
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a surface molecular imprinting polymer based on silica and its application in the identification of nitrocellulose
Yan Yang,
Xiangjun Meng and
Zhenggang Xiao *
*
School of Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: xiaozhg@njust.edu.cn
First published on 8th March 2018
Abstract
A surface molecular imprinting polymer (MIP) based on silica (SiO2/MIP) with excellent selective identification properties towards nitrocellulose (NC) was synthesized with methylacrylic acid as a functional monomer and NC as a template molecule, through simple in situ polymerization. The functional groups of SiO2/MIP were studied through Fourier transform infrared spectroscopy. The morphology, crystalline state and thermostability of SiO2/MIP were investigated respectively by scanning electron microscopy, X-ray diffraction and thermogravimetric analysis. Binding capacity and selectivity studies of SiO2/MIP for NC and its analogues were carried out through ultraviolet-visible spectrophotometry. The thermal analysis and study of crystalline states confirmed the successful imprinting of NC in the polymer networks. The optimized conditions were found to be a polymerization temperature of 45 °C and a functional monomer to cross-linking ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. The adsorption capacity of SiO2/MIP was improved considerably compared with that of polymers prepared by traditional imprinting technology, with a maximum adsorption amount of 1.7 mg mg−1 in 2 mg ml−1 NC solution, compared with an adsorption capacity of about 0.5 mg mg−1 for a traditional MIP. According to the selectivity study, more NC was adsorbed by SiO2/MIP than its analogues; the best adsorption capacity of SiO2/MIP for NC was approaching 5 times that for carboxymethyl cellulose (CMC). The results show that it would be possible to apply SiO2/MIP for the detection of NC, to give improved sensitivity in security checking and improved contaminant adsorption.
:3. The adsorption capacity of SiO2/MIP was improved considerably compared with that of polymers prepared by traditional imprinting technology, with a maximum adsorption amount of 1.7 mg mg−1 in 2 mg ml−1 NC solution, compared with an adsorption capacity of about 0.5 mg mg−1 for a traditional MIP. According to the selectivity study, more NC was adsorbed by SiO2/MIP than its analogues; the best adsorption capacity of SiO2/MIP for NC was approaching 5 times that for carboxymethyl cellulose (CMC). The results show that it would be possible to apply SiO2/MIP for the detection of NC, to give improved sensitivity in security checking and improved contaminant adsorption.
1. Introduction
Nitrocellulose (NC), a cellulose derivative, is a type of explosive material, which is the basic component of single and double base propellants.1 It is also known as a versatile, widely used polymer with numerous civil applications, such as an additive in paints and lacquer coatings for automobile and wood furniture.2,3 Recently, it has been applied in the fabrication of specific biosensors, especially in protein microarrays of biochips for its neutral effect on proteins and immobilization of enzymes.4–7 Nevertheless, most of the terrorist attacks occurring worldwide involve the misuse of NC and other explosives. Further, the preparation of paints and lacquer coatings may cause environmental contamination such as water pollution. Therefore, in order to eliminate the occurrence of terrorist attacks and decrease the pollution of water, it is necessary to find valid methods to detect NC.Many methods have been reported for the detection of NC. Gas chromatography (GC) combined with electron ionization (EI) and mass spectrometry (MS) was adopted by Chajistamatiou and Bakeas to identify NC in bulk explosives.8 However, the application of this method is hindered by the voluminous expensive instruments. MacMillan and co-workers used ion chromatography to determine NC in soil.9 They described the removal of impurities, extraction, hydrolysis and neutralization, followed by the analysis of free nitrate and nitrite through ion chromatography. Obviously, the disadvantage of this method is that it is laborious and time-consuming.
Considering the limitations of the above methods, it is necessary to enhance and improve NC detection technology, and more importantly, to develop a simple, highly-selective and widespread detection technology.10
As a highly selective and highly sensitive detection method for a particular analyte, molecularly imprinted technology (MIT)11,12 has received increasing attention in recent years. Numerous papers in relation to MIT have been published, which involve extensive applied fields including biomedicine,13–16 environmental protection, separation of substances17 and so on. In general, the synthesis of a molecular imprinting polymer (MIP)18–21 involves the polymerization of a functional monomer in the presence of a cross-linking agent and template molecules.22 After the polymerization, the template molecules are removed from the polymer. As a result, the recognition sites, that are complementary to the template molecules in function and shape,23,24 are left in the polymer, which gives the MIP its ability to combine with a particular template molecule or its analogues efficiently and selectively.25
In spite of the synthesis of various imprinted materials, their commercial applications have not been put into practice yet. This results from the fact that the three-dimensional cavities in the synthesized MIP are not easily accessible to template molecules, thus leading to a poor binding capacity, slow binding kinetics and difficult template removal. To improve the traditional MIT, alternative approaches have been explored such as surface molecular imprinting,26,27 magnetic molecular imprinting,28,29 core–shell polymerization30 etc. Surface molecularly imprinted technology (SMIT) has been applied extensively, because the generated cavities are located on the surface of the materials, which decreases the resistance to mass transfer thus facilitating the contact of the template molecules with recognition sites in the polymer matrix. As a result, SMIT overcomes the disadvantages of the traditional MIT.
In this paper, through a simple, time-saving and facile in situ polymerization process, a surface imprinting polymer was synthesized to selectively detect NC, while silica (SiO2) was utilized as a core material. Inspired by the literature,31 3-(trimethoxysilyl) propylmethacrylate (MPS) was used in the form of a modifier to functionalize SiO2 powders with an acrylyl group. Thus through hydrogen bond interactions, it was easier for SiO2 powders to combine with NC molecules. Then the effects of the polymerization temperature and proportions of functional monomer and cross-linking agent on the adsorption capacity of NC were investigated. The surface imprinting polymer based on SiO2 (SiO2/MIP) and surface non-imprinting polymer (SiO2/NIP) were characterized by scanning electron microscopy (SEM), Fourier transform infrared spectroscopy (FT-IR), thermogravimetric analysis (TGA) and X-ray diffraction (XRD). By contrasting SiO2/NIP and a traditional molecular imprinting polymer (TMIP), the adsorption capacity of the prepared SiO2/MIP was studied through ultraviolet-visible (UV-vis) spectrophotometry. Selectivity studies were conducted through attaching the SiO2/MIP to NC and related structural analogues. The results showed that, SiO2/MIP could be applied practically to the highly selective and sensitive determination of NC in security checks and environmental protection.
2. Results and discussion
2.1 Preparation and detection principle of SiO2/MIP
Fig. 1 illustrates the preparation process for poly(methylacrylic acid) (PMAA)/inorganic particle hybrid composites with highly selective properties for NC. Allowing for the scarce hydroxy groups on the surface of the purchased SiO2, the core material SiO2 was prepared directly based on the hydrolyzation of tetraethoxysilicane (TEOS), and silicon-hydroxyl condensation in a solution of ethanol and deionized water with ammonium hydroxide as a catalyst. In order to improve the interface properties, the silane coupling agent MPS was used to increase the surface activity of SiO2 and more importantly to functionalize the SiO2 particles with acrylyl groups for the follow-up experiments. Through ultrasonic dispersion of MPS-SiO2 and NC in the presence of methylacrylic acid (MAA) and ethylene glycol dimethacrylate (EGDMA), hydrogen bonds were formed between the ester group in MPS-SiO2 and the hydroxy group in NC. At the same time, NC was orientated in the mixture by the formation of hydrogen bonds between its nitro group and the carboxy group in MAA. After the addition of 2,2-azobisisobutyronitrile (AIBN), a simple in situ polymerization took place by which NC was assembled and immobilized in the polymer network formed by MAA and EGDMA on the surface of the SiO2 particles. Subsequently, a mixture of methanol and acetic acid was used for the removal of the template NC, owing to the stronger hydrogen bonding interactions formed among carboxy groups in acetic acid, hydroxy groups in methanol and NC, in conditions of excess methanol and acetic acid. The NC was distributed on the surface of the SiO2 owing to its pre-combination with MPS-SiO2, thus benefitting the final removal approach using methanol and acetic acid. After the removal of the template NC, cavities were left on the surface of SiO2, which retained the shape, size and functional groups of NC, thus the resultant SiO2/MIP was able to selectively identify the template NC. | ||
| Fig. 1 Schematic diagram of the imprinting process of nitrocellulose on the surface of SiO2. | ||
TMIP was prepared without the addition of SiO2; hydrogen bonds existed only between the hydroxy groups and nitro groups in NC and carboxy groups in MAA. In consequence, the template NC was stuck in the three-dimensional polymer networks formed after the polymerization process. When methanol and acetic acid were used to try to remove the template NC, access to the chasmal template NC was restricted which resulted in template residue in the polymer. As a result, the adsorption capacity of TMIP was poor in the rebinding test.
2.2 FT-IR analysis
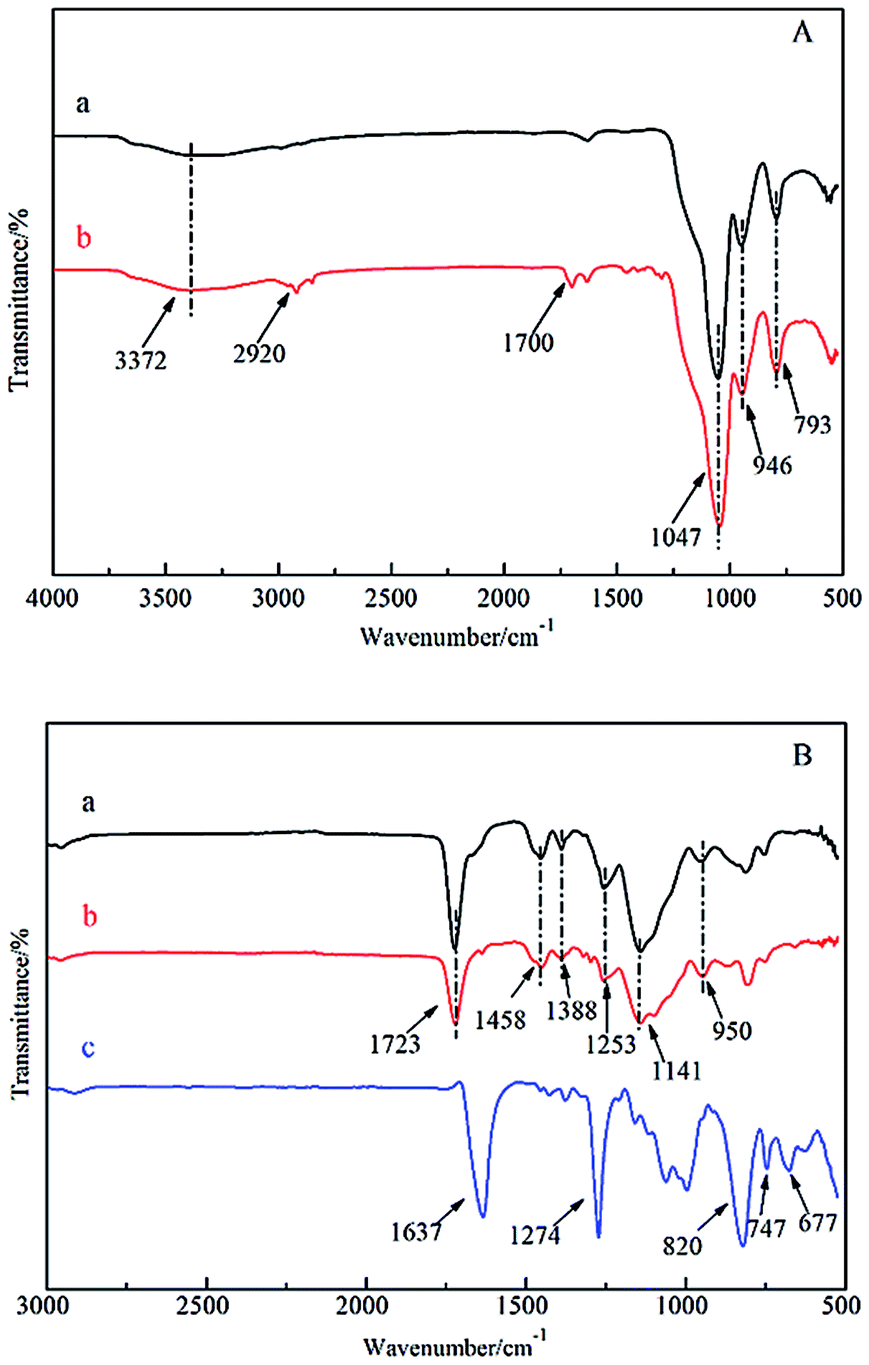
Fig. 2 shows the FT-IR spectra of the prepared surface imprinting polymer based on SiO2 particles. Fig. 2(A) shows the FT-IR measurements of (a) SiO2 and (b) MPS-SiO2. The characteristic absorption peaks at 3372 cm−1 and 946 cm−1 belong to the stretching vibration and bending vibration of Si–OH groups on the surface of SiO2.32 The peaks located at 1047 cm−1 and 793 cm−1 are assigned to Si–O–Si stretching vibrations. The peaks observed at 2920 cm−1 and 1700 cm−1 belong to the stretching vibration of –CH3 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in MPS, which indicates that the SiO2 particles were successfully modified by MPS through a condensation reaction between hydroxy groups on the surface of SiO2 particles and methoxy groups in MPS.
O in MPS, which indicates that the SiO2 particles were successfully modified by MPS through a condensation reaction between hydroxy groups on the surface of SiO2 particles and methoxy groups in MPS.
 | ||
| Fig. 2 FT-IR spectra of the prepared surface imprinting polymers based on silica particles: (A) (a) SiO2, (b) MPS-SiO2, and (B) (a) SiO2/MIP, (b) SiO2/NIP, (c) NC. | ||
Fig. 2(B) shows the FT-IR spectra of SiO2/MIP, SiO2/NIP and NC. From the spectra, it can be seen that the characteristic peaks of SiO2/MIP and SiO2/NIP are highly consistent. The peak at 1723 cm−1 belongs to the stretching vibration of CO in MAA and EGDMA. Peaks at 1458 cm−1, 1388 cm−1 and 1141 cm−1 are assigned to the bending vibrations of C–H in –CH3 and –CH2. Peaks at 1253 cm−1 and 950 cm−1 are attributed to the C–O stretching vibration and –OH in-plane rocking vibration in the carboxy group, respectively. In the spectrum of NC, the strong absorption peaks located at 1637 cm−1 and 1274 cm−1 are assigned to the –NO2 stretching vibration, whereas peaks at 820 cm−1, 747 cm−1 and 677 cm−1 belong to the –NO2 bending vibration.
By contrasting the spectrum of NC, it is clear that there are no remarkable absorption peaks of NC observed in the spectrum of SiO2/MIP, which indicates the thorough removal of the template NC in the process of SiO2/MIP synthesis. This is because the imprinting sites in the cavities in SiO2/MIP are located on the surface of the SiO2 particles, leading to easier access to the NC for the mixture of methanol and acetic acid. On the other hand, the imprinting sites in TMIP are embedded into the three-dimensional polymer network, which results in the difficulty for methanol and acetic acid to bind the NC molecules.
2.3 Morphological analysis
The morphologies of the synthesized SiO2 were studied by SEM as presented in Fig. 3. From Fig. 3(a) and (b), the synthesized SiO2 particles exhibit a spherical shape and smooth surface, while the diameter of the SiO2 particles ranges from 280 nm to 585 nm. It is easy to see that the SiO2 particles are stacked and agglomerated together. This behavior might be due to the high surface free energy of SiO2 particles of small granularity.33 | ||
| Fig. 3 SEM images of the synthesized SiO2 particles: magnification (a) 20000×, (b) 40000×. | ||
Fig. 4 depicts the SEM images of TMIP (a and b), TNIP (c and d), SiO2/MIP (e and f) and SiO2/NIP (g and h). As shown in Fig. 4(a), the polymers in the traditional MIT sample exhibit cross-linked networks in which many cavities are observed, while TNIP is observed to form many large agglomerates. With higher magnification, the TMIP is shown to be made up of irregular spherules with many interstices (Fig. 4(b)), while the surface of TNIP is close-grained and fluctuant (Fig. 4(d)). A polyporous polymer matrix is observed in Fig. 4(e); SiO2 particles appear embedded in the polymer, and some SiO2 particles are on the surface of the polymer (Fig. 4(f)). In Fig. 4(g), many independent blocks are dispersed, and SiO2 particles are submerged into the polymer or on the surface of the polymer (Fig. 4(h)). The SEM images of embedded SiO2 particles in Fig. 4 indicate that the molecular imprinting process of SiO2/MIP takes place on the surface of the SiO2 particles. At the same time, some SiO2 particles are not embedded in the polymer, which might due to the easy agglomeration of SiO2 particles with small granularity. The carbonyl groups and hydroxyl on the surface of MPS-SiO2 are occupied by other SiO2 particles in the process of agglomeration. As a result, this proportion of the SiO2 particles cannot contact the groups in MAA and EGDMA sufficiently. Thus in the polymerization process, it is difficult for agglomerated SiO2 particles to be embedded in the polymer.
 | ||
| Fig. 4 SEM images of TMIP (a and b), TNIP (c and d), SiO2/MIP (e and f), SiO2/NIP (g and h). Magnification (a) 10000×, (b, d, f and h) 40000×, (c) 5000 ×, (e and g) 20000×. | ||
With the reported imprinting process using template NC in the synthesis of MIP, the imprinted sites complementary with NC are left in the MIP once the template is removed, leading to the polyporous structures of TMIP and SiO2/MIP. On the other hand, without the NC imprinting, there are no cavities and imprinted sites in TNIP and SiO2/NIP, which explains the agglomerates and blocks observed in the SEM images of TNIP and SiO2/NIP.
2.4 XRD characterization
Fig. 5 shows the X-ray diffractograms of TMIP, TNIP, SiO2/MIP, SiO2/NIP and SiO2. In the X-ray diffractogram of SiO2 particles, the wide hump in the range of 15° to 30° is typical for amorphous silica.34 The SiO2/MIP does not have diffraction peaks, while peaks in SiO2/NIP, TMIP and TNIP can be seen at an angle of 15°. Meanwhile, the intensity of the peaks decreases progressively according to the sequence of TNIP > SiO2/NIP > TMIP. It can be concluded that the crystallinity of NIP is higher than that of MIP, and with imprinted sites on the surface of SiO2, SiO2/MIP is more amorphous than TMIP. | ||
| Fig. 5 X-ray diffractograms of TMIP, TNIP, SiO2/MIP, SiO2/NIP and SiO2. | ||
The results suggest that the amorphous states of MIP could result from imprinting by NC. In other words, compared with pure PMAA, the imprinting process decreases the regularity of the crystalline region in order to make the template molecule orient in the polymer networks. When the polymers are imprinted with SiO2 as the core material, the amorphous degree of the polymers is at its maximum. This is due to the fact that the polymerization process is severely disorganized when affected by the hydrogen bond interactions among MPS-SiO2, MAA and NC, which makes the polymerization process deviate greatly from the regular pathway of chain growth of a crystalline polymer, thus leading to the formation of amorphous polymers.
2.5 Thermal properties
The TGA curves of TMIP, TNIP, SiO2/MIP and SiO2/NIP in Fig. 6 show scans from 50 °C to 600 °C in an atmosphere of nitrogen. The thermal degradation process of TMIP and SiO2/MIP consists of four stages. The weight loss of TMIP and SiO2/MIP at 200 °C is less than 4%, but it increases steeply between 200 °C and 270 °C. This is followed by a sluggish rise from 270 °C to 457 °C and then the curves reach a plateau. In contrast, TNIP and SiO2/NIP go through three stages with the increase of temperature: TNIP and SiO2/NIP have a weight loss of nearly 6% at 200 °C and show a straightforward increase in weight loss from 330 °C with a final constant weight at 457 °C.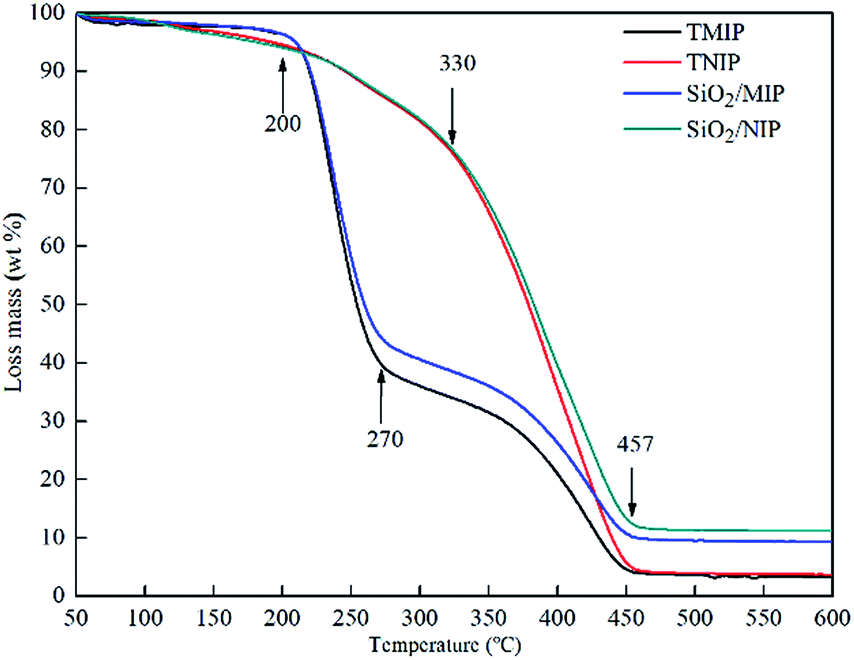 | ||
| Fig. 6 TGA traces showing the thermal decomposition of TMIP, TNIP, SiO2/MIP and SiO2/NIP in a nitrogen atmosphere. | ||
The 4% weight loss of SiO2/MIP (and TMIP) and 6% weight loss of SiO2/NIP (and TNIP) at 200 °C result from adsorbed water. The 50% weight loss of SiO2/MIP (56% for TMIP) from 200 °C to 270 °C is attributed to the formation of six-membered cyclic intramolecular anhydrides.35 The sluggish 30% weight loss of SiO2/MIP (36% for TMIP) from 270 °C to 457 °C derives from degradation to CO2 and H2O. The weight loss of SiO2/NIP (TNIP) from 330 °C is due to the degradation of PMAA.
The residual weights of TMIP, TNIP, SiO2/MIP and SiO2/NIP are presented in Table 1.
| Polymer | Residual weight (%) |
|---|---|
| TMIP | 3.8 |
| TNIP | 4.1 |
| SiO2/MIP | 9.7 |
| SiO2/NIP | 11.5 |
The results show that SiO2/NIP (TNIP) is more stable than SiO2/MIP (TMIP), and that SiO2/MIP is more stable than TMIP in the same way. This is consistent with the morphology observations of the traditional and surface imprinting polymers. SiO2/NIP is made up of many bulk agglomerates, therefore, it is more difficult for SiO2/NIP to transfer the heat when the external temperature is rising, compared with the polyporous polymer networks of SiO2/MIP, which have lower resistance to heat transmission. It ultimately depends on whether the polymers are imprinted by a template molecule or not. At the same time, SiO2/MIP is more stable than TMIP, which confirms the successful imprinting on the surface of SiO2 by NC, and explains the excellent stability of SiO2 in the trial range of 50–600 °C.
2.6 Adsorption capacity
It is essential to study the adsorption capacity of SiO2/MIP and SiO2/NIP for the template NC; this is a critical parameter which can reflect the ability of a material to detect a particular substance. To assess whether surface molecular imprinting technology leads to an excellent adsorption capacity for the template molecule, TMIP and TNIP were compared with SiO2/MIP and SiO2/NIP. Fig. 7 explicitly presents the adsorption capacities (Q values) of SiO2/MIP, SiO2/NIP, TMIP and TNIP for NC. From the adsorption graph (Fig. 7), it is clear that the Q value of SiO2/MIP is far above (approximately 21 times) that of SiO2/NIP. Furthermore, the Q value of TMIP is lower than that of SiO2/MIP. The Q value of SiO2/MIP is about 3.5 times the Q value of TMIP. The results show that SiO2/MIP has an excellent adsorption performance for NC, with a maximum adsorption amount of 1.7 mg mg−1 when the concentration of NC is 2 mg ml−1, and through the surface imprinting process, the adsorption capacity is improved effectively.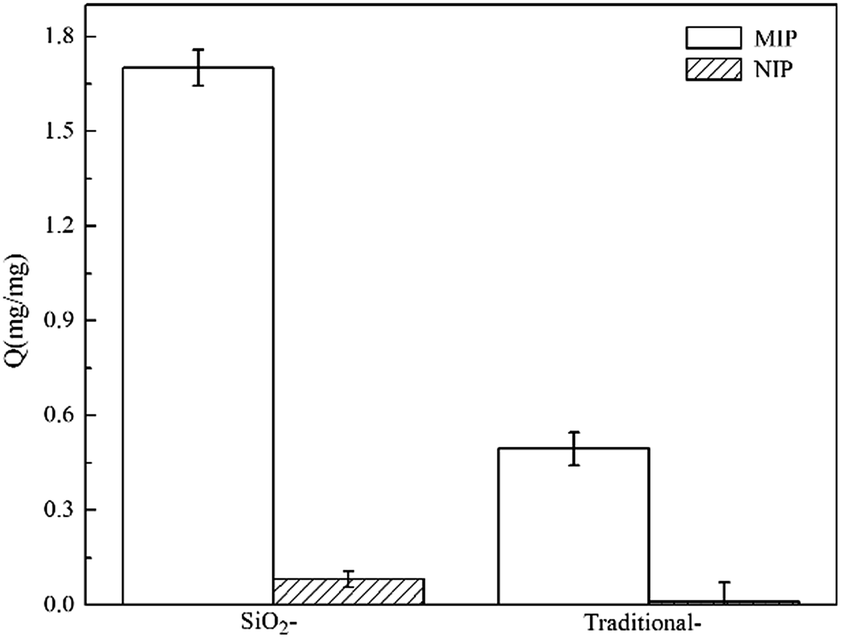 | ||
| Fig. 7 Adsorption graph of SiO2/MIP, SiO2/NIP, TMIP and TNIP. | ||
2.7 Optimization of the polymerization conditions
In order to identify appropriate conditions for the synthesis of SiO2/MIP, the temperature and the proportions of functional monomer and cross-linking agent were studied in this paper. The component proportions and different temperature conditions used in this work are given in Table 2. The adsorption capacities of SiO2/MIP and SiO2/NIP prepared using the different polymerization temperatures and proportions are displayed in Fig. 8.| Variate | NC (g) | MAA (mmol) | EGDMA (mmol) | MPS-SiO2 (g) | AIBN (g) | Acetone (ml) | T (°C) |
|---|---|---|---|---|---|---|---|
| T | 0.1 | 3 | 9 | 0.1 | 0.1642 | 30 | 45 |
| 50 | |||||||
| 55 | |||||||
| Proportion | 0.1 | 3 | 9 | 0.1 | 0.1642 | 30 | 50 |
| 12 | |||||||
| 15 |
 | ||
| Fig. 8 Influence of polymerization temperature (A) and the ratio of monomer to cross-linker (B) for SiO2/MIP and SiO2/NIP on the adsorption of NC. | ||
The polymerization temperature is important for the formation of a polymer network. With a lower external temperature, the polymerization process is difficult to initiate. However, when the temperature is too high, the polymerization advances very quickly, even developing into an implosion. The proportions of the functional monomer and cross-linking agent are another important parameter. A lower amount of cross-linking agent is beneficial for increasing the cross-linking index of the final polymers and speeding up the process of polymerization. However, excessive cross-linking agent can result in the formation of a rigid polymer, which is detrimental to the adsorption capacity for NC.
In consideration of the explosion hazard of NC at higher temperatures and the reactive ability of MAA at low temperatures, the temperatures of 45 °C, 50 °C and 55 °C were chosen for polymerization. As shown in Fig. 8(A), when the polymerization temperature was controlled at 45 °C, the resulting SiO2/MIP realized the best adsorption performance. With a temperature of 45 °C, the Q value of SiO2/MIP was 20.9 times that of SiO2/NIP, 1.2 times that of SiO2/MIP prepared at 50 °C, and 3.7 times that of SiO2/MIP prepared at 55 °C. This indicates that the polymerization is exactly initiated at the temperature of 45 °C. The decomposition of the initiator is an endothermal reaction, and the decomposition rate of AIBN is slow. When the temperature is increased, more free radicals can be produced by AIBN. Plenty of free radicals attack the monomers and accelerate the process of chain propagation. Chain propagation is an exothermic process. So at higher temperatures, the reaction balance deviates to the depolymerization processes, which results in the formation of a polymer with a low polymerization degree. The polymer networks of SiO2/MIP with a low polymerization degree are loose and cannot immobilize NC molecules sufficiently. However, the SiO2/MIP prepared at 45 °C has the appropriate cross-linking network density to fix more NC molecules, and is not too rigid to hinder the access of NC to the imprinted sites during the recombination process. The results show that increasing the temperature is favorable for the polymerization process, but it is adverse to the adsorption of NC.
From the adsorption capacities graph of SiO2/MIP with different proportions of functional monomer and cross-linking agent (Fig. 8(B)), the SiO2/MIP prepared with the ratio of 1:3 has the best adsorption capacity and specificity, while with the ratio of 1:4, the specificity of SiO2/MIP is inferior. The adsorption capacity of SiO2/MIP for NC is 8.3 times and 857 times that of SiO2/NIP, when prepared with ratios of 1:3 and 1:5, respectively, while the adsorption capacity of SiO2/MIP is 1.6 times that of SiO2/NIP when prepared with a ratio of 1:4. It is concluded that the maximum adsorption capacities can be reached when the temperature is 45 °C and the ratio of functional monomer to cross-linking agent is 1:3.
2.8 Selectivity of SiO2/MIP
The structures of NC and its analogues are shown in Fig. 9. NC, and its analogues methyl cellulose (MC), carboxymethyl cellulose (CMC) and hydroxypropyl cellulose (HPC), are derivatives of cellulose, which are formed through substitution reactions of hydroxyl groups with nitric ester, methoxyl, carboxymethyl and hydroxypropyl groups, respectively. The SiO2/MIP and SiO2/NIP were prepared with a polymerization temperature of 45 °C and a ratio of monomer to cross-linking agent of 1:3. The adsorption capacities of SiO2/MIP and SiO2/NIP particles for the three analogues of NC in aqueous solutions are shown in Fig. 10. Meanwhile, the corresponding UV-vis spectra and standard curves are depicted in Fig. 11. In comparison with SiO2/NIP, it is obvious that SiO2/MIP possesses higher adsorption capacities for the analogues of NC. This results from the fact that the imprinting cavities and bigger surface area inside the SiO2/MIP make it easier for it to combine with analogues of NC, while no imprinting sites are available inside the SiO2/NIP. However, the adsorption capacities of SiO2/MIP towards MC, CMC and HPC are very weak. The adsorption capacity of SiO2/MIP for NC is approaching 5 times more than that for CMC, which can be ascribed to the unsuitable imprinting cavities inside SiO2/MIP for MC, CMC and HPC in terms of shape, size, and functional distribution. The results confirm the particular selectivity of SiO2/MIP for NC.
 | ||
| Fig. 9 Structures of NC (A) and its analogues: MC (B), CMC (C) and HPC (D). | ||
 | ||
| Fig. 10 Binding amounts of different cellulose derivatives. | ||
 | ||
| Fig. 11 UV-vis spectra of NC (A) and its analogues: MC (B), CMC (C), and HPC (D). The insets show the standard curves for each substance. The solvent for NC in this work was acetone; deionized water was used as the solvent for the other analogues, without any exception. | ||
3. Conclusions
This paper introduces a novel detection method for NC using surface molecularly imprinted technology. The synthesized SiO2/MIP consists of a polyporous and amorphous polymer matrix on the surface of SiO2 particles. Through FT-IR analysis, the removal of NC is shown to be thorough; there is no residual NC in imprinted materials. The thermal analysis and study of crystalline states confirm the successful imprinting of NC in the polymers. The prepared SiO2/MIP exhibits a high adsorption capacity and an excellent selectivity for NC, which opens a new pathway for the detection of NC. This method could also be applied to the detection of other flammable explosives. The SiO2/MIP material could be further used in security checks and contaminant adsorption.4. Experimental
4.1 Reagents and materials
Nitrocellulose (NC, A grade) was provided by North Sichuan Nitrocellulose Co., China. 2,2-Azobisisobutyronitrile (AIBN, 98.5%), 3-(trimethoxysilyl) propylmethacrylate (MPS, 97%), ethanol (99.7%), acetic acid (99.5%) and deionized water were purchased from Nanjing Chemical Reagent Co., Nanjing, China. Methylacrylic acid (MAA, 98%), tetraethoxysilicane (TEOS), acetone (99.7%) and methanol (99.5%) were obtained from Sinopharm Chemical Reagent Co., Nanjing, China. Ammonium hydroxide was provided by Shanghai Lingfeng Chemical Reagent Co., China. Ethylene glycol dimethacrylate (EGDMA, 98%) was purchased from Aladdin Reagent Co., Shanghai, China. Methyl cellulose (MC, 15 mPa s) was purchased from Meryer Chemical Technology Co., Shanghai, China. Carboxymethyl cellulose (CMC, 600–1000 mPa s, USP grade) and hydroxypropyl cellulose (HPC, M. W.100000) were purchased from Macklin Biochemical Co., Shanghai, China. AIBN was recrystallized from ethanol before use, and all other chemicals were used directly without purification.
4.2 Surface pre-treatment of SiO2 particles
SiO2 particles were prepared according to the previously reported method.23 Then MPS was used to improve the affinity of the SiO2 particles for NC. Briefly, SiO2 particles (0.5 g), deionized water (5 ml), aqueous ammonia (6 ml), and MPS (3 ml) were ultrasonically dispersed in ethanol (50 ml) for 1 h.36 Then the mixture was transferred into a 150 ml round-bottom flask and stirred at 75 °C in a dry nitrogen atmosphere for 4 h. After the reaction, the products were centrifuged and washed with ethanol three times to remove the unreacted MPS. Finally, the resultant MPS-SiO2 particles were obtained after a vacuum drying procedure.4.3 Preparation of SiO2/MIP
SiO2/MIP particles were prepared through in situ polymerization in which MAA was used as a functional monomer and EGDMA was used as a cross-linking agent. Briefly, NC (0.1 g) was dissolved in acetone (30 ml) and then MAA (3 mmol), EGDMA (9 mmol), and MPS-SiO2 particles (0.1 g) were dispersed in the NC solution by ultrasonication for 10 min. Subsequently, AIBN (0.1642 g) was added and the above solution was transferred to a 150 ml round-bottom flask to react at 50 °C with continuous stirring in a dry nitrogen atmosphere for about 4 h. Finally, the polymers were added to methanol/acetic acid (4:1, v/v) solution with ultrasonication for 2 h to remove the NC. The resultant SiO2/MIP particles were obtained by vacuum drying after washing with ethanol and deionized water and centrifuging three times to remove residual methanol and acetic acid. As a control, the corresponding SiO2/NIP was synthesized using an identical method but without the addition of NC.
4.4 Preparation of TMIP
TMIP particles were synthesized for comparison; the preparation procedure was as follows: NC (0.1 g) was dissolved in acetone (30 ml). MAA (3 mmol) and EGDMA (9 mmol) were added to the solution above following ultrasonication for 10 min. Subsequently, AIBN (0.1642 g) was added, the mixture was transferred to a 150 ml round-bottom flask and the reaction was processed at 50 °C with continuous stirring in a dry nitrogen atmosphere for about 4 h. After the final removal of NC and vacuum drying, the resultant TMIP particles were obtained.The TNIP particles were synthesized using the same method but in the absence of the NC template.
4.5 Adsorption capacities of SiO2/MIP and SiO2/NIP
Adsorption capacities were studied using the following procedures: 10 mg SiO2/MIP (SiO2/NIP, TMIP or TNIP) were added into a 10 ml 2 mg ml−1 NC solution. After ultrasonication for 2 h, the supernatant liquid was collected through centrifugation. The concentration of supernatant liquid was measured by UV-vis spectrophotometry. The NC amount bound by SiO2/MIP was calculated by the following equation:| Q = (c0 − ce)V/m | (1) |
4.6 Effects of polymerization conditions on adsorption capacity
To evaluate the effects of polymerization temperature and the proportions of functional monomer and cross-linking agent on the adsorption capacity of SiO2/MIP, the polymerization process was conducted at different temperatures and monomer to cross-linking ratios.Temperature: NC (0.1 g) was dissolved in acetone (30 ml) and then MAA (3 mmol), EGDMA (9 mmol), and MPS-SiO2 particles (0.1 g) were dispersed in the NC solution by ultrasonication for 10 min. Subsequently, AIBN (0.1642 g) was added, the mixture was transferred to a 150 ml round-bottom flask and the reaction was processed at either 45 °C or 55 °C with continuous stirring in a dry nitrogen atmosphere for 4 h. After the final removal of NC and vacuum drying, the resultant MIP particles were obtained. The amount of bound NC was calculated according to eqn (1).
Proportions: NC (0.1 g) was dissolved in acetone (30 ml) and then MAA (3 mmol), EGDMA (12 mmol or 15 mmol), and MPS-SiO2 particles (0.1 g) were dispersed in NC solution by ultrasonication for 10 min. Subsequently, AIBN (0.1642 g) was added, the mixture was transferred to a 150 ml round-bottom flask and the reaction was processed at 50 °C with continuous stirring in a dry nitrogen atmosphere for 4 h. After the final removal of NC and vacuum drying, the resultant MIP particles were obtained. The amount of bound NC was calculated according to eqn (1).
4.7 Selectivity performance of SiO2/MIP and SiO2/NIP
20 mg MC (CMC or HPC) were dissolved into 10 ml deionized water, after the addition of 10 mg SiO2/MIP (or SiO2/NIP). After ultrasonication for 2 h, the supernatant liquid was obtained by centrifugation. The concentration of MC (CMC or HPC) in the supernatant liquid was measured by UV-vis spectrophotometry. The amount of bound MC (CMC or HPC) was calculated according to eqn (1).4.8 Characterization
The functional groups of pure SiO2, MPS-SiO2, SiO2/MIP, SiO2/NIP and NC were confirmed by Fourier transform infrared (FT-IR, Nicolet iS10, America) spectroscopy in the range of 4000–500 cm−1. The morphological structures of SiO2/MIP particles were obtained from scanning electron microscopy (SEM, FEG-250, Quanta, America, operating at 40 kV). The crystalline states of SiO2 and SiO2/MIP were determined through X-ray diffraction (XRD, D8 Advance, Germany) with conditions as follows: Cu Kα radiation (λ = 1.5406), 40 kV, 40 mA, 2θ scanning range of 10–90°, step size of 0.05° and step time of 19.2 s. The thermogravimetric analysis (TGA) was performed on a SDTQ600 thermal analyzer (PerkinElmer Pyris Diamond, America) under a nitrogen atmosphere at a heating rate of 10 °C min−1. The adsorption capacity of SiO2/MIP particles was analyzed by ultraviolet-visible (UV-vis) spectrophotometry (Evolution 220, America).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).References
- W. Tomaszewski, K. Cieślak and A. Zygmunt, Polym. Degrad. Stab., 2015, 111, 169–175 CrossRef CAS.
- X. Su, Q. Zhao, D. Zhang and W. Dong, Appl. Surf. Sci., 2015, 356, 610–614 CrossRef CAS.
- Z. Xiao, S. Ying, W. He, F. Xu and P. Sun, J. Appl. Polym. Sci., 2007, 105, 510–514 CrossRef CAS.
- L.-T. Yin, Y.-T. Lin, Y.-C. Leu and C.-Y. Hu, Sens. Actuators, B, 2010, 148, 207–213 CrossRef CAS.
- M. Cretich, V. Sedini, F. Damin, M. Pelliccia, L. Sola and M. Chiari, Anal. Biochem., 2010, 397, 84–88 CrossRef CAS PubMed.
- L. Hecht, D. van Rossum and A. Dietzel, Microelectron. Eng., 2016, 158, 52–58 CrossRef CAS.
- S. Ma, G. Song and N. Feng, Carbohydr. Polym., 2012, 89, 36–40 CrossRef CAS PubMed.
- A. S. Chajistamatiou and E. B. Bakeas, Talanta, 2016, 151, 192–201 CrossRef CAS PubMed.
- D. K. MacMillan, C. R. Majerus, R. D. Laubscher and J. P. Shannon, Talanta, 2008, 74, 1026–1031 CrossRef CAS PubMed.
- H. Rao, M. Chen, H. Ge, Z. Lu, X. Liu, P. Zou, X. Wang, H. He, X. Zeng and Y. Wang, Biosens. Bioelectron., 2017, 87, 1029–1035 CrossRef CAS PubMed.
- M. Huang, T. Zhou, X. Wu and J. Mao, Chin. J. Chem. Eng., 2015, 23, 1698–1704 CrossRef CAS.
- Z. F. Cai, H. J. Dai, S. H. Si and F. L. Ren, Appl. Surf. Sci., 2008, 254, 4457–4461 CrossRef CAS.
- G. Erturk and B. Mattiasson, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1021, 30–44 CrossRef CAS PubMed.
- M. Perez, R. Concu, M. Ornelas, M. N. Cordeiro, M. Azenha and A. F. Silva, Spectrochim. Acta, Part A, 2016, 153, 661–668 CrossRef CAS PubMed.
- J. Li, S. M. Li and Z. G. He, Mater. Sci. Eng., C, 2017, 71, 999–1003 CrossRef CAS PubMed.
- A. Sorribes-Soriano, F. A. Esteve-Turrillas, S. Armenta, M. de la Guardia and J. M. Herrero-Martinez, J. Chromatogr. A, 2017, 1481, 23–30 CrossRef CAS PubMed.
- Y. Zhang, J. Wan and X. Cao, Process Biochem., 2016, 51, 517–527 CrossRef CAS.
- C. K. M. Faizal and T. Kobayashi, Polym. Eng. Sci., 2008, 48, 1085–1093 CAS.
- B. R. Hart and K. J. Shea, Macromolecules, 2002, 35, 6192–6201 CrossRef CAS.
- D. Oliveira, C. P. Gomes, R. C. S. Dias and M. R. P. F. N. Costa, React. Funct. Polym., 2016, 107, 35–45 CrossRef CAS.
- C. Baggiani, C. Giovannoli, L. Anfossi, C. Passini, P. Baravalle and G. Giraudi, J. Am. Chem. Soc., 2012, 134, 1513–1518 CrossRef CAS PubMed.
- M. Torkashvand, M. B. Gholivand and G. Malekzadeh, Sens. Actuators, B, 2016, 231, 759–767 CrossRef CAS.
- R. Gao, J. Zhang, X. He, L. Chen and Y. Zhang, Anal. Bioanal. Chem., 2010, 398, 451–461 CrossRef CAS PubMed.
- A. M. Shrivastav, S. P. Usha and B. D. Gupta, Biosens. Bioelectron., 2016, 79, 150–157 CrossRef CAS PubMed.
- W. Tan, Q. Yu, X. Ruan and X. Huang, Sens. Actuators, B, 2015, 212, 47–54 CrossRef CAS.
- H. Duan, X. Wang, Y. Wang, J. Li, Y. Sun and C. Luo, Sens. Actuators, B, 2016, 236, 44–51 CrossRef CAS.
- Y. Liu, B. Cao, P. Jia, J. An, C. Luo, L. Ma, J. Chang and K. Pan, J. Phys. Chem. A, 2015, 119, 6661–6667 CrossRef CAS PubMed.
- B. Yuan, X. Q. Yang, L. W. Xue, Y. N. Feng and J. H. Jiang, Bioresour. Technol., 2016, 222, 14–23 CrossRef CAS PubMed.
- H. Duan, X. Wang, Y. Wang, Y. Sun, J. Li and C. Luo, Anal. Chim. Acta, 2016, 918, 89–96 CrossRef CAS PubMed.
- Z. Wang and X. Cao, Process Biochem., 2015, 50, 1136–1145 CrossRef CAS.
- C. G. Xie, B. H. Liu, Z. Y. Wang, D. M. Gao, G. J. Guan and Z. P. Zhang, Anal. Chem., 2008, 80, 437–443 CrossRef CAS PubMed.
- N. Sun, X. Meng and Z. Xiao, Ceram. Int., 2015, 41, 13830–13835 CrossRef CAS.
- Z. Xiao, P. Guo and N. Sun, J. Appl. Polym. Sci., 2017, 134, 44377 Search PubMed.
- N. Bogdanchikova, A. Pestryakov, M. H. Farias, J. A. Diaz, M. Avalos and J. Navarrete, Solid State Sci., 2008, 10, 908–914 CrossRef CAS.
- A. Asano and T. Kurotu, Polym. Degrad. Stab., 2002, 78, 137–141 CrossRef CAS.
- X. Wang, J. Yu, Q. Kang, D. Shen, J. Li and L. Chen, Biosens. Bioelectron., 2016, 77, 624–630 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |