
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiol–ene photoimmobilization of chymotrypsin on polysiloxane gels for enzymatic peptide synthesis†
Meng Wang,
Jun Xing,
Yu-Tang Sun,
Ling-Xiang Guo,
Bao-Ping Lin and
Hong Yang*
and
Hong Yang*
School of Chemistry and Chemical Engineering, Jiangsu Province Hi-Tech Key Laboratory for Bio-medical Research, Jiangsu Key Laboratory for Science and Application of Molecular Ferroelectrics, Southeast University, Nanjing, 211189, China. E-mail: yangh@seu.edu.cn
First published on 27th March 2018
Abstract
Chemical incorporation of enzymes onto polymeric materials has recently attracted intense scientific attention. Cross-linked polysiloxane gels as a typical super-hydrophobic support, are a good candidate for supporting enzymes in low-water organic medium to efficiently catalyze peptide synthesis because the hydrophobic polysiloxane matrix can prevent water from attacking the acyl-enzyme intermediate, which is beneficial for the shift in equilibrium to peptide formation. In this work, we develop a facile strategy to photoimmobilize olefin-functionalized chymotrypsin onto cross-linked polysiloxane gels via UV-initiated thiol–ene click chemistry. The impacts of water addition amount, heat-treatment and recyclability of the immobilized chymotrypsin influencing the peptide synthesis efficiency are investigated. Compared with the native chymotrypsin, polysiloxane-immobilized chymotrypsin showed advantageous catalytic activity, higher thermal stability and superior recyclability.
Introduction
Enzymes are regarded as highly efficient and multi-functional biocatalysts, which can be widely applicable and useful tools for organic chemical reactions, particularly in chemo-, regio-, and enantio-selective processes.1–4 Nowadays, due to the large and growing interest in peptides in market-oriented applications for the food and pharmaceutical industries,5 enzymatic synthesis of peptides has become a typical bioprocess with many advantages, including high efficiency, high selectivity, high product yields, mild reaction conditions, and minimal side chain protection requirements.6,7Specifically, enzymatic peptide synthesis is a kinetically-controlled two-step reaction, which includes the formation of acyl-enzyme intermediate, and the competition between water which leads to hydrolysis and the nucleophile which leads to peptide synthesis, as illustrated in Fig. 1a.8 Hitherto, much work has discovered the fact that low-water organic media were necessary for retaining enzymes' native conformation, sufficient flexibility, and catalytic activity in enzyme-catalyzed peptide synthesis.9–11 However, the application of enzymes in their native forms for biotransformation in organic media is plagued by several drawbacks, such as autolysis, structural denaturation, low operational stability and poor recyclability, etc.12–14 In order to overcome these limitations, it is necessary to adopt proper strategies for enzyme stabilization in organic solvents, for example, by physisorption, covalent immobilization, entrapment, or encapsulation of enzymes in various solid supports.15–22
 | ||
| Fig. 1 (a) Kinetically-controlled enzymatic peptide synthesis mechanism. (b) Schematic illustration of the thiol–ene photoimmobilization approach towards polysiloxane-gel-bonded chymotrypsin. | ||
Among various support media and immobilization techniques, chemically bonding of enzymes onto polymeric materials23–27 offers several advantages due to its good resistance to organic solvents, excellent processability, biocompatibility, and inertness to microbial attack. In particular, cross-linked polysiloxane gels as typical super-hydrophobic supports, have garnered so much excitement for their advantageous properties, such as excellent chemical resistance, good thermal stability, and extraordinary permeability in organic solvents, which render it a good candidate for supporting enzymes in organic media to efficiently catalyze peptide synthesis. In addition, the enzyme leakage and poor reusability can be avoided due to the strong chemical bonds between enzymes and polymer network. Although several organic and inorganic small molecule catalysts have been immobilized onto polysiloxane gels,28,29 occlusion of biocatalysts into polysiloxane gels has also been reported,30 there was barely a literature describing chemically incorporation of enzymes onto polysiloxane gels, possibly because the widely used platinum-catalyzed hydrosilylation reaction of polymethylhydrosiloxanes (PMHS) and olefin-terminated molecules required platinum as the catalyst and a relatively high reaction temperature, which might severely harm the enzyme activity. To this end, the light-stimulated thiol–ene31–33 photoimmobilization, which utilizes multifunctional mercapto and vinyl monomers to form polymer networks, provides an alternative approach as a mild, efficient and green method for surface immobilization purposes.34–36
Herein, we report a facile and fast thiol–ene photoimmobilization of olefin-terminated enzyme onto poly[3-mercaptopropylmethylsiloxane] (PMMS).37–40 Chymotrypsin (CT) is chosen as a modal biocatalyst for peptide synthesis in this study because it possesses many accessible amino groups of side-chain-exposed amino acids (such as Lys, Ser, Gly, Cys, and Tyr residues) which can be chemically modified.41 As shown in Fig. 1b, native chymotrypsin was firstly functionalized with terminal olefin groups by a “grafting to” strategy in aqueous solution. The vinyl-decorated chymotrypsin was then immobilized onto PMMS gels by UV-initiated thiol–ene click chemistry reaction, to provide the desired polysiloxane-gel-bonded chymotrypsin (PMMS-CT).
Previously, considerable research efforts have been devoted to the conjugates of poly(ethylene glycol) (PEG) derivatives and chymotrypsin for peptide synthesis.42,43 Inspired by these pioneering works, we used hydrophilic PEG diacrylate as the cross-linker, in order to reserve a tiny amount of water to retain the catalytic activity of chymotrypsin for peptide synthesis. Meanwhile, the hydrophobic polysiloxane matrice will prevent water from attacking the acyl-enzyme intermediate, which is beneficial for the shift in equilibrium to peptide formation in low-water organic media.
Experimental section
Materials and instruments
Poly[3-mercaptopropylmethylsiloxane] (PMMS, SMS-992, MW 4000–7000, 95 cst) was purchased from Gelest Inc. Poly(ethylene glycol)diacrylate (average Mn ∼ 700) was purchased from Aldrich Inc. 2,2-Dimethoxy-2-phenylacetophenone (DMPA), chymotrypsin (from bovine pancreas, Type II, lyophilized powder, 48 units per mg protein; 1.0 unit represents that 1.0 mg of protein hydrolyzes 1.0 μmol of N-benzoyl-L-tyrosine ethyl ester per minute at pH ∼ 7.8 and 25 °C), N-benzoyl-L-tyrosine ethyl ester (Bz-Tyr-OEt), N-acetyl-L-phenylalanine ethyl ester (Ac-Phe-OEt), N-acetyl-L-tryptophan ethyl ester (Ac-Trp-OEt), L-phentlalaninamide (Phe-NH2), L-tyrosine amide (Tyr-NH2), glycinamide (Gly-NH2), alaninamide (Ala-NH2), anhydrous organic solvents (hexane, THF, toluene, DMF, DMSO) were purchased from Aladdin (Shanghai) Inc. The detailed information of instrumentation, synthetic procedures, NMR spectra and ESI-MS data are listed in the ESI.†Enzyme modification
Chymotrypsin (2.00 g, 0.08 mmol of protein, 1.04 mmol of –NH2 groups) was dissolved in 200 mL of phosphate buffer (0.1 M, pH 8.0). 10-Undecenoyl chloride (3.24 g, 16.0 mmol, the molar ratio of enzyme to 10-undecenoyl chloride was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200) dissolved in 20 mL of dichloromethane was added dropwise into the above phosphate buffer solution. The two phase reaction mixture was vigorously stirred at 25 °C for 36 h and the pH value was maintained at 7.5–8.0 (pH 7.5–8.0 was chosen to increase the nucleophilicity of the amino groups of lysine fragments in the protein towards 10-undecenoyl chloride) by the addition of 0.1 M sodium hydroxide solution during the whole process. Then, the vinyl-modified proteins were purified by dialysis against phosphate buffer (0.1 M, pH 8.0) for 48 h using a dialysis cube with molecular weight cut-off (MWCO) of 5000 Da, followed by lyophilization for another 48 h.
:200) dissolved in 20 mL of dichloromethane was added dropwise into the above phosphate buffer solution. The two phase reaction mixture was vigorously stirred at 25 °C for 36 h and the pH value was maintained at 7.5–8.0 (pH 7.5–8.0 was chosen to increase the nucleophilicity of the amino groups of lysine fragments in the protein towards 10-undecenoyl chloride) by the addition of 0.1 M sodium hydroxide solution during the whole process. Then, the vinyl-modified proteins were purified by dialysis against phosphate buffer (0.1 M, pH 8.0) for 48 h using a dialysis cube with molecular weight cut-off (MWCO) of 5000 Da, followed by lyophilization for another 48 h.
Typical preparation procedure of PMMS-CT
Vinyl-modified proteins chymotrypsin (50 mg), PMMS (100 mg, 0.75 mmol of –SH groups), poly(ethylene glycol) diacrylate (average Mn ∼ 700, 140 mg, 0.2 mmol, as cross-linker), DMPA (10 mg, as photo-initiator), and dichloromethane (2 mL) were added in a 10 mL transparent vial tube. The mixture was ultrasonicated for 2 min to ensure a homogeneous dispersion, and then UV irradiated at 365 nm at room temperature for 2 min. The biocatalyst gel was carefully crushed to small particles, washed 3 times with dichloromethane to remove any unreacted chymotrypsin and DMPA, and dried in a fume hood overnight, to provide the polysiloxane-based biocatalyst gel PMMS-CT which was stored for future uses.Enzymatic synthesis of dipeptide Bz-Tyr-Phe-NH2
In a 10 mL round-bottom flask, acyl donor N-benzoyl-L-tyrosine ethyl ester (Bz-Tyr-OEt, 250.8 mg, 0.8 mmol), and acyl acceptor L-phenylalaninamide (Phe-NH2, 262.4 mg, 1.6 mmol), distilled water (40 μL, 1.0 vol%), PMMS-CT (100 mg, ca. 17.2 mg chymotrypsin immobilized) were added in 4.0 mL of hexane/THF (v/v = 1:1). The reaction mixture was stirred at 37 °C for 3 h under nitrogen atmosphere. The crude product mixture was dissolved in MeOH (40 mL) and PMMS-CT was separated by filtration. The resulting solvent was evaporated and the crude product was first well-dispersed in benzene, then filtered, washed with benzene and water, and finally dried under vacuum to give the dipeptide Bz-Tyr-Phe-NH2.
Results and discussion
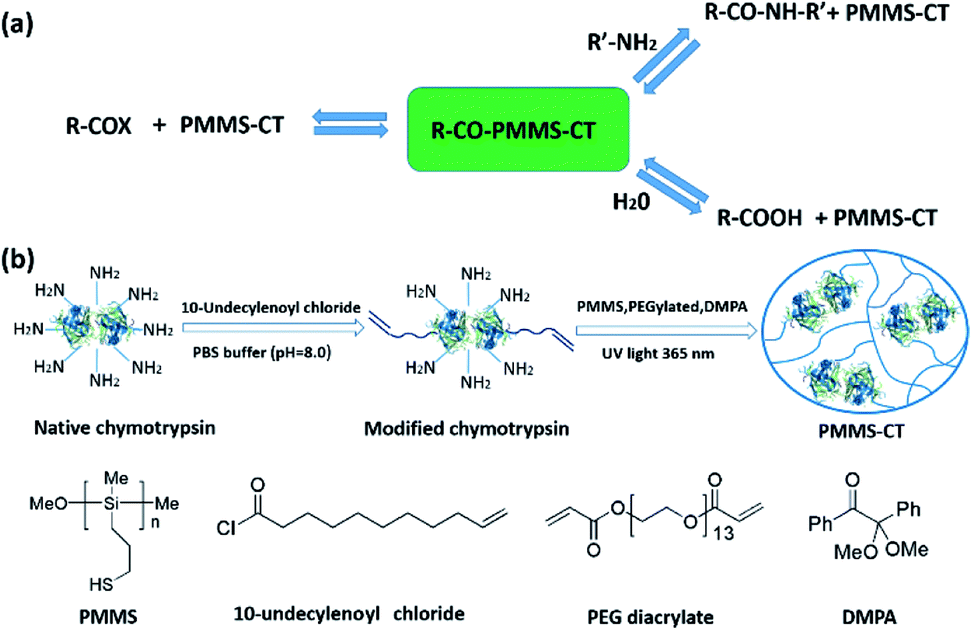
In order to synthesize the designed polysiloxane-gel-bonded enzyme PMMS-CT, we first functionalized the native chymotrypsin molecules with terminal olefin groups by a “grafting to” strategy in aqueous solution. Previously, Russell group44 has used 2-bromoisobutyryl bromide to modify lysine sites of chymotrypsin which resulted in the conjugation of 3-7 ATRP initiators per protein molecule, and further discovered that the larger and denser conjugation chains would sterically hinder the access of substrates to the enzyme active site. Following this pioneering study, native chymotrypsin dissolved in 0.1 M phosphate buffer (pH ∼ 8.0) was first treated with an excess amount of 10-undecenoyl chloride dissolved in dichloromethane. The two-phase amidation reaction was vigorously stirred at 25 °C and pH was maintained at 7.5–8.0. The crude product was purified by a dialysis-lyophilization process to provide the vinyl-modified chymotrypsin. The attachment of 10-undecenoyl group onto native chymotrypsin was confirmed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry and Fourier transform infrared spectroscopy (FTIR) analysis of the native chymotrypsin and the modified chymotrypsin samples. As can be seen from MALDI-TOF MS spectra (Fig. 2a), the maximum ion peaks of native chymotrypsin and modified chymotrypsin samples exhibit at 25456 and 25923 respectively, which indicates that roughly 2–3 moieties of 10-undecenoyl group have been successfully installed on the amino groups of chymotrypsin. Furthermore, as shown in Fig. 2b, compared with the curve of the native chymotrypsin, a new characteristic IR peak of the modified chymotrypsin sample vibrated at 3070 cm−1 indicates the existence of terminal olefin groups on the surface of chymotrypsin.
 | ||
| Fig. 2 (a) MALDI-TOF MS spectra of the native chymotrypsin and vinyl-modified chymotrypsin. (b) FTIR spectra of the native chymotrypsin, vinyl-modified chymotrypsin and PMMS-CT samples. | ||
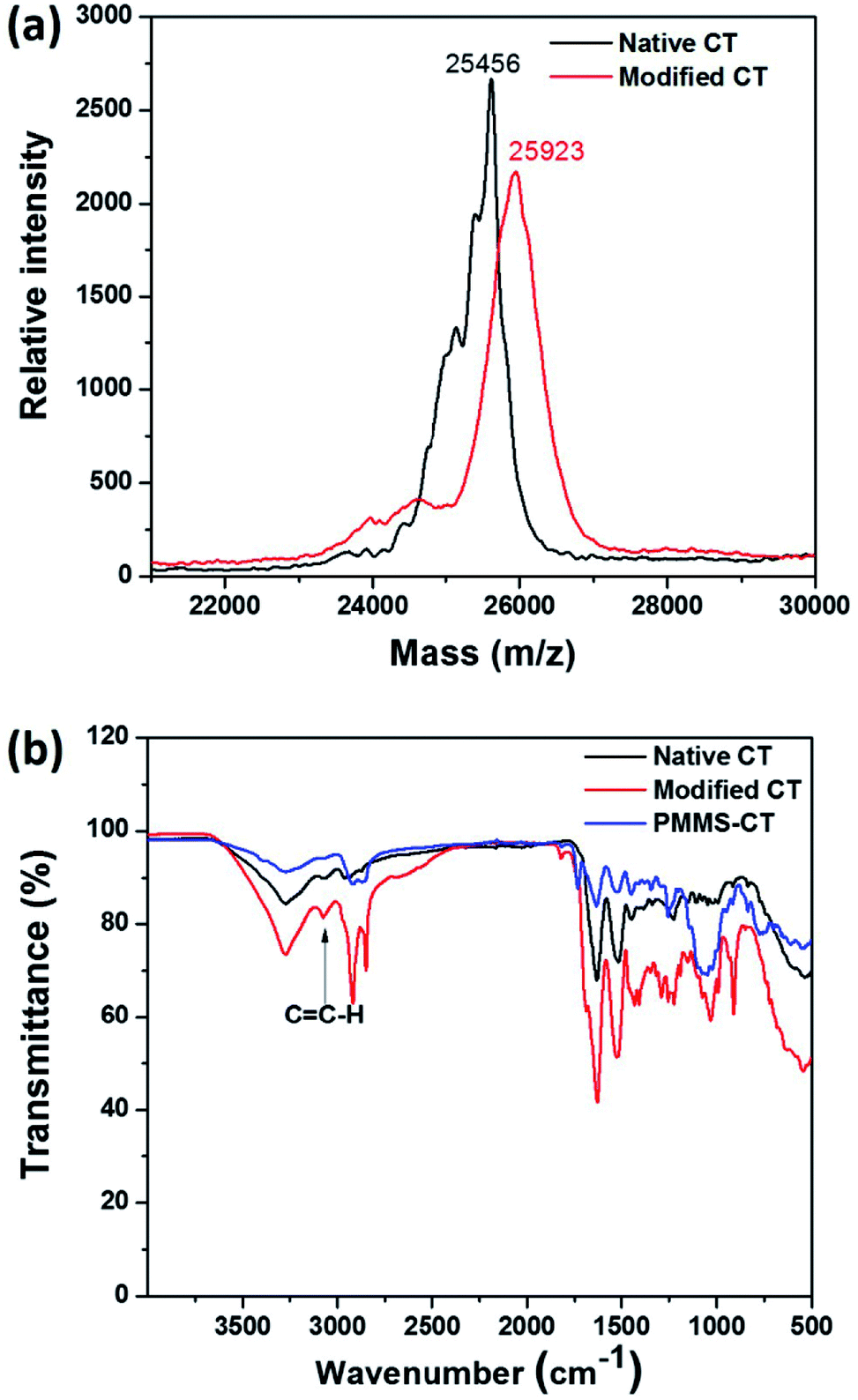
To prepare the polysiloxane-gel-bonded chymotrypsin PMMS-CT, mercapto-functionalized polysiloxane PMMS, vinyl-modified chymotrypsin, the crosslinker PEG diacrylate and the photoinitiator dimethoxyphenylacetophenone (DMPA) were first mixed in 2 mL anhydrous dichloromethane, the reaction solution was ultrasonicated for 2 min to ensure a homogeneous dispersion and then irradiated by UV light (365 nm, 15.53 mW cm−2) at room temperature for 2 min, which transformed the solution into a crosslinked gel. Fig. 2b presented FTIR spectra of the native chymotrypsin, vinyl-modified chymotrypsin and PMMS-CT samples. The absorption peak at 3070 cm−1 of the vinyl-modified chymotrypsin ascribed to the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H stretching vibration disappeared in PMMS-CT sample, which indicated that the CC groups of the vinyl-modified chymotrypsin have been completely consumed by the mercapto groups of PMMS and chymotrypsin were successfully immobilized.
C–H stretching vibration disappeared in PMMS-CT sample, which indicated that the CC groups of the vinyl-modified chymotrypsin have been completely consumed by the mercapto groups of PMMS and chymotrypsin were successfully immobilized.
Moreover, for comparison purpose, we also physically occluded native chymotrypsin in polysiloxane gels by replacing vinyl-modified chymotrypsin with the native chymotrypsin in the above experimental protocol, the resulting chymotrypsin-occluded polysiloxane product was named as PMMS@CT. Scanning electron microscope (SEM) was used to examine the microscopic distributions of chymotrypsin inside PMMS-CT and PMMS@CT gels respectively. As shown in Fig. 3a, there were apparently some aggregated enzyme particles on the surface of PMMS@CT sample, which indicated that chymotrypsin was physically embedded inside pores of polysiloxane matrix, whereas the smooth surface of PMMS-CT sample (Fig. 3b) indicated that the vinyl-modified chymotrypsin has been at least partially chemically-bonded onto the crosslinked polysiloxane gel. Moreover, PMMS-CT and PMMS@CT samples were dispersed in hexanes and stirred for 3 h. As illustrated in Fig. 3c, PMMS@CT solution became more and more turbid after 3 h of swelling, whereas PMMS-CT solution (Fig. 3d) during the same incubation period remained transparent and contained no suspended particles. This comparative experiment demonstrated that physical occlusion of chymotrypsin into polymeric matrices would induce spontaneous enzyme leakage when dispersed in organic solvents. On the contrary, thiol–ene photoimmobilization method could chemically incorporate vinyl-modified enzymes onto polysiloxane matrices in an efficient way.
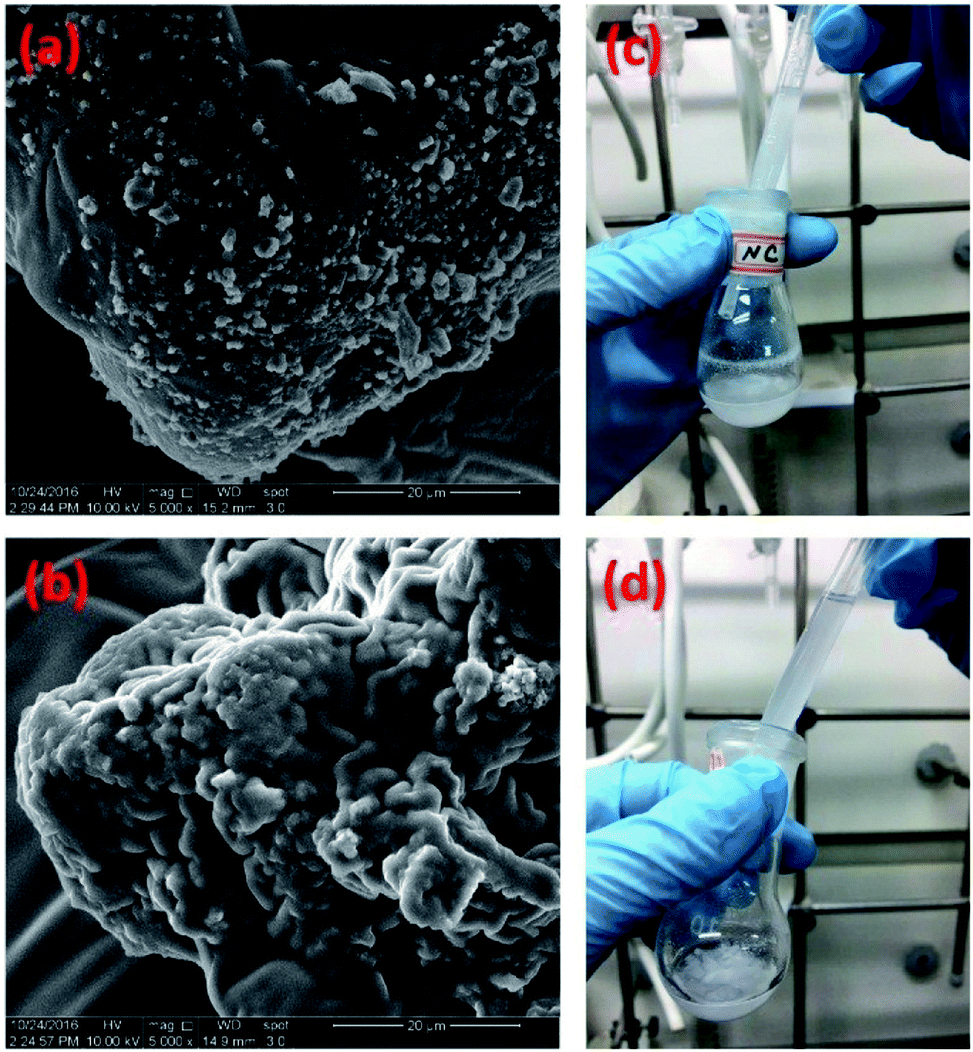 | ||
| Fig. 3 SEM images of polysiloxane gels with (a) physically occluded native chymotrypsin and (b) chemically bonded vinyl-modified chymotrypsin. Photo-images of (c) PMMS@CT and (d) PMMS-CT samples dispersed in hexanes. | ||
Enzymatic dipeptide synthesis of L-phenylalaninamide (Phe-NH2) and N-benzoyl-L-tyrosine ethyl ester (Bz-Tyr-OEt) providing N-benzoyl-tyrosyl phenylalaninamide (Bz-Tyr-Phe-NH2, Scheme 1), was one of the most typical enzymatic peptide synthesis reactions in low-water organic medium, because the dipeptide product Bz-Tyr-Phe-NH2 was totally insoluble in most of organic solvents, easily precipitated out, and could be efficiently separated from its two starting reagents which had good solubility contrarily.45,46 In general, when the reaction was finished, a large quantity of methanol was first added into the reaction mixture to dissolve the product precipitates, then the catalyst PMMS-CT gels were filtered off, and the resulting solution was concentrated to provide crude product powders which were substantially rinsed by benzene and water to remove the unreacted amino acid derivatives. The conversion yield could be calculated based on the weight of the dried product Bz-Tyr-Phe-NH2. Meanwhile, the side reactions (ester hydrolysis of Bz-Tyr-OEt and peptide hydrolysis of Bz-Tyr-Phe-NH2) could also happen which would give the byproduct N-benzoyl-L-tyrosine (Bz-Tyr-OH). The detailed experimental procedures are described in ESI.† The effects of water addition amount, heat-treatment and recyclability of PMMS-CT influencing on the peptide synthesis efficiency were investigated herein.
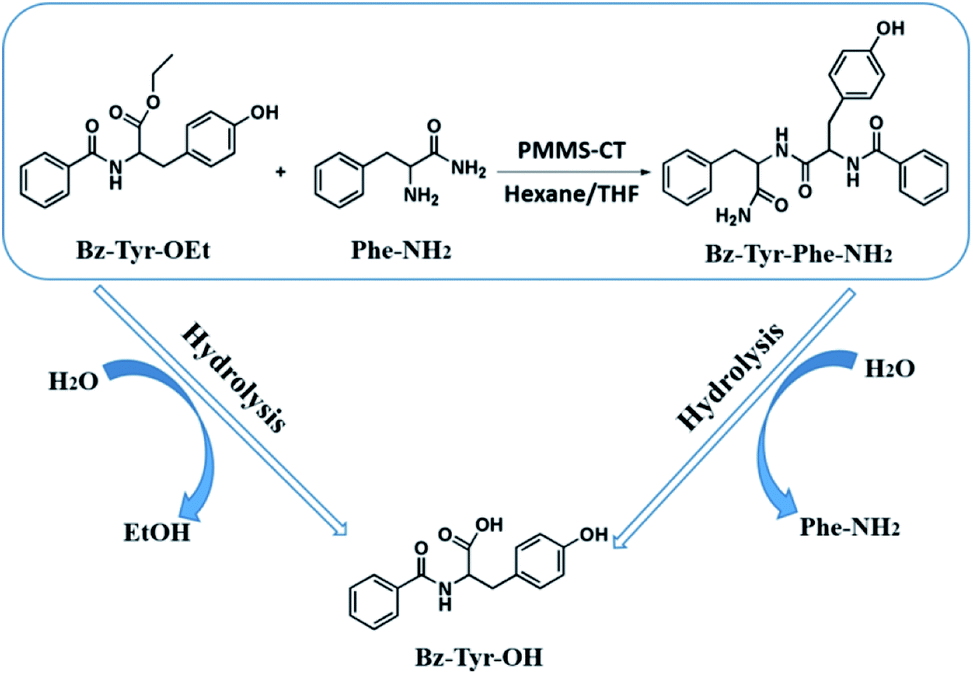 | ||
| Scheme 1 Enzymatic dipeptide synthesis of Bz-Tyr-Phe-NH2 is examined as the PMMS-CT catalyzed model reaction. | ||
In conventional enzymatic peptide synthesis, a small amount of water was required to be added into organic solvents to retain the catalytic activities of enzymes, completely anhydrous organic medium would result in failure of peptide formation. In order to explore for the optimal water addition amount, we added 0, 0.25, 0.5, 0.75, 1.0, 3.0, 5.0, 10, 20, 50 vol% water into the reaction mixture and examined the reaction yields, respectively. As shown in Fig. 4, without the addition of water, Bz-Tyr-Phe-NH2 could not be obtained in either native chymotrypsin or PMMS-CT catalytic system. When the addition of water was below 0.75 vol%, the native chymotrypsin had no catalytic activity in peptide synthesis. In a sharp contrast, adding 0.25 vol% water into PMMS-CT catalytic system could provide more than 93% yield of Bz-Tyr-Phe-NH2, which might benefit from the hydrophilic crosslinker poly(ethylene glycol) diacrylate preserving a trace amount of water inside the polymeric matrice during the catalyst preparation procedure. The highest outcome achieved at the addition of 1.0 vol% water for both PMMS-CT catalyst and native chymotrypsin. Further increasing the water content although accelerated the reaction process, would reduce the preparation yield of the dipeptide due to the shift in equilibrium to hydrolysis of Bz-Tyr-Phe-NH2. It was interesting to note that in comparison with the native enzyme, PMMS-CT showed a significantly improved tendency in favor of dipeptide synthesis instead of hydrolysis, which might derive from the super-hydrophobicity of the polysiloxane shell blocking the entrance of water molecules to some extent.
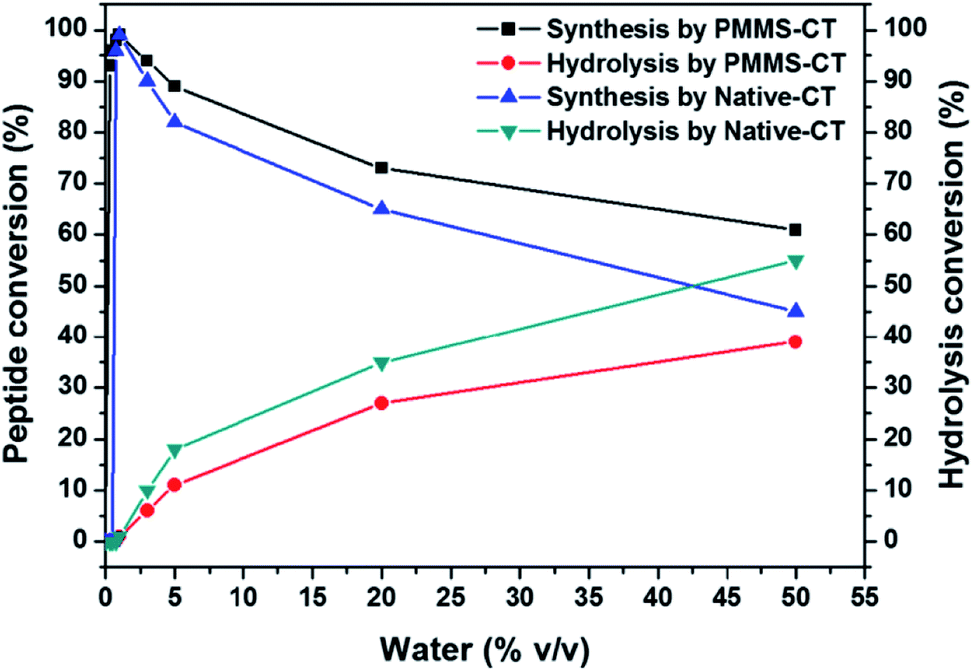 | ||
| Fig. 4 The dipeptide conversion and hydrolysis conversion versus water content diagram showing the impact of water content (vol% = 0%, 0.25%, 0.5%, 0.75%, 1%, 3%, 5%, 10%, 20%, 50%) of 4 mL hexane/tetrahydrofuran (v/v = 1/1) mixture on the enzymatic peptide synthesis using native chymotrypsin or PMMS-CT. The model reaction carried on between L-phenylalaninamide (Phe-NH2) and N-benzoyl-tyrosine ethyl ester (Bz-Tyr-OEt). Reaction conditions: Bz-Tyr-OEt (0.8 mmol), Phe-NH2 (1.6 mmol), PMMS-CT (100 mg) or native chymotrypsin (50 mg), 37 °C, 3 h. | ||
The thermo-stability of PMMS-CT catalyst was investigated by evaluating the dipeptide synthesis efficiency after preheat-treatment of PMMS-CT catalyst in neat conditions at various temperatures for 2 h. As shown in Fig. 5, PMMS-CT apparently had a much better thermal stability than the native chymotrypsin. In a temperature range from ambient temperature to 40 °C (the reaction temperature was 37 °C), both enzymes preserved the most effective catalytic activities and gave negligible hydrolysis byproducts. When heat-treatment temperature was raised beyond 40 °C, the dipeptide synthetic yields catalyzed by PMMS-CT and native chymotrypsin gradually decreased to 89% and 82% respectively at 60 °C while the hydrolysis conversions slightly increased. Above 60 °C, the catalytic activity of native chymotrypsin acceleratingly decreased and eventually reached to zero at 90 °C, whereas PMMS-CT had a higher jumping point at 65 °C and could still provide 34% yield of dipeptide and 1% conversion of the hydrolysis byproduct at even 100 °C. The higher stability of the immobilized chymotrypsin might be caused by the restriction effect of polysiloxane-gel microcavities on the enzyme molecules, which could not only decrease the variations in chymotrypsin conformational structure and the protein unfolding rate, but also prevent the functional groups of chymotrypsin molecules from contacting with each other to diminish autolysis, eventually resulting in the increase of the enzyme stability.47,48
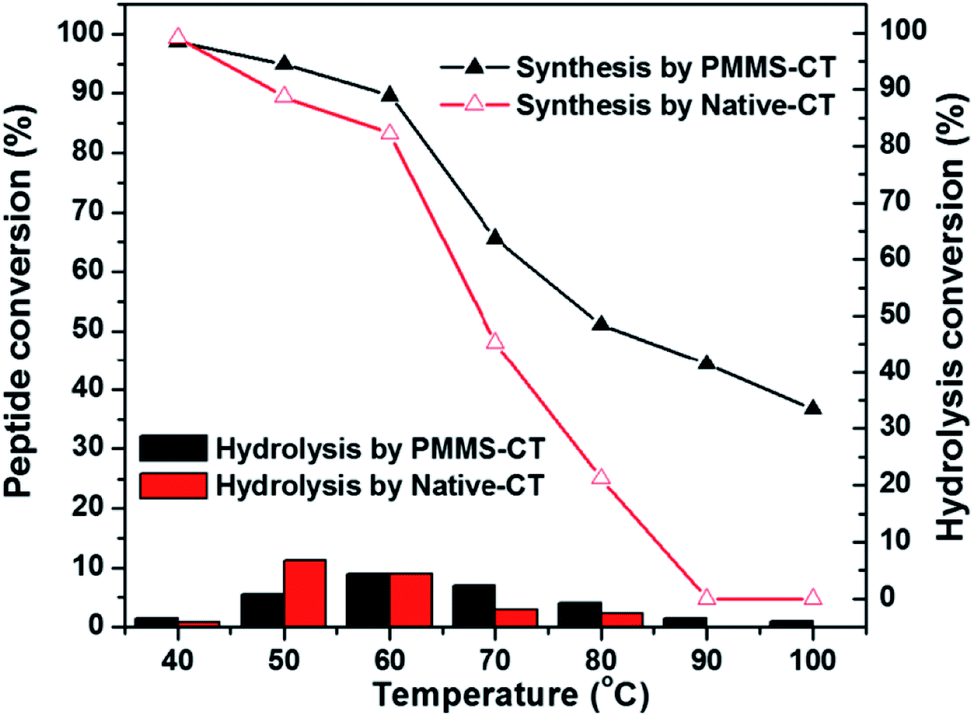 | ||
| Fig. 5 The dipeptide conversion and hydrolysis conversion versus heat-treatment temperature diagram showing the impact of heat-treatment on PMMS-CT catalyzed enzymatic peptide synthesis. The model reaction was performed in 4 mL low-water organic medium (hexane/tetrahydrofuran, v/v = 1/1) containing a trace amount of distilled water (40 μL, 1.0 vol%), Bz-Tyr-OEt (0.8 mmol), Phe-NH2 (1.6 mmol), PMMS-CT (100 mg) or native chymotrypsin (50 mg), 37 °C, 3 h. Both PMMS-CT and native chymotrypsin were heated at 40, 50, 60, 70, 80, 90 or 100 °C for 2 h before catalysing the reaction. | ||
Recyclability is one of the most vital properties of biocatalysts when applying to the synthetic processes and industrial manufacturing.49 Accordingly, we performed the recyclability experiment of PMMS-CT on catalysing the synthesis of Bz-Tyr-Phe-NH2. As shown in Fig. 6, PMMS-CT catalysts have been used 9 times, the enzymatic peptide synthesis efficiency decreased gradually with the increasing cycle number. Moreover, lengthening the reaction time could improve the synthetic yield to some extent. For example, PMMS-CT could achieve about 45% production yield during a period of 8 h in the ninth cycle, whereas a ca. 70% yield of dipeptide Bz-Tyr-Phe-NH2 in the ninth cycle was obtained when the reaction time extending to 24 h. On the contrary, the hydrolysis conversion slightly increased to 6–8% in the first four cycles and then dropped to almost zero in the next five cycles. Another interesting point was that poor recyclability of PMMS-CT catalyst was observed when applied in some organic solvents, including toluene, acetonitrile, and menthol, possibly due to the structural denaturation of enzyme in these solvents, whereas chymotrypsin exhibited good catalytic efficiency and recyclability in hexanes and tetrahedrofuran.50 Nonetheless, these results confirmed that thiol–ene photoimmobilization of enzymes on polysiloxane gels has led to an excellent reusability of chymotrypsin for peptide synthesis and showed a dramatic improvement of reaction yield compared to the native chymotrypsin.
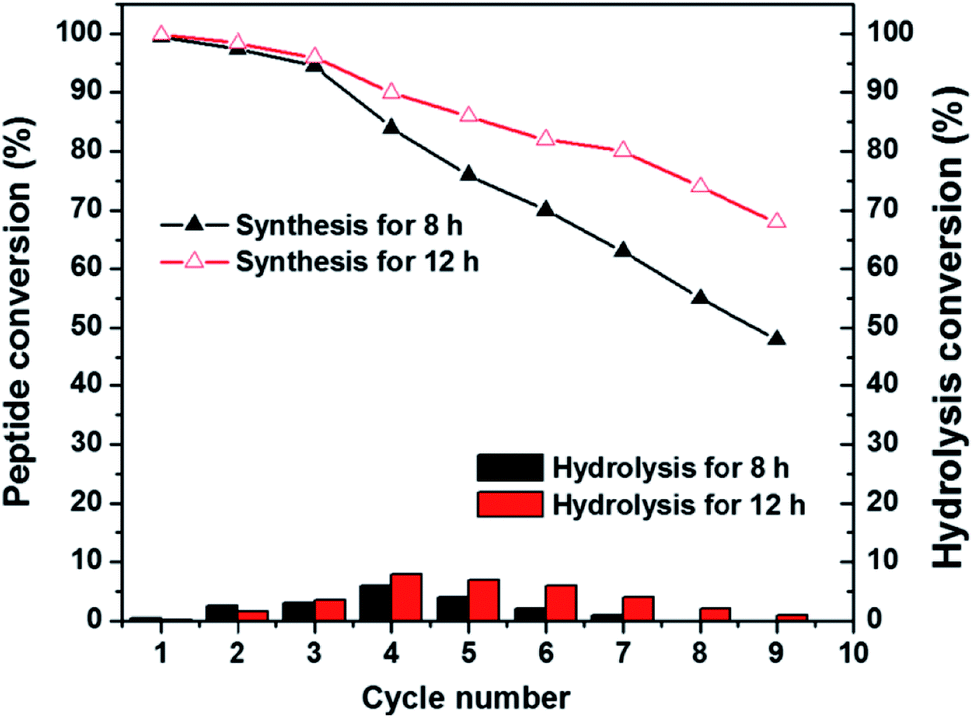 | ||
| Fig. 6 The dipeptide conversion and hydrolysis conversion versus reaction cycle number diagram showing the recyclability of PMMS-CT used in the enzymatic peptide synthesis. The model reaction was performed in 4 mL low-water organic medium (hexane/tetrahydrofuran, v/v = 1/1) containing a trace amount of distilled water (40 μL, 1.0 vol%), Bz-Tyr-OEt (0.8 mmol), Phe-NH2 (1.6 mmol), and PMMS-CT (100 mg) at 37 °C for 8 h and 24 h respectively. | ||
In order to examine the wide applicability of PMMS-CT in peptide synthesis, several dipeptides were synthesized by using PMMS-CT catalyst under the above-mentioned conditions. Since water-soluble chymotrypsin reflected the P1 specificity for hydrolytic reaction,51 high yields of peptide bond formation would be realized when hydrophobic and bulky amino acid amides (Phe-NH2, Tyr-NH2) as nucleophile were used in this study. As shown in Table 1, although hydrolytic reactions of eater donors still existed, the catalytic activities of PMMS-CT for peptide bond formation were satisfactory for providing nearly 80% reaction yields. In addition, PMMS-CT exhibited no catalytic activity when treated with non-aromatic hydrophilic amino acids such as Gly-NH2, Ala-NH2 and Leu-NH2, which indicated that these hydrophilic amino acids in organic solvents could not efficiently traverse hydrophobic polysiloxane matrice to reach the enzyme active sites, so that the reaction yields were not satisfactory. Besides, PMMS-CT still showed no activity in transforming non-aromatic amino acids to peptides even by using hydrophilic organic solvents including DMSO, DMF, acetonitrile, DMSO/hexane, DMF/hexane or acetonitrile/hexane. Apparently, the polysiloxane-gel-bonded biocatalyst was advantageous in catalyzing peptide synthesis when using aromatic amino acids, particularly in the cases of Tyr-Phe, Phe-Phe, Trp-Phe, and Trp-Try, etc.
| Ester donora | Nucleophile | Reaction timeb (h) | Hydrolysis yieldc (%) | Dipeptide yieldc (%) |
|---|---|---|---|---|
| a The reaction was carried out in 4 mL low-water organic medium (hexane/tetrahydrofuran, v/v = 1/1) containing a trace amount of distilled water (40 μL, 1.0 vol%), 0.8 mmol ester donor and 1.6 mmol nucleophile, and PMMS-CT (100 mg) at 37 °C. Ac and Bz represent N-acetyl and N-benzoyl, respectively.b The reaction time was monitored by TLC for ester donor.c The crude products were purified by flash column chromatography and confirmed by 1H NMR analysis. | ||||
| Bz-Tyr-OEt | Phe-NH2 | 2 | 1 | 98 |
| Ac-Trp-OEt | Phe-NH2 | 4 | 11 | 82 |
| Bz-Tyr-OEt | Tyr-NH2 | 6 | 10 | 87 |
| Ac-Phe-OEt | Tyr-NH2 | 4 | 9 | 83 |
| Ac-Trp-OEt | Tyr-NH2 | 6 | 8 | 82 |
| Ac-Phe-OEt | Phe-NH2 | 4 | 7 | 79 |
| Bz-Tyr-OEt | Gly-NH2 | 12 | 99 | 0 |
| Bz-Tyr-OEt | Ala-NH2 | 12 | 99 | 0 |
| Bz-Tyr-OEt | Leu-NH2 | 12 | 99 | 0 |
Conclusions
In summary, chymotrypsin has been successfully immobilized onto cross-linked polysiloxane gels by facile thiol–ene photo-click chemistry. The hydrophobic polysiloxane matrice could prevent water from attacking the acyl-enzyme intermediate, which was beneficial for the shift in equilibrium to peptide formation. The prepared polysiloxane-immobilized chymotrypsin could efficiently catalyze peptide synthesis in low water organic medium. Compared with the native chymotrypsin, polysiloxane-immobilized chymotrypsin showed advantageous catalytic activity, higher thermal stability and superior recyclability. We hope this work will provide a new perspective for enzyme immobilization and enzymatic peptide synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by National Natural Science Foundation of China (21374016), Jiangsu Provincial Natural Science Foundation of China (BK20170024), Priority Academic Program Development of Jiangsu Higher Education Institutions, the Fundamental Research Funds for the Central Universities and the Open Research Fund of State Key Laboratory of Bioelectronics, Southeast University.Notes and references
- E. T. Hwang, B. Lee, M. Zhang, S. H. Jun, J. Shim, J. Lee, J. Kim and M. B. Gu, Green Chem., 2012, 14, 1884–1887 RSC.
- H. E. Schoemaker, D. Mink and M. G. Wubbolts, Science, 2003, 299, 1694–1697 CrossRef CAS PubMed.
- A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–261 CrossRef CAS PubMed.
- K. M. Koeller and C. H. Wong, Nature, 2001, 409, 232–240 CrossRef CAS PubMed.
- V. Nithya, P. S. K. Murthy and P. M. Halami, J. Appl. Microbiol., 2013, 115, 475–483 CrossRef CAS PubMed.
- A. L. Margolin and A. M. Klibanov, J. Am. Chem. Soc., 1987, 109, 3802–3804 CrossRef CAS.
- H. D. Jakubke, P. Kuhl and A. Könnecke, Angew. Chem., Int. Ed., 1985, 24, 85–93 CrossRef.
- H. Gaertner, T. Watanabe, J. V. Sinisterra and A. Puigserver, J. Org. Chem., 1991, 56, 3149–3153 CrossRef CAS.
- T. Miyazawa, S. Nakajo, M. Nishikawa, K. Hamahara, K. Imagawa, E. Ensatsu, R. Yanagihara and T. Yamada, J. Chem. Soc., Perkin Trans. 1, 2000, 32, 82–86 Search PubMed.
- Z. Maugeri, W. Leitner and P. D. D. María, Eur. J. Org. Chem., 2013, 20, 4223–4228 CrossRef.
- R. V. Ulijn, B. Baragaña, P. J. Halling and S. L. Flitsch, J. Am. Chem. Soc., 2002, 124, 10988–10989 CrossRef CAS PubMed.
- J. J. Roy and T. E. Abraham, Chem. Rev., 2004, 104, 3705–3722 CrossRef CAS PubMed.
- A. M. Klibanov, Trends Biotechnol., 1997, 15, 97–100 CrossRef CAS PubMed.
- S. Sharma, P. Kaur, A. Jain, M. R. Rajeswari and M. N. Gupta, Biomacromolecules, 2003, 4, 330–336 CrossRef CAS PubMed.
- V. Stepankova, S. Bidmanova, T. Koudelakova, Z. Prokop, R. Chaloupkova and J. Damborsky, ACS Catal., 2013, 3, 2823–2836 CrossRef CAS.
- L. Wilson, L. Betancor, G. Fernández-Lorente, M. Fuentes, A. Hidalgo and J. M. Guisán, Biomacromolecules, 2004, 5, 814–817 CrossRef CAS PubMed.
- B. D. Malhotra, R. Singhal, A. Chaubey, S. K. Sharma and A. Kumar, Current Applied Physics, 2005, 5, 92–97 CrossRef.
- Y. Wang and F. Caruso, Chem. Mater., 2005, 17, 953–961 CrossRef CAS.
- H. R. Luckarift, J. C. Spain, R. R. Naik and M. O. Stone, Nat. Biotechnol., 2004, 22, 211–213 CrossRef CAS PubMed.
- S. C. Davis, V. C. Sheppard, G. Begum, Y. Cai, Y. Fang, J. D. Berrigan, N. Kröger and K. H. Sandhage, Adv. Funct. Mater., 2013, 23, 4611–4620 CrossRef CAS.
- M. Sevilla, P. Valle-Vigón, P. Tartaj and A. B. Fuertes, Carbon, 2009, 47, 2519–2527 CrossRef CAS.
- F. Lopez-Gallego and L. Yate, Chem. Commun., 2015, 51, 8753–8756 RSC.
- X. Li, X. Wang, G. Ye, W. Xia and X. Wang, Polymer, 2010, 51, 860–867 CrossRef CAS.
- Z. G. Wang, L. S. Wan, Z. M. Liu, X. J. Huang and Z. K. Xu, J. Mol. Catal. B: Enzym., 2009, 56, 189–195 CrossRef CAS.
- Y. Okada, H. Suzuki, T. Nakae, S. Fujita, H. Abe, K. Nagano, T. Yamada, N. Ebata, S. Kim and K. Chiba, J. Org. Chem., 2013, 78, 320–327 CrossRef CAS PubMed.
- S. Iimura, K. Manabe and S. Kobayashi, Org. Lett., 2003, 34, 101–104 CrossRef PubMed.
- F. A. Lage, J. J. Bassi, M. C. Corradini, L. M. Todero, J. H. Luiz and A. A. Mendes, Enzyme Microb. Technol., 2016, 84, 56–67 CrossRef CAS PubMed.
- M. T. Mwangi, M. B. Runge and N. B. Bowden, J. Am. Chem. Soc., 2006, 128, 14434–14435 CrossRef CAS PubMed.
- H. Yang, M. Xu, L. X. Guo, H. F. Ji, J. Y. Wang, B. P. Lin, X. Q. Zhang and Y. Sun, RSC Adv., 2015, 5, 7304–7310 RSC.
- S. Cao and B. Liu, Macromol. Biosci., 2009, 9, 361–368 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- G. Franc and A. K. Kakkar, Chem. Soc. Rev., 2010, 39, 1536–1544 RSC.
- A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC.
- P. Jonkheijm, D. Weinrich, M. Köhn, H. Engelkamp, P. C. M. Christianen, J. Kuhlmann, J. C. Maan, D. Nusse, H. Schroeder, R. Wacker, R. Breinbauer, C. M. Niemeyer and H. Waldmann, Angew. Chem., Int. Ed., 2008, 120, 4421–4424 CrossRef PubMed.
- D. Weinrich, M. Köhn, P. Jonkheijm, U. Westerlind, L. Dehmelt, H. Engelkamp, P. C. M. Christianen, J. Kuhlmann, J. C. Maan, D. Nusse, H. Schrçder, R. Wacker, E. Voges, R. Breinbauer, H. Kunz, C. M. Niemeyer and H. Waldmann, ChemBioChem, 2010, 11, 235–247 CrossRef CAS PubMed.
- M. Buhl, B. Vonhören and B. J. Ravoo, Bioconjugate Chem., 2015, 26, 1017–1020 CrossRef CAS PubMed.
- L. M. Campos, I. Meinel, R. G. Guino, M. Schierhorn, N. Gupta, G. D. Stucky and C. J. Hawker, Adv. Mater., 2008, 20, 3728–3733 CrossRef CAS.
- L. M. Campos, T. T. Truong, D. E. Shim, M. D. Dimitriou, D. Shir, I. Meinel, J. A. Gerbec, H. T. Hahn, J. A. Rogers and C. J. Hawker, Chem. Mater., 2009, 21, 5319–5326 CrossRef CAS.
- L. Wang, W. Liu, L. X. Guo, B. P. Lin, X. Q. Zhang, Y. Sun and H. Yang, Polym. Chem., 2017, 8, 1364–1370 RSC.
- H. Yang, J. J. Liu, Z. F. Wang, L. X. Guo, P. Keller, B. P. Lin, Y. Sun and X. Q. Zhang, Chem. Commun., 2015, 51, 12126–12129 RSC.
- C. Capasso, M. Rizzi, E. Menegatti and P. Ascenzi, J. Mol. Recognit., 1997, 10, 26–35 CrossRef CAS PubMed.
- N. Askinadze, E. Gluz, O. Ziv, D. M. Mizrahi and S. Margel, Polymer, 2013, 54, 2926–2934 CrossRef CAS.
- H. C. Chiu, S. Zalipsky, P. Kopecková and J. Kopecek, Bioconjugate Chem., 1993, 4, 290–295 CrossRef CAS PubMed.
- B. S. Lele, H. Murata, K. Matyjaszewsk and A. J. Russell, Biomacromolecules, 2005, 6, 3380–3387 CrossRef CAS PubMed.
- M. Erbeldinger, X. Ni and P. J. Halling, Enzyme Microb. Technol., 1998, 23, 141–148 CrossRef CAS.
- P. Clapés, L. Espelt, A. N. Ma and C. Solans, J. Chem. Soc., Perkin Trans. 2, 2001, 2, 1394–1399 RSC.
- N. Ortega, M. P. Mateos, M. C. Pilar and M. D. Busto, J. Agric. Food Chem., 2009, 57, 109–115 CrossRef CAS PubMed.
- V. V. Mozhaev and K. Martinek, Adv. Drug Delivery Rev., 1990, 4, 387–419 CrossRef CAS.
- S. W. Lu, F. Khan and J. Micklefield, Chem. Rev., 2009, 109, 4025–4053 CrossRef PubMed.
- V. M. Paradkar and J. S. Dordick, J. Am. Chem. Soc., 1994, 116, 5009–5010 CrossRef CAS.
- K. Morihara and T. Oka, Biochem. J., 1977, 163, 531–534 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and instrumentation, synthetic procedures, NMR spectra and ESI-MS data. See DOI: 10.1039/c7ra13320k |
| This journal is © The Royal Society of Chemistry 2018 |