrate-capped AuNPs and polyelectrolyte-passivated AuNPs. Each data point represents mean ± standard deviation (SD) of triplicate experiments. Scale bar in the TEM images represents 50 nm. | ||
When these polyelectrolyte-passivated AuNPs were incubated in normal human serum, the generation of SC5b-9 as an endpoint biomarker showed complement activation regardless of the activation pathway.3,4,8 Here, elevated levels of SC5b-9 were detected in all AuNPs-treated sera (Fig. 2), indicating activation of the complement system. Prior to surface passivation by polyelectrolytes, we observed an expected complement activation by AuNP-citrate, although the level was significantly lower than our positive control zymosan (a well-known complement activator derived from the wall of yeast cell). Also, Au40-citrate induced more SC5b-9 than Au20-citrate and AuNR-citrate (Fig. 2).
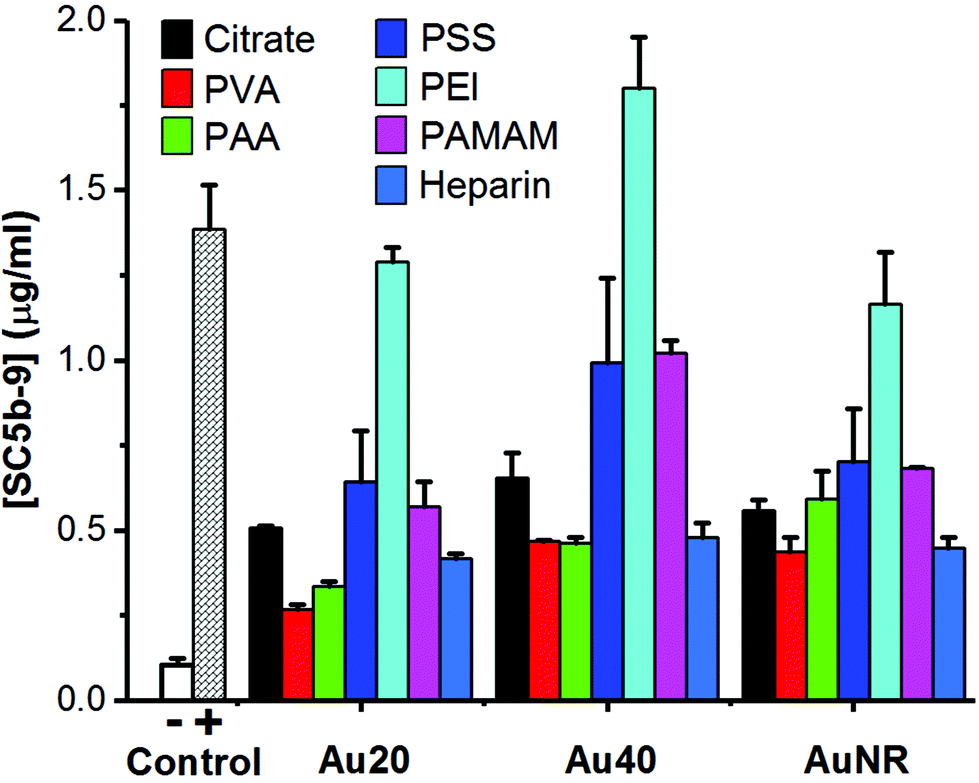 | ||
| Fig. 2 Detection of endpoint product of complement activation, SC5b-9, using ELISA kit. 1× PBS and zymosan (10 mg ml−1) were used as negative (−) and positive (+) controls, respectively. Each data point represents the mean ± standard deviation of triplicate experiments. | ||
The level of complement activation was dependent on both the AuNP core and polyelectrolyte ligand. Amongst the polyelectrolytes, we observed the highest level of complement activation from PEI, comparable or even higher than zymosan (Fig. 2). This agreed with previously published results where positively charged polymers carrying primary amino groups were shown to interact strongly with complement proteins to activate the complement system.18,21
Between the three AuNP cores, Au40-PEI had the highest level of complement activation ([SC5b-9] = 1.80 μg ml−1), followed by Au20-PEI ([SC5b-9] = 1.29 μg ml−1) and AuNR-PEI ([SC5b-9] = 1.16 μg ml−1), which had comparable levels of complement activation (Fig. 2). The same trend was true not only for AuNPs-PEI but also other polyelectrolyte-passivated AuNPs. While complement activation by nanomaterials have been shown to depend on their sizes and shapes,8,29 the differences observed here were more likely attributed to the amount of PEI adsorbed on the surface of AuNPs, which was in turn dictated by their surface area, since PEI by itself has been shown to activate the complement system in concentration-dependent manner.18 Here, Au20 and AuNR had comparable surface areas (1256 and 1413 nm2, respectively) and hence similar levels of complement activation, while Au40 had the largest surface area (5024 nm2), thus accounting for the highest level of complement activation (Fig. 2).
Unlike PEI, AuNPs passivated by both PVA and PAA induced the lowest levels of SC5b-9 (Fig. 2). While PVA is widely known as a biocompatible ligand on AuNRs,23 its complement activation was not totally avoided probably due to its nucleophilic hydroxyl groups,16,17 similar to PEI with nucleophilic amine groups. However, its complement activation level was much lower, and was likely due to its near neutral surface charge which did not promote interaction with many negatively charged complement proteins unlike the positively charged PEI.
Similarly, the highly negative charge AuNP-PAA due to high density of carboxyl groups did not promote their interaction with negatively charged complement proteins, thus inducing an equally low level of complement activation as AuNP-PVA. Therefore, PAA is one of the most widely used water-soluble polyelectrolyte, superabsorbent polymer as well as food additive. Nonetheless, PAA conjugated to IgG has been found to specifically interact with positively charged C1q complement protein to activate the classical pathway.30,31 Hence, AuNP-PAA has been found to promote inflammation via activation with fibrinogen.32 In this study, the AuNP-PAA could have activated the complement system via the classical pathway due to their interactions with C1q complement protein as reported.30,31
Owing to its nature as an anticoagulant, heparin has been reported to minimize complement activation.33–35 However, we found that AuNP-heparin still activated the complement system although their level of complement activation was comparable to PVA and PAA, and much lower than PEI, PSS and PAMAM.
Interestingly, despite the presence of nucleophilic amine groups on PAMAM similar to PEI, AuNP-PAMAM activated the complement system at much lower levels compared to AuNP-PEI. PAMAM dendrimers were known to be generation-dependent complement activators with stronger complement activation observed in higher generations.15 Here, we used PAMAM of generation 2.0 with aminoethanol surface and reasoned that coupling of amine with hydroxyl group helped to reduce activation level of AuNP-PAMAM. In fact, the activation level of AuNP-PAMAM was between that of AuNP-PVA with hydroxyl groups, and AuNP-PEI with amine groups. Furthermore, since primary amine groups adsorbed more C3b, a major component of complement proteins in serum, than secondary or tertiary amino groups,20 the lack of primary amine group in PAMAM also explained its weaker complement activation than PEI which possesses a mixture of primary, secondary, and tertiary amine groups.
The amount of SC5b-9 induced by AuNP-PSS was comparable to that of AuNP-PAMAM (Fig. 2). PSS is a widely used polyelectrolyte building block in layer-by-layer assembly,36 and we used it as an intermediate ligand to prepare AuNR-citrate (Fig. S2, ESI†). PSS by itself interacted with complement proteins of the classical pathway to activate the complement system.19 Our results not only confirmed complement activation by PSS passivation but also highlight the potential side effects of block-copolymer containing PSS commonly used as drug delivery platform in activating the complement system.
Since the hydrophilicity of nanoparticles has been shown to modulate non-specific protein adsorption37 and dictate immune response,38 we further examined for possible correlation between the hydrophilicity of polyelectrolyte-passivated AuNPs and their level of complement activation. We measured relative hydrophilicity of polyelectrolyte-passivated AuNPs by dye absorption using a hydrophilic dye Nile Blue which interacted with a hydrophilic moiety (see Experimental section, ESI). On mixing the dye with AuNPs passivated with different polyelectrolyte ligands, we determined the partitioning quotient (PQ) as the ratio of dye bound on nanoparticles surface to the amount of free dye. In a plot of PQ versus surface area of nanoparticles, the slope of this linear regression line represented relative surface hydrophilicity.39
We observed different levels of hydrophilic dye adsorption on all polyelectrolyte-passivated AuNPs after 3 h incubation, indicating differences in their hydrophilicities (Fig. S8–10, ESI†). AuNPs-PEI was the least hydrophilic (Fig. 3) as determined from the smallest slope of the linear regression line (Table S1, ESI†), while AuNPs passivated with PVA, PAA, and heparin are amongst the most hydrophilic as given by the larger slope of the PQ vs. nanoparticle surface area plot (Fig. 3 and Table S1, ESI†).
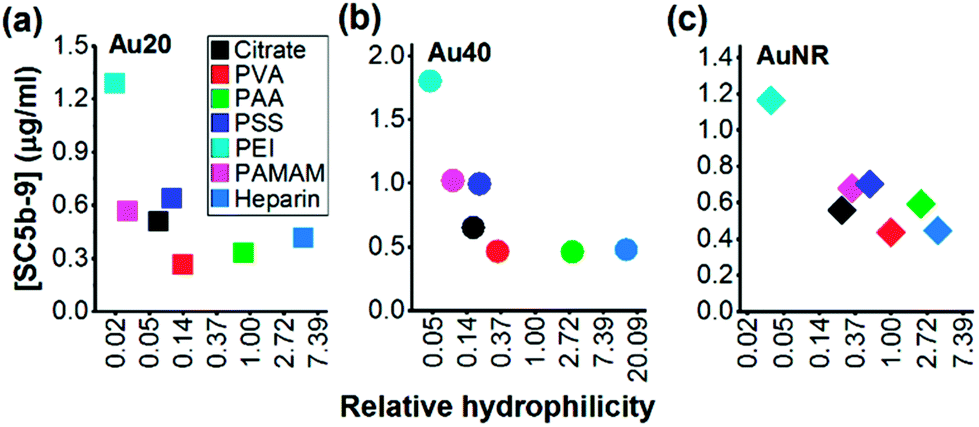 | ||
| Fig. 3 Negative correlation between surface hydrophilicity and complement activation by polyelectrolyte-passivated (a) Au20, (b) Au40 and (c) AuNR. X scale bar was plotted as ln of the value of relative hydrophilicity. | ||
We also observed a negative correlation between surface hydrophilicity and complement activation level across all AuNPs regardless of shape or size (Fig. 3). However, this correlation was only moderate with Pearson correlation coefficient, r = −0.2730, −0.4101, and −0.5489 for Au20, Au40 and AuNR, respectively (Fig. 3). Since Moyano et al. reported a positive correlation between nanoparticle surface hydrophobicity and their immune response previously,38 the similarity in our observations suggested that the complement system could be a potential mediator between surface hydrophilicity/hydrophobicity and downstream immune response. Nonetheless, our results also demonstrated the complexity of complement activation by engineered nanomaterials, as it was not dependent solely on any one physical property such as shape, size, surface charge, surface functional group or surface hydrophilicity, but the interplay of these properties to influence the complement proteins adsorption.
In summary, AuNPs passivated with different polyelectrolyte ligands activated the complement system at different levels, as characterized by the presence of endpoint product of complement activation, SC5b-9. The surface area of AuNPs appeared as a major determinant of complement activation level as it determined the amount of adsorbed polyelectrolytes. Although a moderate negative correlation between AuNPs surface hydrophilicity and their activation level was observed, the surface charge and functional group of polyelectrolyte ligands also influenced the final complement activation level. These findings provide new insights to rational selection and design guidelines for the use of polyelectrolytes to either suppress complement activation and downstream immune response for nanoparticulate drug delivery systems or to enhance complement activation and immune response for vaccine development.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The funding used to support the research of the manuscript was from the Ministry of Education (MOE) AcRF Tier 2 Grant. We acknowledge Laurent Bekale for useful discussion on the preparation of various polyelectrolyte-passivated AuNPs. Additional description of the experimental methodology and data on the characterization of polyelectrolyte-passivated AuNPs, concentration calibration curves of SC5b-9 and Nile Blue dye and hydrophilicity measurements with Nile Blue dye are available as ESI.†References
- N. Bertrand and J.-C. Leroux, J. Controlled Release, 2012, 161, 152–163 CrossRef PubMed.
- D. Ricklin, G. Hajishengallis, K. Yang and J. D. Lambris, Nat. Immunol., 2010, 11, 785–797 CrossRef CAS PubMed.
- A. J. Andersen, S. H. Hashemi, T. L. Andresen, A. C. Hunter and S. M. Moghimi, J. Biomed. Nanotechnol., 2009, 5, 364–372 CrossRef CAS PubMed.
- N. S. Merle, S. E. Church, V. Fremeaux-Bacchi and L. T. Roumenina, Front. Immunol., 2015, 6, 262 Search PubMed.
- J. R. Dunkelberger and W. C. Song, Cell Res., 2010, 20, 34–50 CrossRef CAS PubMed.
- H. Rus, C. Cudrici and F. Niculescu, Immunol. Res., 2005, 33, 103–112 CrossRef CAS PubMed.
- F. R. Toapanta and T. M. Ross, Immunol. Res., 2006, 36, 197–210 CrossRef CAS PubMed.
- S. M. Moghimi, A. J. Andersen, D. Ahmadvand, P. P. Wibroe, T. L. Andresen and A. C. Hunter, Adv. Drug Delivery Rev., 2011, 63, 1000–1007 CrossRef CAS PubMed.
- B. Nilsson, K. N. Ekdahl, T. E. Mollnes and J. D. Lambris, Mol. Immunol., 2007, 44, 82–94 CrossRef CAS PubMed.
- J. Szebeni, Toxicology, 2005, 216, 106–121 CrossRef CAS PubMed.
- Q. H. Quach and J. Chen Yong Kah, Nanotoxicology, 2017, 1–39 Search PubMed.
- J. Shan and H. Tenhu, Chem. Commun., 2007, 4580–4598 RSC.
- L. Chen and H.-A. Klok, Soft Matter, 2013, 9, 10678–10688 RSC.
- O. S. Muddineti, B. Ghosh and S. Biswas, Int. J. Pharm., 2015, 484, 252–267 CrossRef CAS PubMed.
- A. Åkesson, M. Cárdenas, G. Elia, M. P. Monopoli and K. A. Dawson, RSC Adv., 2012, 2, 11245–11248 RSC.
- Y. Arima, M. Kawagoe, M. Furuta, M. Toda and H. Iwata, Biomaterials, 2010, 31, 6926–6933 CrossRef CAS PubMed.
- Y. Arima, M. Kawagoe, M. Toda and H. Iwata, ACS Appl. Mater. Interfaces, 2009, 1, 2400–2407 CAS.
- O. M. Merkel, R. Urbanics, P. Bedőcs, Z. Rozsnyay, L. Rosivall, M. Toth, T. Kissel and J. Szebeni, Biomaterials, 2011, 32, 4936–4942 CrossRef CAS PubMed.
- Y. Murakami, H. Iwata, E. Kitano, H. Kitamura and Y. Ikada, J. Biomater. Sci., Polym. Ed., 2005, 16, 685–697 CrossRef CAS PubMed.
- M. Toda, T. Kitazawa, I. Hirata, Y. Hirano and H. Iwata, Biomaterials, 2008, 29, 407–417 CrossRef CAS PubMed.
- M. Toda and H. Iwata, ACS Appl. Mater. Interfaces, 2010, 2, 1107–1113 CAS.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS PubMed.
- C. Kinnear, D. Burnand, M. J. Clift, A. F. Kilbinger, B. Rothen-Rutishauser and A. Petri-Fink, Angew. Chem., Int. Ed., 2014, 53, 12613–12617 CAS.
- W. Leng, P. Pati and P. J. Vikesland, Environ. Sci.: Nano, 2015, 2, 440–453 RSC.
- E. L. L. Yeo, J. U.-J. Cheah, B. Y. Lim, P. S. P. Thong, K. C. Soo and J. C. Y. Kah, ACS Biomater. Sci. Eng., 2017, 3(6), 1039–1050 CrossRef.
- J. G. Mehtala, D. Y. Zemlyanov, J. P. Max, N. Kadasala, S. Zhao and A. Wei, Langmuir, 2014, 30, 13727–13730 CrossRef CAS PubMed.
- W. Haiss, N. T. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef PubMed.
- J. C. Y. Kah, Nanomaterial Interfaces in Biology: Methods and Protocols, 2013, pp. 119–126 Search PubMed.
- M. A. Dobrovolskaia and S. E. McNeil, Nat. Nanotechnol., 2007, 2, 469–478 CrossRef CAS PubMed.
- G. Schmer, R. A. Henderson and W. Müller, J. Biomater. Sci., Polym. Ed., 1995, 6, 67–78 CrossRef.
- R. J. Klauser, G. Schmer, W. L. Chandler and W. Müller, Biochim. Biophys. Acta, Mol. Cell Res., 1990, 1052, 408–415 CrossRef CAS.
- Z. J. Deng, M. Liang, M. Monteiro, I. Toth and R. F. Minchin, Nat. Nanotechnol., 2011, 6, 39–44 CrossRef CAS PubMed.
- R. Biran and D. Pond, Adv. Drug Delivery Rev., 2017, 112, 12–23 CrossRef CAS PubMed.
- M. Pekna, R. Larsson, B. Formgren, U. Nilsson and B. Nilsson, Biomaterials, 1993, 14, 189–192 CrossRef CAS PubMed.
- J. P. Rosengren-Holmberg, J. Andersson, J. R. Smith, C. Alexander, M. R. Alexander, G. Tovar, K. N. Ekdahl and I. A. Nicholls, Biomater. Sci., 2015, 3, 1208–1217 RSC.
- A. S. Angelatos, K. Katagiri and F. Caruso, Soft Matter, 2006, 2, 18–23 RSC.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. W. Chan, J. Am. Chem. Soc., 2012, 134, 2139–2147 CrossRef CAS PubMed.
- D. F. Moyano, M. Goldsmith, D. J. Solfiell, D. Landesman-Milo, O. R. Miranda, D. Peer and V. M. Rotello, J. Am. Chem. Soc., 2012, 134, 3965–3967 CrossRef CAS PubMed.
- Y. Xiao and M. R. Wiesner, J. Hazard. Mater., 2012, 215, 146–151 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, 10 supporting figures (Fig. S1–S10), and 1 supporting table (Table S1). See DOI: 10.1039/c7ra13325a |
| This journal is © The Royal Society of Chemistry 2018 |