
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew bioactive cyclopeptide alkaloids with rare terminal unit from the root bark of Ziziphus cambodiana†
Natthakaln Lomchoey a,
Panomwan Panseetaab,
Pornthip Boonsria,
Nuttapon Apiratikula,
Samran Prabpaic,
Palangpon Kongsaereec and
Sunit Suksamrarn*a
a,
Panomwan Panseetaab,
Pornthip Boonsria,
Nuttapon Apiratikula,
Samran Prabpaic,
Palangpon Kongsaereec and
Sunit Suksamrarn*a
aDepartment of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Srinakharinwirot University, Bangkok 10110, Thailand. E-mail: sunit@g.swu.ac.th
bDepartment of Chemistry, Chulachomklao Royal Military Academy, Nakornnayok 26001, Thailand
cDepartment of Chemistry and Center for Excellence in Protein Structure and Function, Faculty of Science, Mahidol University, Bangkok 10400, Thailand
First published on 17th May 2018
Abstract
Six new 14-membered ring cyclopeptide alkaloids, cambodines A–F (1–6), and two known compounds, frangufoline (7) and lotusanine B (8), were isolated from the root bark extract of Ziziphus cambodiana Pierre. Their structures and configurations were established based on 1D and 2D NMR, HRMS, ECD, and X-ray crystallographic data. Compounds 1 and 3 are rare 5(14)-type cyclopeptide alkaloids that possess an imidazolidin-4-one ring in the terminal unit. The cyclopeptides were tested for their in vitro antiplasmodial, antitubercular, and cytotoxic effects against three cancer cell lines. Compound 3 showed significant antiplasmodial activity against the malarial parasite Plasmodium falciparum, with an IC50 value of 6.09 μM.
Introduction
Ziziphus, a genus of almost 100 species in the Rhamnaceae family, is found in the warmer parts of the world,1 of which Thailand exhibits a diversity of 9 species.2 Ziziphus plants are recognized to produce triterpenoids and their saponins,3–5 flavonoids6 and cyclopeptide alkaloids7–9 and have been used in indigenous medicine for the treatment of various diseases.10,11 We previously reported the characterization of several bioactive natural products from Thai Ziziphus plants.12–14 Z. cambodiana Pierre is a thorny scandent that is commonly found in the north-east of Thailand and mainly in Southeast Asia and has traditionally been used for its anti-infective properties.15,16 The bioactive substances obtained from this plant include flavonoids and their glycosides with neuraminidase inhibitory activity.17 Triterpenoids with antiplasmodial, antimycobacterial,12 and hedgehog/GLi-mediated transcriptional inhibitors18 were also reported. To date, no study has been published on the cyclopeptide alkaloids from this plant species. Herein, the isolation and structural elucidation of six new 14-membered ring cyclopeptides, cambodines A–F (1–6), along with two known compounds, 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 and 8,20 from the root bark of Z. cambodiana are reported. Compounds 1 and 3 are additional representatives of the (5)14-membered ring cyclopeptide series, with a terminal imidazolidinone ring. Interestingly, these 8 cyclopeptides pose a challenging problem in the determination of the absolute configuration. Electronic circular dichroism (ECD) have been widely used in the determination the absolute configuration, and a great number of chiral compounds have been well investigated via theoretical methods.21–26 Among the techniques employed to compute electronic transitions, TD-DFT is the most widely used and most efficient method for the study of excited states and the prediction of ECD spectra. It has been successfully applied for the description of the conformational structures and the electronic spectra of natural products, metal complexes and other organic molecules.27–31 In the present work, the DFT and TD-DFT methods were applied to simulate the ECD spectra in order to confirm the structure assignment obtained from NMR spectroscopy and the observed ECD spectra. Finally, the in vitro antimalarial, antimycobacterial, and cytotoxic activities of the isolated compounds are also described.
19 and 8,20 from the root bark of Z. cambodiana are reported. Compounds 1 and 3 are additional representatives of the (5)14-membered ring cyclopeptide series, with a terminal imidazolidinone ring. Interestingly, these 8 cyclopeptides pose a challenging problem in the determination of the absolute configuration. Electronic circular dichroism (ECD) have been widely used in the determination the absolute configuration, and a great number of chiral compounds have been well investigated via theoretical methods.21–26 Among the techniques employed to compute electronic transitions, TD-DFT is the most widely used and most efficient method for the study of excited states and the prediction of ECD spectra. It has been successfully applied for the description of the conformational structures and the electronic spectra of natural products, metal complexes and other organic molecules.27–31 In the present work, the DFT and TD-DFT methods were applied to simulate the ECD spectra in order to confirm the structure assignment obtained from NMR spectroscopy and the observed ECD spectra. Finally, the in vitro antimalarial, antimycobacterial, and cytotoxic activities of the isolated compounds are also described.
Results and discussion
Chemical investigations of the EtOAc and MeOH extracts of Z. cambodiana root bark resulted in the isolation of three 14-membered ring cyclopeptide alkaloids, cambodines A (1), B (4), and C (2), and known compounds 7 and 8. The removal of tannins from the MeOH extract with NaCl solution (1% v/v) followed by partitioning with EtOAc yielded a detannified EtOAc-soluble fraction that was column chromatographed to furnish three additional cyclopeptides: cambodines D (5), E (6), and F (3) (Fig. 1).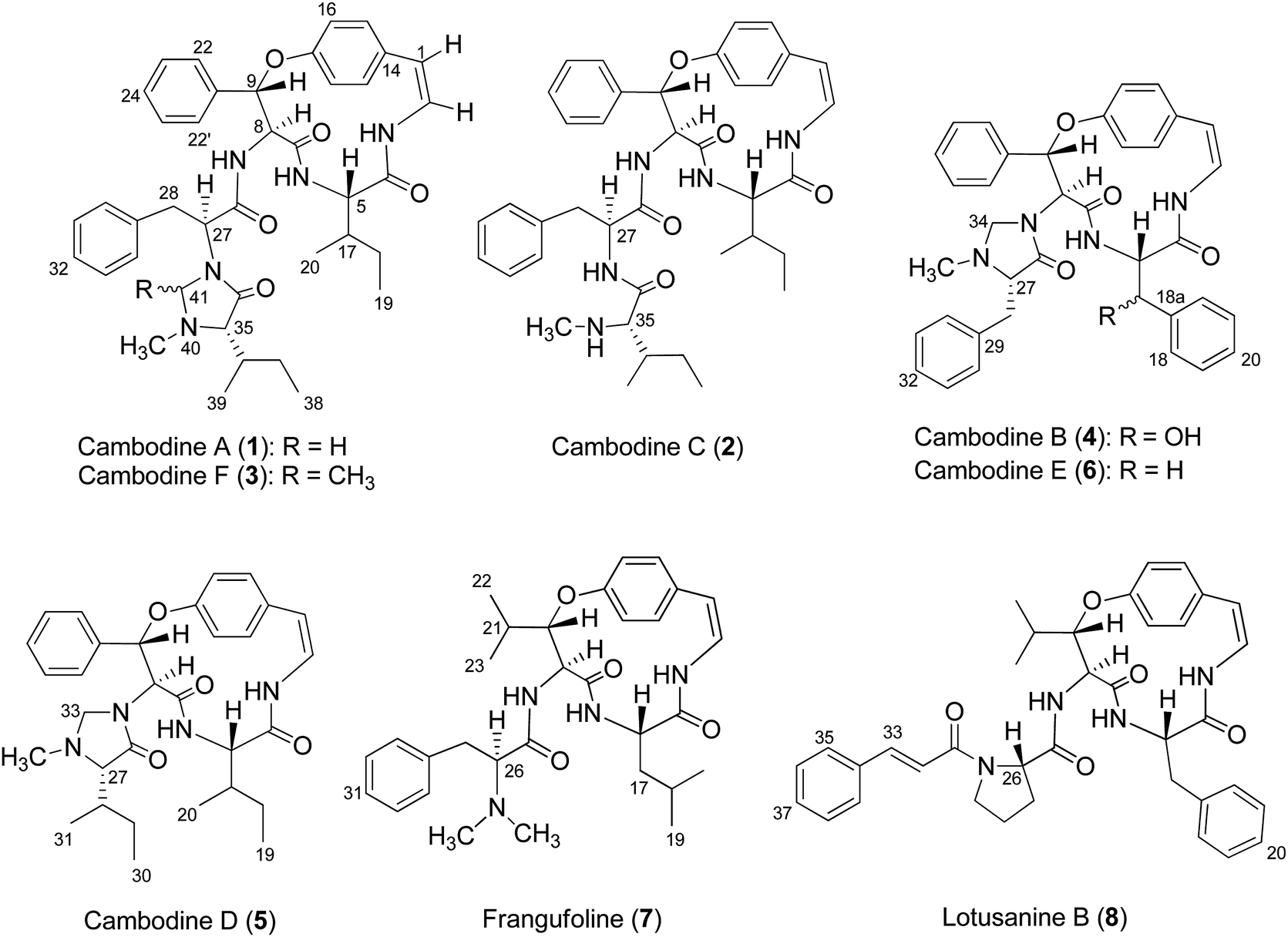 | ||
| Fig. 1 Chemical structures of compounds 1–8. | ||
Several common chemical and spectroscopic characteristics were evident for compounds 1–8. They displayed a blue colour upon staining with anisaldehyde-H2SO4 reagent on TLC.13 Their IR spectra showed absorption bands for amide (3266–3338 and 1632–1685 cm−1) and aryl ether (1221–1237 cm−1) functionalities. The ECD spectra of compounds 1–8 showed intense negative and weak positive Cotton effects in the 236–244 and 279–286 nm regions, respectively, consistent with the (5S,8S,9S)-configurations.32 Their 1H and 13C NMR data agreed well with the published values for 5(14)-scutianine A-type (1–3), 4(14)-integerrine-type (4–6), 4(14)-frangulanine-type (7), and 5(14)-neutral-type cyclopeptide alkaloid (8).
Compound 1 was isolated as a colourless amorphous solid. The HRESITOFMS data showed a protonated ion at m/z 680.3808 [M + H]+, in accordance with a molecular formula C40H49N5O5. The 13C NMR and DEPT data in DMSO-d6 (Table 1) revealed 40 carbon resonances, which were classified as four methyls (δC 10.8, 12.0, 14.5, 15.4), an N-methyl (δC 40.5), four methylenes (δC 24.0, 24.3, 34.2, 66.3), 23 methines (six aliphatic at δC 35.0, 35.5, 53.4, 56.0, 57.4, 69.7, one oxygenated at δC 80.8, two olefinic at δC 125.8 and 126.7 and 14 aromatic at δC 119.5, 121.8, 126.2, 127.6 × 2, 127.9 × 3, 128.4 × 2, 128.8 × 2, 129.5, 130.0), four amide carbonyls (δC 167.4, 168.9, 169.9, 170.7), three aromatic quaternary (δC 131.5, 136.7, and 137.8), and an oxygenated tertiary (δC 154.7). Interpretation of the 1H, 13C NMR, and 2D NMR spectroscopic data and the IR absorption frequencies at 3289, 1682, 1636, and 1241 cm−1 led to the conclusion that 1 was a 5(14)-type cyclopeptide alkaloid in which the cyclic part comprised a phenylalanine, an p-oxystyrylamine with a Z double bond, an isoleucine moiety, in addition to the coupled phenylalanine and N-methylisoleucine moiety as the terminal unit (Fig. 1). The assembly of these fragments was made possible on the basis of correlations in the COSY, HMBC, and NOESY experiments (Fig. 2). The HMBC spectrum showed correlations of the olefinic H-2 (δH 6.23, dd, J = 7.3, 4.5 Hz) to the C-4 carbonyl carbon resonance (δC 168.9) and of the oxygenated methine H-9 (δH 5.84, d, J = 8.1 Hz) and isoleucyl H-5 (δH 3.81, t, J = 8.5 Hz) to C-7 (δC 169.9), in addition to the NOESY cross-peaks between H-9 and the aromatic resonances H-12 (δH 7.14) and H-22 (δH 7.44, dd, J = 7.7, 2.2 Hz) established the linkage of the p-oxystyrylamine moiety to the phenylalanine unit and the location of this dimeric moiety in the macrocyclic ring. The H-8 methine signal at δH 4.78 (brt, J = 8.8 Hz) showed an HMBC cross-peak to the phenylalanine carbonyl C-26 (δC 167.4), indicating the connection between the phenylalanine units. A prominent fragment ion at m/z 622 and a base peak at m/z 155 in the EIMS spectrum of 1 (the molecular ion of which is presented as 1′ in Fig. 3) indicated the fragmentations of the corresponding N-methylimidazolidin-4-one to produce 1′a and the terminal residue 1′b,33,34 respectively (Fig. 3). The relatively low field chemical shifts of the two diastereotopic protons at δH 3.99 and 3.11 (each d, J = 4.4 Hz, H-41a and H-41b, respectively), δC 66.3 (C-41), an N-methyl (δH 2.13), a sec-butyl group (δH 1.23, m, H-36, δC 35.0; δH 0.99, m, H-37, δC 24.3; δH 0.67, t, J = 6.3 Hz, H-38, δC 12.0; and δH 0.41, d, J = 6.8 Hz, H-39, δC 14.5), and an amide carbonyl carbon at δC 170.7 (C-34) further supported the presence of a 3-substituted 5-(sec-butyl)-1-methylimidazolidin-4-one ring. Furthermore, the correlations of H-8, NH-25 (δH 8.23, d, J = 9.4 Hz), and H-27 (δH 4.58, dd, J = 10.7 and 5.1 Hz) to CO-26 (δC 167.4) in the HMBC spectrum along with the interactions of H-27 to H-30 (δH 7.02), of H-41a to H-28 (δH 2.61 m) and H-30, and of N–CH3 to H-41b in the NOESY experiments permitted the assignment of the linkage between both phenylalanine units to the imidazolidinone group. The HMBC cross-peaks of H-41a to CO-34, C-35 (δC 69.7), and N–CH3 and of H-35 to CO-34 and C-36 (δC 35.0) and the NOESY interaction between H-35 and N–CH3 were also observed. Thus, the structure of cyclopeptide 1, cambodine A, was deduced as a new member of the 5(14)-scutianine A-type cyclopeptides.
| No. | δC | δH | ||||||
|---|---|---|---|---|---|---|---|---|
| 1a | 2a | 2b | 3b | 1a | 2a | 2b | 3b | |
| a Recorded in DMSO-d6.b Recorded in CDCl3. | ||||||||
| 1 | 125.8 | 127.5 | 117.5 | 117.5 | 6.60, d (7.3) | 6.63, d (7.3) | 6.44, d (7.1) | 6.45, d (7.4) |
| 2 | 126.7 | 126.7 | 125.5 | 125.4 | 6.23, dd (7.3, 4.5) | 6.14, dd (7.3, 3.4) | 6.65, dd (9.6, 7.1) | 6.65, dd (9.2, 7.4) |
| 4 | 168.9 | 169.0 | 167.2 | 167.2 | ||||
| 5 | 57.4 | 57.3 | 59.3 | 59.3 | 3.81, t (8.5) | 3.81, t (8.9) | 4.08, dd (8.3, 4.1) | 3.98, dd (8.0, 3.9) |
| 7 | 169.9 | 170.2 | 171.0 | 171.0 | ||||
| 8 | 56.0 | 55.9 | 56.7 | 57.2 | 4.78, brt (8.8) | 4.83, dt (9.8, 7.9) | 4.78, dd (8.9, 6.7) | 4.70, dd (8.4, 7.1) |
| 9 | 80.8 | 80.9 | 81.5 | 81.6 | 5.84, d (8.1) | 5.84, d (7.9) | 6.10, d (6.7) | 6.17, d (7.1) |
| 11 | 154.7 | 154.8 | 155.1 | 155.2 | ||||
| 12 | 121.8 | 118.1 | 123.1 | 122.9 | 7.14, overlap | 7.16, overlap | 7.32, overlap | 7.35, dd (8.4, 2.1) |
| 13 | 129.5 | 129.4 | 130.1 | 130.1 | 6.96, d (6.9) | 6.96, overlap | 7.15, overlap | 7.17, overlap |
| 14 | 131.5 | 131.2 | 132.5 | 132.2 | ||||
| 15 | 130.0 | 129.9 | 131.9 | 131.8 | 6.93, d (6.9) | 6.95, overlap | 7.11, overlap | 7.12, overlap |
| 16 | 119.5 | 121.1 | 122.6 | 122.5 | 7.19, overlap | 7.06, overlap | 7.34, overlap | 7.40, dd (8.4, 2.1) |
| 17 | 35.5 | 37.2 | 35.2 | 35.3 | 1.58, m | 1.45, m | 2.11, m | 2.04, m |
| 18 | 24.0 | 24.5 | 23.9 | 23.9 | a 1.16, m | a 1.15, m | a 1.25, m | a 1.15, m |
| b 0.99, m | b 0.80, m | b 0.80, m | b 0.85, m | |||||
| 19 | 10.8 | 11.3 | 11.9 | 11.9 | 0.66, t (7.1) | 0.64, overlap | 0.84, t (7.2) | 0.79, t (7.2) |
| 20 | 15.4 | 15.2 | 15.8 | 16.4 | 0.65, t (7.3) | 0.64, overlap | 0.75, d (6.8) | 0.67, d (6.9) |
| 21 | 137.8 | 137.5 | 136.8 | 137.2 | ||||
| 22,22′ | 128.4 | 128.6 | 128.1 | 128.2 | 7.44, dd (7.7, 2.2) | 7.49, d (6.9) | 7.51, dd (7.5, 1.0) | 7.67, brd (7.2) |
| 23,23′ | 127.6 | 127.7 | 128.8 | 128.8 | 7.28, overlap | 7.27, t (6.9) | 7.40, overlap | 7.48, brt (7.4) |
| 24 | 127.9 | 127.8 | 128.9 | 128.9 | 7.26, overlap | 7.22, overlap | 7.45, overlap | 7.31, overlap |
| 26 | 167.4 | 169.5 | 170.9 | 170.0 | ||||
| 27 | 53.4 | 53.0 | 54.7 | 63.0 | 4.58, dd (10.7, 5.1) | 4.23, dt (10.8, 7.9) | 4.20, dd (10.4, 4.8) | 3.49, dd (11.9, 4.4) |
| 28 | 34.2 | 37.5 | 36.6 | 33.2 | 2.61, m | a 2.33, dd (11.2, 7.9) | a 2.88, dd (14.2, 4.8) | a 2.29, brt (12.6) |
| b 2.13, brt (11.2) | b 2.45, dd (14.2, 10.4) | b 2.19, dd (12.6, 4.4) | ||||||
| 29 | 136.7 | 137.7 | 136.3 | 136.6 | ||||
| 30,30′ | 128.8 | 129.0 | 128.9 | 129.2 | 7.02, overlap | 6.99, overlap | 6.98, dd (8.4, 2.1) | 6.93, dd (7.6, 1.1) |
| 31,31′ | 127.9 | 127.5 | 128.6 | 128.5 | 7.08, overlap | 7.10, overlap | 7.18, overlap | 7.22, overlap |
| 32 | 126.2 | 126.0 | 127.0 | 127.0 | 7.07, overlap | 7.08, overlap | 7.22, overlap | 7.24, overlap |
| 34 | 170.7 | 172.2 | 174.2 | 174.8 | ||||
| 35 | 69.7 | 69.1 | 69.1 | 71.0 | 2.45, brs | 2.35, brs | 2.50, brs | 2.57, brs |
| 36 | 35.0 | 36.1 | 37.6 | 36.5 | 1.23, m | 1.52, m | 1.51, m | 1.50, m |
| 37 | 24.3 | 24.1 | 24.2 | 24.8 | 0.99, m | a 1.15, m | a 0.86, m | 1.28, m |
| b 0.82, m | b 0.70, m | |||||||
| 38 | 12.0 | 10.9 | 11.7 | 12.2 | 0.67, t (6.3) | 0.64, overlap | 0.65, t (6.0) | 0.85, t (7.3) |
| 39 | 14.5 | 15.4 | 15.5 | 14.9 | 0.41, d (6.8) | 0.46, d (6.7) | 0.62, d (6.9) | 0.77, d (6.8) |
| 41 | 66.3 | 78.9 | a 3.99, d (4.4) | 2.41, q (5.4) | ||||
| b 3.11, d (4.4) | ||||||||
| 42 | 19.5 | 0.92, d (5.4) | ||||||
| 3-NH | 7.84, brd (4.5) | 8.05, brd (3.4) | 6.73, d (9.6) | 6.52, d (9.2) | ||||
| 6-NH | 7.54, brd (8.5) | 7.62, d (8.9) | 6.19, d (8.3) | 5.92, d (8.0) | ||||
| 25-NH | 8.23, d (9.4) | 7.93, d (8.9) | 6.84, d (8.9) | 8.73, d (8.4) | ||||
| 33-NH | 7.64, d (7.9) | |||||||
| NMe | 40.5 | 34.9 | 36.5 | 39.6 | 2.13, s | 1.79, s | 2.11, s | 1.85, s |
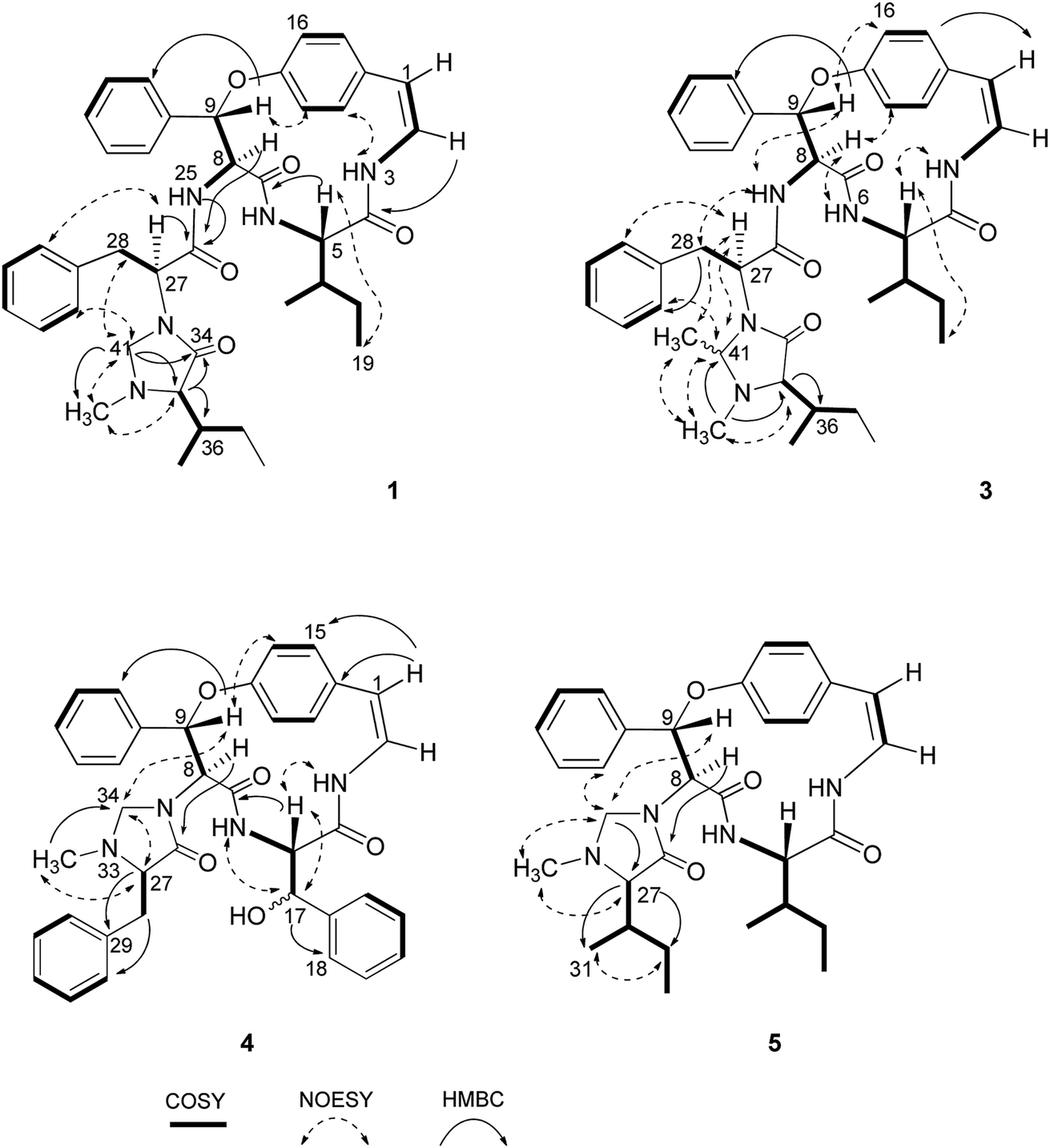 | ||
| Fig. 2 Selected COSY, HMBC and NOESY interactions for compounds 1, 3, 4 and 5. | ||
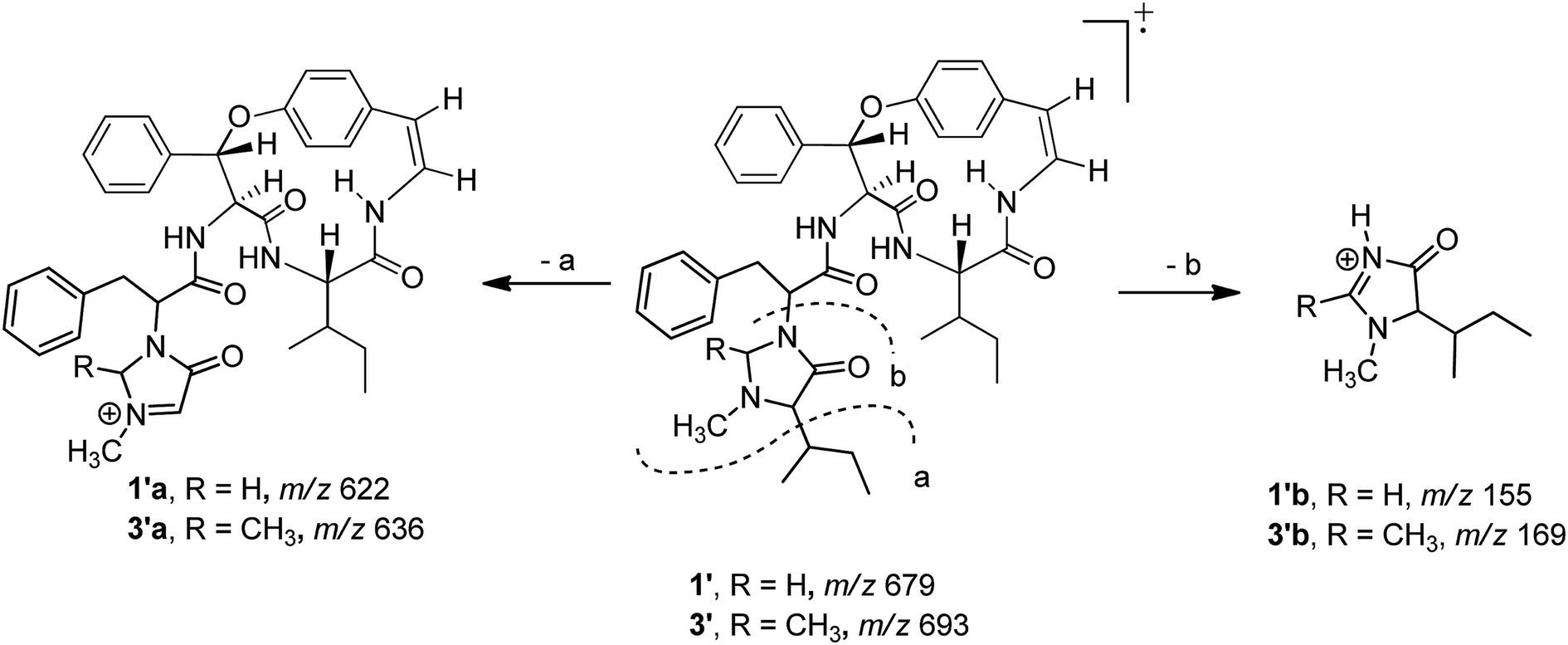 | ||
| Fig. 3 EI fragmentations of compounds 1 and 3. | ||
Compound 2 was also obtained as a colourless amorphous solid. Its molecular formula was determined by the HRESITOFMS ion at m/z 668.3807 [M + H]+ and 13C NMR spectroscopic data as C39H49N5O5. Its 1H and 13C NMR data in DMSO-d6 (Table 1) were similar to those of 1, except for the absence of the two geminal protons at C-41 in 1 and the presence of an amide proton NH-33 at δH7.64 (1H, d, J = 7.9 Hz) in 2, which are in agreement with the structural change in the terminal residue (Fig. 1). The NMR data of 2 recorded in CDCl3 (Table 1) were similar to those observed in DMSO-d6, except for the C-1 chemical shift. Information from the HMBC association of H-8 (δH 4.83, dt, J = 9.8, 7.9 Hz) to CO-26 (δC 169.5) and of NH-33 to CO-34 (δC 172.2) in addition to the NOESY interactions of H-27 (δH 4.23, dt, J = 10.8, 7.9 Hz) to NH-25 (δH 7.93, d, J = 8.9 Hz) and H-30 (δH 6.99) and of H-35 (δH 2.35, brs) to NCH3 (δH 1.79, s) (Fig. 2) provided evidence for the exocyclic phenylalanine moiety connecting to the macrocyclic ring at N-25 and the terminal N-methylisoleucine unit. The structure of 2, cambodine C, was therefore also defined as a new 5(14)-scutianine A-type compound.
Compound 3 displayed a sodium adduct molecular ion at m/z 716.3792 [M + Na]+ in the HRESITOFMS, corresponding to the molecular formula C41H51N5O5. Its 1H and 13C NMR data in CDCl3 (Table 1) showed signals similar to those observed for compounds 1 and 2 in the same NMR solvent, except for one fewer methylene carbon and the additional methyl (δC 19.5) and methine resonances (δC 78.9) observed in the DEPT spectra for 3. Similar to those of 1, the observed fragment peaks at m/z 636 and 169 in the EIMS data of 3 suggest the presence of phenylalanine and a 5-(sec-butyl)-1,2-dimethylimidazolidin-4-one as the respective exocyclic units (Fig. 3), which is supported by a molecular mass 14 amu greater than that of 1. The NOE associations of NH-25 (δH 8.73, d, J = 8.4 Hz) to H-9 (δH 6.17, d, J = 7.1 Hz) and H-28a (δH 2.29, brt, J = 12.6 Hz), of H-41 (δH 2.41, q, J = 5.4 Hz) to H-27 (δH 3.49, dd, J = 11.9, 4.4 Hz), H-30 (δH 6.93, dd, J = 7.6, 1.1 Hz), and N–CH3 (δH1.85), and of the latter N–CH3 to H-35 (δH 2.57 brs), together with connectivities of H-28 to C-30 (δC 129.2), of N–CH3 to C-41 (δC 78.9) and C-35 (δC 71.0), and of H-35 to C-36 (δC 36.5) in the HMBC spectrum also supported the linkage between the acyclic part and the macrocyclic ring (Fig. 2). The NOESY associations of NH-6 (δH 5.92, d, J = 8.0 Hz)/H-8 and H-9/H-16 (δH7.40, dd, J = 8.4, 2.1 Hz) confirmed the location of the phenylalanine unit next to the p-oxystyrylamine moiety and the isoleucine fragments in the macrocyclic system. The significantly different C-27 (δC 63.0) and C-41 (δC 78.9) chemical shifts compared to those of 1 (C-27, δC 53.4 and C-41, 66.3) and 2 (C-27, δC 54.7) could be attributed to the presence of the C-41 methyl group. However, the existing data did not permit the establishment of the configuration at C-41. Thus, the structure of 3, cambodine F, was deduced as a new member of the 5(14)-scutianine A-type cyclopeptides possessing a 5-(sec-butyl)-1,2-dimethylimidazolidin-4-one moiety.
The J values of H-1 and H-2 ranging from 7.1–7.4 Hz accounted for the Z geometry of the double bond in compounds 1–3. The intense negative (236 nm) and weak positive (283 nm) Cotton effects present in the CD spectrum of 1, similar to those of 2 and 3, and indicative of the (5S,8S,9S) configuration.32 The H-8/H-9 vicinal coupling constants of 8.1 Hz for 1, 6.7 for 2, and 7.1 for 3 and the relatively shielded shift of the 13C NMR resonance at approximately δC 81 ppm for C-9 permitted assignment of the erythro, i.e., (8S,9S) configuration.13 The 13C NMR chemical shifts of the phenylalanine and the N-methyl isoleucine terminal amino acid units for 1–3, particularly those of stereocenters [1: δC 53.4 (C-27), 69.7 (C-35); 2: δC 53.0 (C-27), 69.1 (C-35); 3: δC 63.0 (C-27), 71.0 (C-35)] were comparable, except for C-27 in 3 as indicated above, with the same terminal dipeptide in paliurine B [δC 51.5 (C-27), 69.9 (C-35)], a 13-membered ring cyclopeptide obtained from Paliurus ramossisimus.35 The stereochemical assignment of the exocyclic dipeptide in paliurine B was determined by the 13C NMR data comparison with those of the comparable synthesized model dipeptide-L-Phe(OMe)-L-Ile(NMe2),35 the L amino acids, or 27S and 35S, in the acyclic part attached to the macrocycles in 1–3 were then suggested, though the DD-dipeptide cannot be ruled out due to no NMR information available. Furthermore, amino acids in Rhamnaceous cyclopeptide alkaloids generally present in L-form whilst the D-amino acid only rarely found. For instances, the D-erythro (8R,9R) of scutianine E,36 and D-threo (8R,9S) of scutianene L,37 were isolated from Scutia buxifolia; whereas epimauritine A and its N-oxide obtained from Z. apetala showed the R-configuration at the terminal unit.38 Formation of the imidazolidinone ring in cyclopeptide has been implicated from the reaction between the amino acid residue and aldehyde.34,39 The imidazolidinone ring in compounds 1 and 3 could therefore arise through the condensation of 2 with the respective corresponding carbonyl compounds without an alteration in the amino acid stereocenter.
The two diagnostic Cotton effect bands at about 240 (intense negative) and 280 nm (weak positive) displayed in the ECD spectrum which could be attributed to the transition of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond conjugation with benzene ring or adjacent amide group, and characterized for the existence of L-amino acids in the 14-membered ring cyclopeptide.32,40,41 Comparison of the experimental ECD spectrum of 1 to the qualitative calculated ECD data of (5S,8S,9S,17S,27S,35S,36S)-1 and the (5R,8R,9R,17R,27R,35R,36R)-1 (Fig. 4A), indicated 1 to be in agreement with that of all S configuration on the amino acid residues. Calculation of the ECD spectra of individual epimer of 1 were also performed (Fig. 4B) and revealed that the major Cotton effect contribution with opposite sign observed at the band near 238 nm for 9S/9R and at around 245 nm for 8S/8R epimer pair. Similar Cotton effects were found for compounds 2 (negative 236 nm and positive 283 nm bands) and 3 (negative 236 nm and positive 282 nm bands) and comparable to the theoretical ECD ones (Fig. 4C) allowed the same absolute configuration assignment as for 1. From these evidences, including their negative specific rotations, the stereochemical structures of compounds 1–3 were then deduced.
C double bond conjugation with benzene ring or adjacent amide group, and characterized for the existence of L-amino acids in the 14-membered ring cyclopeptide.32,40,41 Comparison of the experimental ECD spectrum of 1 to the qualitative calculated ECD data of (5S,8S,9S,17S,27S,35S,36S)-1 and the (5R,8R,9R,17R,27R,35R,36R)-1 (Fig. 4A), indicated 1 to be in agreement with that of all S configuration on the amino acid residues. Calculation of the ECD spectra of individual epimer of 1 were also performed (Fig. 4B) and revealed that the major Cotton effect contribution with opposite sign observed at the band near 238 nm for 9S/9R and at around 245 nm for 8S/8R epimer pair. Similar Cotton effects were found for compounds 2 (negative 236 nm and positive 283 nm bands) and 3 (negative 236 nm and positive 282 nm bands) and comparable to the theoretical ECD ones (Fig. 4C) allowed the same absolute configuration assignment as for 1. From these evidences, including their negative specific rotations, the stereochemical structures of compounds 1–3 were then deduced.
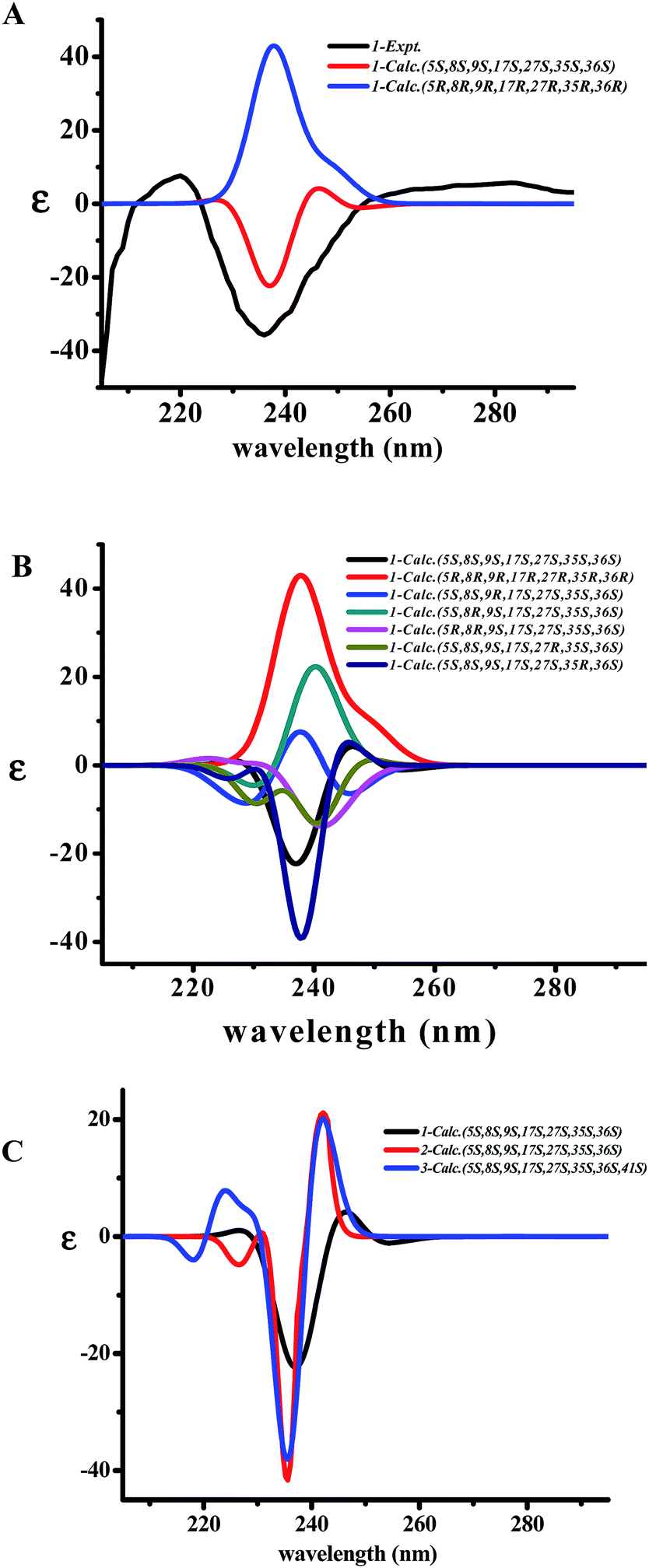 | ||
| Fig. 4 The superimpose of calculated ECD of (5S,8S,9S,17S,27S,35S,36S)-1 and the (5R,8R,9R,17R,27R,35R,36R)-1 compared to the observed ECD spectra (A); the ECD spectra of each epimer for 1 were also calculated for the stereochemical structures (B) and the theoretical spectra of 1–3 were calculated at the M062X/6-311G(d,p)//M062X/6-311G level of theory in the MeOH solution and their superimpose ECD (C). | ||
Compound 4, a minor compound from the same fraction of compound 1, was isolated as a colourless amorphous solid and given a molecular formula C37H36N4O5, as deduced from its positive ion HRESITOFMS at m/z 617.2751 [M + H]+. The 13C NMR and DEPT spectra (CDCl3) disclosed 37 carbon resonances, consisting of an N-methyl (δC 39.8), two methylenes (δC 37.5, 67.1), 23 methines (two olefinic at δC 121.2, 125.2, two oxymethine at δC 73.1, 81.6, 19 aromatic at δC 121.9, 123.2, 126.3, 126.9 × 2, 128.2 × 4, 128.6 × 2, 128.7 × 3, 129.1 × 3, and 130.2), four aromatic quaternary (δC 131.9, 136.0, 138.1, 139.0), an oxygenated tertiary (δC 155.3), three amide carbonyls (δC 167.7, 167.8 and 172.1) and no signal observed in the aliphatic region (Table 2, Fig. 1). A 1D and 2D NMR extensive data analysis in addition to a comparison with previously described values evidenced the spin system of a p-oxystyrylamine, a β-hydroxyphenylalanine, a phenylalanine, and a 1-methylimidazolidin-4-one, which is derived from the phenylalanine of the 4(14)-integerrine-type cyclopeptide alkaloid. Interestingly, a broad three-bond singlet at δH1.59 was assigned to the styrylamine double bond H-1, H-2, and NH-3 by the HMBC of H-1 to C-2 (δC 125.5), C-14 (δC 131.9) and C-15 (δC 131.0) and of H-2 to C-14 and the NOESY of H-3 to H-5 (δH 4.51, brt, J = 8.1 Hz) connectivities (Fig. 2). The presence of the β-hydroxyphenylalanine was characterized by the OH absorption frequency at 3447 cm−1 in the IR data, along with the correlations from the oxymethine doublet resonance H-17 (δH 4.94, J = 7.6 Hz) to a carbonyl carbon C-4 at δC 167.8 and an aromatic carbon at δC 126.9 (C-18) in the HMBC spectrum, in addition to the NOESY connectivities of H-17 to NH-6 (δH 6.37, brd, J = 8.5 Hz). As for other cyclopeptides, the HMBC correlations of H-1 to C-14 and C-15, of H-5 to CO-7 (δC 167.7), and of H-9 (δH 5.90, d, J = 7.3 Hz) to C-22 (δC 128.2) and of the NOESY interactions of H-5 to NH-3 and of H-9 to H-16 (δH 7.29) suggested that the phenylalanine and β-hydroxyphenylalanine were placed next to each other and attached to the oxystyrylamine in the cyclic ring. A 5-benzyl-1-methylimidazolidin-4-one group was determined by the analysis of the 1D and 2D NMR spectra of 4: the presence of the two diastereotopic protons at δH 3.66 and 2.70 (each d, J = 4.3 Hz, H-34a and H-34b), δC 67.1; an N-methyl (δH 1.64, s), a ring multiplet methine proton resonance at δH 2.69 (H-27), δC 66.5; a benzyl group (δH 2.67, m, H-28, δC 37.5; δH 7.24–7.47, ArH, δC 126.3–138.1) and an amide carbonyl at δC 172.1 (C-26). A series of connectivities was observed: a diastereotopic proton at δH 3.66 showed NOESY correlations to H-9 and H-27 and of H-27 to N–CH3, and the HMBC connections of H-8 (δH 4.87, d, J = 7.3 Hz) to carbonyl carbon C-26, of H-27 to C-29 and of H-28 to C-30 showed that the imidazolidin-4-one ring was connected to the macrocyclic ring at C-8 of the phenylalanine. The structure of 4, cambodine B, was thus elucidated as a new member of the 4(14)-integerrine-type cyclopeptides.
| No. | δC | δH | ||||
|---|---|---|---|---|---|---|
| 4 | 5 | 6 | 4 | 5 | 6 | |
| 1 | 121.2 | 117.8 | 116.9 | 6.59, brs | 6.46, d (7.6) | 6.47, d (7.6) |
| 2 | 125.2 | 125.4 | 125.6 | 6.59, brs | 6.66, dd (9.5, 7.6) | 6.37, brt (7.6) |
| 4 | 167.8 | 166.8 | 166.5 | |||
| 5 | 57.5 | 59.3 | 55.4 | 4.51, brt (8.1) | 4.01, dd (8.5, 4.4) | 4.44, ddd (11.2, 8.1, 3.4) |
| 7 | 167.7 | 168.2 | 168.1 | |||
| 8 | 57.8 | 57.9 | 57.6 | 4.87, d (7.3) | 5.09, d (7.6) | 4.95, d (7.2) |
| 9 | 81.6 | 80.7 | 81.7 | 5.90, d (7.3) | 5.99, d (7.6) | 5.91, d (7.2) |
| 11 | 155.3 | 155.0 | 155.4 | |||
| 12 | 123.2 | 123.3 | 123.2 | 7.26, overlap | 7.32, overlap | 7.34, overlap |
| 13 | 130.2 | 130.2 | 130.4 | 7.05, dd (6.8, 1.3) | 7.13, t (7.1) | 7.14, overlap |
| 14 | 131.9 | 132.3 | 132.2 | |||
| 15 | 131.0 | 131.6 | 131.7 | 7.10, dd (6.8, 1.3) | 7.10, t (7.1) | 7.14, overlap |
| 16 | 121.9 | 122.8 | 122.5 | 7.29, overlap | 7.34, overlap | 7.34, overlap |
| 17 | 73.1 | 34.4 | 36.7 | 4.94, d (7.6) | 2.16, m | a 3.40, dd (14.8, 3.4) |
| b 2.61, dd (14.8, 11.2) | ||||||
| 18a | 139.0 | 136.9 | ||||
| 18,18′ | 126.9 | 23.9 | 128.8 | 7.24, overlap | 1.17, m, 0.88, m | 7.09, d (8.7) |
| 19,19′ | 128.7 | 11.7 | 128.5 | 7.03, overlap | 0.81, t (6.3) | 7.19, brt (8.7) |
| 20 | 128.7 | 16.5 | 127.1 | 7.24, overlap | 0.70, d (6.8) | 7.07, overlap |
| 21 | 136.0 | 136.2 | 136.0 | |||
| 22,22′ | 128.2 | 128.2 | 128.2 | 7.46, overlap | 7.52, dd (7.7, 1.9) | 7.46, overlap |
| 23,23′ | 128.6 | 128.8 | 128.8 | 7.19, overlap | 7.39, t (6.9) | 7.39, overlap |
| 24 | 129.1 | 129.2 | 129.0 | 7.39, overlap | 7.41, overlap | 7.11, overlap |
| 26 | 172.1 | 172.8 | 172.3 | |||
| 27 | 66.5 | 70.4 | 66.1 | 2.69, m | 2.48, d (2.6) | 2.39, m |
| 28 | 37.5 | 35.8 | 37.6 | 2.67, m | 1.29, m | a 2.66, dd (13.8, 8.9) |
| b 1.72, dd (13.8, 11.2) | ||||||
| 29 | 138.1 | 24.7 | 138.2 | 1.08, m | ||
| 30 | 129.1 | 11.9 | 129.1 | 7.39, overlap | 0.77, t (7.4) | 7.00, brd (6.1) |
| 31 | 128.2 | 14.7 | 128.2 | 7.24, overlap | 0.55, d (6.8) | 7.26, overlap |
| 32 | 126.3 | 126.2 | 7.47, overlap | 7.22, overlap | ||
| 33 | 67.6 | a 3.83, br d (3.9) | ||||
| b 3.22, br d (3.9) | ||||||
| 34 | 67.1 | 67.0 | a 3.66, d (4.3) | a 3.56, d (4.7) | ||
| b 2.70, d (4.3) | b 2.43, d (4.7) | |||||
| 3-NH | 6.59, brs | 6.50, d (9.5) | 6.51, d (9.9) | |||
| 6-NH | 6.37, brd (8.5) | 5.81, d (8.5) | 6.40, d (8.1) | |||
| NMe | 39.8 | 41.1 | 39.5 | 1.64, s | 2.11, s | 1.48, s |
Compound 5 was also obtained as a colourless amorphous solid from the detannified EtOAc-soluble fraction of Z. cambodiana. Its molecular formula was deduced as C31H40N4O4 from its positive ion HRESITOFMS at m/z 555.2929 [M + H]+. A detailed analysis of the 1D, DEPT and 2D spectroscopic data suggested that 5 also possessed a 4(14)-type cyclopeptide containing the same macrocyclic ring and terminal unit as for 1 (Fig. 1). The main differences in their 13C NMR data (Table 2) are the absence of the intermediate phenylalanine resonances in 5 compared to that of 1. A series of correlations of H-8 (δH 5.09, d, J = 7.6 Hz) to C-26 (δC 172.8), of H-33a (δH 3.83, brd, J = 3.9 Hz) to C-27 (δC 70.4), and of H-27 (δH 2.48, d, J = 2.6 Hz) to CO-26, C-29 (δC 24.7) and C-31 (δC 14.7) displayed in the HMBC spectrum, together with the interactions of H-33a to H-9 (δH 5.99, d, J = 7.6 Hz), H-22′ (δH 7.52, dd, J = 7.7, 1.9 Hz) and N–CH3 (δH 2.11) and of H-27 to N–CH3, H-29 (δH 1.08, m) and H-31 (δH 0.55, d, J = 6.8 Hz) observed in the NOESY spectrum indicated that the 5-(sec-butyl)-1-methylimidazolidin-4-one ring was the end fragment of the system (Fig. 2). The structure of 5, cambodine D, was thus established as an additional member of the 4(14)-integerrine-type cyclopeptides.
Compound 6 was isolated as colourless needles and displayed a sodium adduct molecular ion [M + Na]+ at m/z 623.2606 in the HRESITOFMS, corresponding to a molecular formula of C37H36N4O4. Its 1H and 13C NMR data (Table 2, Fig. 1) were almost identical to those of compound 4, with the difference being the presence of the two germinal proton resonances at δH 3.40 (dd, J = 14.8 and 3.4 Hz, H-17a) and δH 2.61 (dd, J = 14.8 and 11.2 Hz, H-17b) (δC 36.7) of the ring-bound amino acid in 6, instead of the oxymethine signal in 4. In the HMBC spectrum, the connectivities of the resonance at δH 2.61 to C-5 (δC 55.4) and a carbonyl carbon at δC 166.5 (C-4), of NH-6 at δH 6.40 (d, J = 8.1 Hz) to C-5, and of an aromatic signal at δH 7.09 (d, J = 8.7 Hz, H-18) to C-17 confirmed that the phenylalanine was bound to p-oxystyrylamine and β-hydroxyphenylalanine in the cyclic structure. Its EIMS spectrum showed the base peak at m/z 509 and a fragment ion peak at m/z 227, indicating the existence of a 5-benzyl-1-methylimidazolidin-4-one unit as the end residue. This was supported by the presence of two geminal protons at δH 3.56 (d, J = 4.7 Hz, H-34a) and δH 2.43 (d, J = 4.7 Hz, H-34b), a singlet N–CH3 at δH 1.48 (δC 39.5), and a multiplet methine resonance of H-27 at δH 2.39 (δC 66.1), in addition to a carbonyl carbon signal at δC 172.3 (C-26) and a benzyl group in its NMR data. Compound 6 exhibited similar 2D NMR (HMBC and NOESY) correlations, both at the cyclic and the terminal ring, to those of compound 4. A remarkable upfield-shifted N–CH3 resonance at δH 1.48 in 6 could be due to an anisotropic effect arising from the benzyl group when compared with compound 5 (δH 2.11) that has a sec-butyl moiety. The structure of 6, cambodine E, was thus elucidated as an analogue of cyclopeptide 4.
The X-ray crystal structure of 6 supported a skeleton comprised of a Z-styrylamine, two phenylalanines and a 5-benzyl-1-methylimidazolidin-4-one subunit and also confirmed the trans arrangement between H-8 and H-9 displayed in the macrocyclic motif (Fig. 5). The ECD spectrum of 6, as well as the spectra of the other cyclopeptides indicated above, provided the 5S, 8S and 9S configuration assignments at the amino acid residues of the cyclic part. The imidazolidinone configuration at 27S was then subsequently decisively assigned. The similarities in the NMR data on the terminal ring of 6 compared with those of 4 and 5, associated with the calculated ECD spectra of 4–6 (Fig. 6) were in accordance with those observed ECD value have led to the conclusion that cyclopeptides 4–6 contribute the same stereochemistry both at the macrocyclic ring and at the imidazolidinone unit.
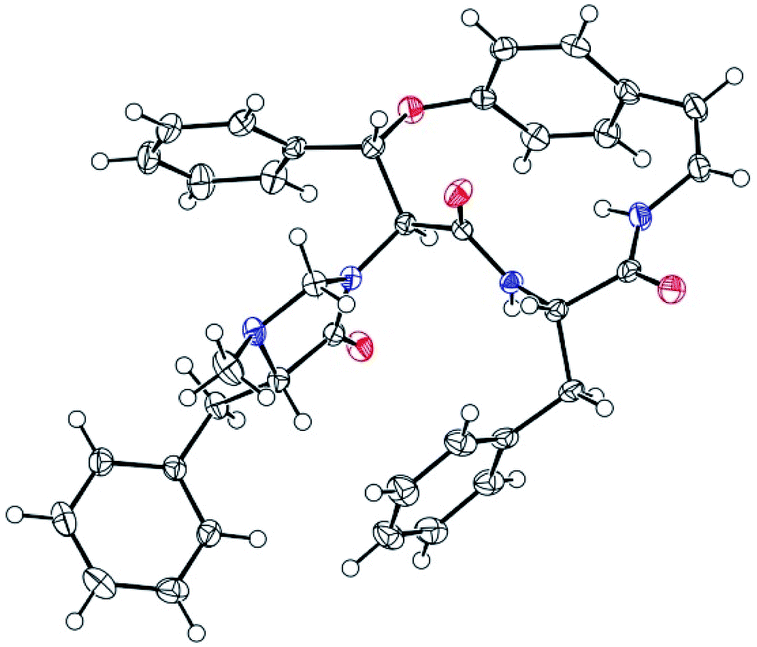 | ||
| Fig. 5 ORTEP plot of the X-ray crystal structure for compound 6. | ||
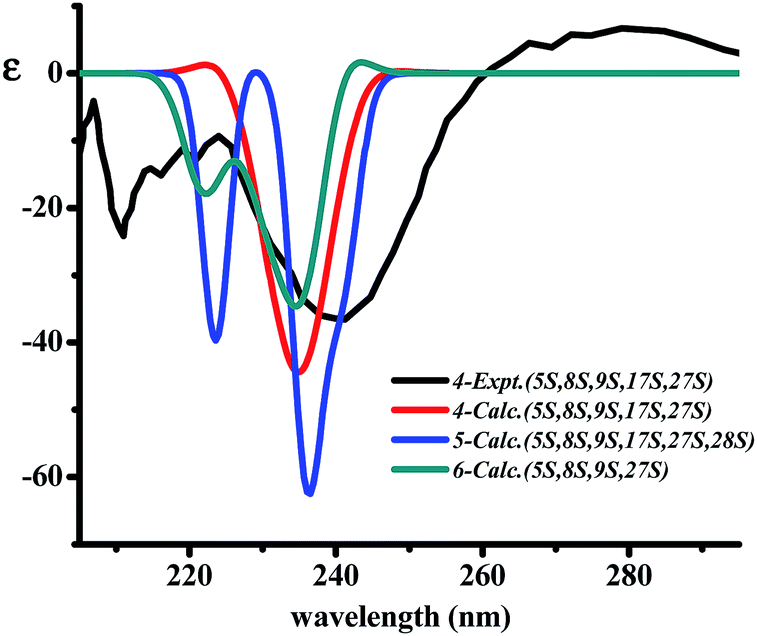 | ||
| Fig. 6 The superimpose of calculated ECD for 4–6 in comparison to the observed ECD spectra of 4. | ||
The two cyclopeptides 7 and 8 were identified as the 4(14)-type cyclopeptide alkaloids, frangufoline19 or daechuine S,32 or sanjoinine A42 and lotusanine B,20 respectively, by detailed examinations of their 1D and 2D NMR and MS spectroscopic data along with comparison to their reported values (Fig. 1, ESI†). The 13C NMR resonances of the chiral carbons observed for 7 were in good agreement with the literature data for frangufoline whose stereochemistry was proven to be all S configurations by the analysis of each amino acid residue in its acid hydrolysate and confirmed by total synthesis.19,43 The ECD spectrum of 7 exhibited strong negative (239 nm) and positive (287 nm) Cotton effects that also supported the same (5S,8S,9S)-configurations at the 14-membered ring and its negative specific rotation ([α]39D −218.5), which led to the conclusion that 7 has the same structure and configuration as the reported frangufoline. In a manner similar to that observed for the other cyclopeptide alkaloids, the 13C NMR chemical shifts at the chiral carbons of 8 are almost identical to those of amaiouine, a structurally related cyclopeptide isolated from Amaioua guianensi that was different from 8 by having a phenylalanine in place of a leucine amino acid in the macrocyclic ring. The absolute configurations of amaiouine were established to be all S according to its X-ray crystallographic data.44 Therefore, our observed ECD spectrum (strong negative and weak positive bands at 237 and 283 nm, respectively) was also in accordance with the all S configurations at the amino acid residues for 8. It should be noted that compound 8 showed negative optical activity, although the previously reported lotusanine B was a racemate. Frangufoline has been isolated from many plants belonging to the Rhamnaceae, Celastraceae and Sterculiaceae families,32 whereas lotusanine B was obtained from Ziziphus lotus.20
The in vitro antimalarial effect against Plasmodium falciparum of compounds 1–8 was evaluated.45,46 Only cambodine F (3) showed interesting antiplasmodial activity, with an IC50 of 6.09 μM, which could be accounted for by the importance of the rare 5-(sec-butyl)-1,2-dimethylimidazolidin-4-one unit. All compounds were considered inactive against Mycobacterium tuberculosis,47 except for cambodines B (4) and C (2), and lotusanine B (8) which showed weak activity, with MICs of 81.0, 149.7 and 93.8 μM, respectively. All compounds were also tested in vitro for cytotoxic properties against three human cancer cell lines: epidermoid carcinoma of the mouth (KB), breast cancer (BC-1), and small cell lung cancer (NCI-H187) cells.48 Of the tested compounds, cambodine A (1) was moderately active against the BC cells at IC50 11.1 μM, and cambodines C–E (2, 5, 6) exhibited weak activity towards the NCI cells, with IC50 values of 20.7, 36.7 and 35.0 μM, respectively. None of the compounds were toxic to the non-cancerous Vero cells, except for 6, which showed weak action with an IC50 of 48.2 μM. To date, only two studies on the in vitro cytotoxicity of natural rhamnaceous cyclopeptide alkaloids have been reported,38,49 although the weak activity of some representative synthetic 13- and 15-membered ring cyclopeptides was described recently.50
Experimental section
General experimental procedure
Melting points were determined using a Griffin melting point apparatus and are uncorrected. Optical rotations at the sodium D line were measured on a JASCO-1020 digital polarimeter. ECD spectra were recorded on a Jasco J-810 spectropolarimeter. IR spectra were measured on a Perkin-Elmer FT-IR Spectrum BX spectrophotometer, with νmax given in cm−1. NMR spectra were measured at 300 MHz (1H) and 75 MHz (13C) on a Bruker AVANCE 300 FT-NMR spectrometer using TMS or residual non-deuterated solvent signals as an internal standard (CDCl3: δH 7.24, δC 77.00; DMSO-d6: δH 2.49 and δC 39.5 for 1H and 13C NMR spectra, respectively). EIMS were recorded on a Thermo Finnigan Polaris Q mass spectrometer at 70 eV (probe). ESIMS were obtained on a Finnigan LC-Q mass spectrometer. The HRESITOFMS were measured on a Bruker micrOTOF-QII mass spectrometer. The X-ray crystallographic data analysis was carried out with a Bruker-Nonius kappaCCD diffractometer with a graphite monochromator, MoKα radiation (λ = 0.71073 Å) at 298(2) K. Silica gel (finer than 0.063 mm, Merck) and Sephadex LH-20 (Pharmacia) were used for column chromatography. TLC analyses were carried out on plates that had been precoated with silica gel F254 from Merck and visualized under a UV light at 254 or 365 nm and by spraying with 5% anisaldehyde-H2SO4 solution followed by heating.Plant materials
Shade-dried root bark of Z. cambodiana was collected from Chamni District, Burirum Province, Thailand, in March 2007. The material was identified by James F. Maxwell of the Faculty of Science, Chiang Mai University, Thailand. A voucher specimen (Wichan Wisetsri 002) has been deposited at the Laboratory of the Natural Product Research Unit, Srinakharinwirot University, Thailand.Extraction and separation
The powdered root bark of Z. cambodiana (10.0 kg) was extracted successively with EtOAc (20 L x 3) and MeOH (20 L x 3) at 50 °C for 48 h for each solvent. The combined extract was evaporated under reduced pressure at temperature 40–45 °C to yield EtOAc (144.7 g) and MeOH (1.4 kg) extracts. The crude extract was subjected to chromatographic separation, and only fractions that showed blue spots upon staining with anisaldehyde-H2SO4 reagent on TLC were selected for further separation and purification. A portion of the EtOAc soluble extract (80 g) was fractionated by quick column chromatography on silica gel (160 g), eluting with a gradient system of hexanes, CH2Cl2, EtOAc and MeOH (5% increment of the more polar component, each 300 mL) to provide nine major fractions (Fr. A–I) on the basis of TLC analysis. Fr. F (8.9 g) was separated on a silica gel column (100 g), employing solvent gradient EtOAc–CH2Cl2 (5:95–50:50), and eight subfractions (F1–F8) were collected. Subfraction F5 (2.1 g) was further chromatographed on a silica gel column (60 g), eluting with EtOAc-CH2Cl2 (5:95–50:50), and nine subfractions (F5/1–F5/9) were collected. Subfractions F5/6 (5 mg) and F5/7 (73 mg) were combined and rechromatographed on a silica gel column (9 g), eluting with EtOAc–CH2Cl2 (1:10), and subfraction 6 was proved to be cambodine A (1) (48 mg). Subfraction 8 (22 mg, 50–100% (v/v) EtOAc–CH2Cl2) was further separated on a Sephadex LH-20 column, eluting with MeOH, to give cambodine B (4) (6.6 mg). Subfraction F7 (535 mg) was further chromatographed on a silica gel column (19 g), eluting with a gradient system of hexanes, CH2Cl2 and MeOH, and six subfractions (F7a–F7f) were collected to give cambodine C (2) (4.4 mg). Subfraction 3 (130 mg) was further chromatographed on a silica gel column (5 g), eluting with CH2Cl2–MeOH (80:20) to obtain more of 2 (12.8 mg).
A portion of the MeOH extract (30.7 g) was fractionated by quick column chromatography (silica gel, 170 g), eluting with EtOAc–CH2Cl2 (50:50), EtOAc, MeOH and H2O–MeOH (50:50) to yield four major fractions (Fr. J–M). Fr. J (0.8 g) was applied to a silica gel column (20 g) and eluted with a MeOH–CH2Cl2 (1:99–30:70) gradient system to yield 10 subfractions (J1–J10). Subfraction J2 was purified by CC over silica gel (eluted with MeOH–CH2Cl2, 3:97–15:85 gradient system) followed by a Sephadex LH-20 column (eluted with MeOH) to yield 7 (11 mg) as colourless needles. Fr. K (11 g) was chromatographed on a silica gel column (16 g) to give six subfractions (K1–K6). Subfraction K2 (163 mg) was separated on a silica gel column (7 g) eluting with EtOAc–CH2Cl2 (5:95–15:85) to give more of frangufoline (7) (16.3 mg) and lotusanine B (8) (7.5 mg).
Another portion of the MeOH extract (225 g) was suspended in 1% NaCl (100 mL) and then partitioned with EtOAc (6 × 150 mL), combined and concentrated to give a detannified EtOAc soluble fraction (12.7 g) as a pale brown paste. This detannified EtOAc soluble fraction was chromatographed on a silica gel column (150 g) to give six major fractions (Fr. N–S). Fr. O (1.2 g) was further separated on a gel column (29 g), eluted with EtOAc–CH2Cl2 (5:95–13:87), to provide 17 subfractions. Compound 5 (29.4 mg) was separated after the subfraction O12 (85 g) was rechromatographed on silica gel (8 g), eluting with EtOAc–CH2Cl2 (10:90). Subfraction O14 (203 mg) was separated on a Sephadex LH-20 column, eluting with MeOH–CH2Cl2, 50:50 to give compounds 3 (7.0 mg) and 6 (35.2 mg).
938/7921, number of observations [I > 2σ(I)] 7250, final R indices [I > 2σ (I)]: R1 = 0.0427, wR2 = 0.0893. The structure was solved by the direct method using SIR9751 and refined with a full-matrix least-squares calculation on F2 using SHELXL-97.52 Crystallographic data have been deposited at the Cambridge Crystallographic Data Centre under the reference number CCDC 1469415.Calculation of ECD spectra
Conformational analysis of all structures were carried out, the ground state geometries were computed at the M062X/6-311G level of theory. Excited states were performed by TD-DFT using M062X/6-311G(d,p) method. Geometry optimization and TD-DFT computations were both carried out with CPCM solvation model in MeOH solution. All quantum chemical calculations were performed with Gaussian 09 programs.53 The ECD spectra were simulated with overlapping Gaussian functions with sigma (σ) = 0.25 eV fitting parameter using Gauss Sum program.54 Analysis of the excited states was carried out with Gauss Sum program.Bioassay procedure
The antimalarial activity was assayed against the parasite Plasmodium falciparum (K1, multidrug resistant strain), which was cultured continuously using the method of Trager and Jensen.45 An in vitro quantitative assessment of the antiplasmodial activity was performed by means of the microculture radioisotope technique based upon the method described by Desjardins.46 The inhibitory concentration that causes a 50% reduction in parasite growth was indicated by the in vitro uptake of 3[H]-hypoxanthine by P. falciparum. Under the same test system, the standard compound, dihydroartemisinin, showed an IC50 value of 4.29 nM. By employing the microplate Alamar blue assay described by Collins and Franzblau,47 the antimycobacterial activity was evaluated against Mycobacterium tuberculosis H37Ra (purchased from ATCC). The standard drugs for the antimycobacterial assay, isoniazid and kanamycin sulfate, showed MICs of 0.44 and 4.29 μM, respectively. By using the previously described colorimetric method,48 the cytotoxicity of the sample was determined. The reference substance, ellipticine, exhibited cytotoxic activity against human epidermod carcinoma (KB, ATCC CCL-87), human breast cancer (BC-1, ATCC11778), and human small cell lung cancer (NCI-H187, ATCC CRL-5804) cells, with IC50 values of 5.39, 5.92, and 1.58 μM, respectively. The cytotoxicity against an African green monkey kidney (Vero) cell line was evaluated by green fluorescent protein (GFP) detection, and ellipticine was used as a positive control.55Conclusions
The present study revealed that the root bark of Z. cambodiana is a rich source of 14-membered ring cyclopeptide alkaloids. Six new 14-membered cyclopeptide alkaloids, cambodines A–F (1–6) along with two known cyclopeptides, frangufoline and lotosanine B were obtained. Compounds 1 and 3 are rare 5(14)-type alkaloids possessing an imidazolidin-4-one ring in the terminal unit. Interestingly, the in vitro antimalarial assay disclosed that only the cyclopeptide with the 5-(sec-butyl)-1,2-dimethylimidazolidin-4-one exhibited significant activity. Some of the isolated alkaloids displayed antimycobacterial and cytotoxic properties, and most of them were nontoxic to Vero cells. Other bioactivity evaluations of these Ziziphus constituents are under active investigation.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by The Royal Golden Jubilee PhD (RGJ) Program (PHD/0326/2552), The Thailand Research Fund and the Center of Excellence for Innovation in Chemistry (PERCH-CIC), Office of the Higher Education Commission. Partial support from the Faculty of Science, Srinakharinwirot University is gratefully acknowledged. P. K. thanks Mahidol University for financial support. We are indebted to the Bioassay Research Facility of BIOTEC for the bioactivity assays and to the Department of Chemistry, Ramkhamhaeng University for the measurements of the ESIMS and HRESIMS spectra. The authors would like to acknowledge National e-Science Infrastructure Consortium for providing computing resources that have partly contributed to the research results reported within this paper.References
- B. Bhattachachryya and B. M. Johri, Flowering Plants, Taxonomy and Phylogeny, Narosa Publishing House, New Delhi, 1998, pp. 326–328 Search PubMed.
- T. Smitinand, Thai plant names (Revised edition), The Forest Herbarium, Bangkok, 2014, pp. 564–565 Search PubMed.
- K. B. Kang, H. W. Kim, J. W. Kim, W. K. Oh, J. Kim and S. H. Sung, J. Nat. Prod., 2017, 80, 1048–1054 CrossRef PubMed.
- S. R. Wang and W. S. Fang, Curr. Top. Med. Chem., 2009, 9, 1581–1596 CrossRef PubMed.
- Q.-H. Gao, C.-S. Wu and M. Wang, J. Agric. Food Chem., 2013, 61, 3351–3363 CrossRef PubMed.
- B. Yang, H. Yang, F. Chen, Y. Huad and Y. Jiang, Analyst, 2013, 138, 6881–6888 RSC.
- E. Tuenter, V. Exarchou, S. Apers and L. Pieters, Phytochem. Rev., 2017, 16, 623–637 CrossRef.
- H. R. El-Seedi, M. H. Zahra, U. Goransson and R. Verpoorte, Phytochem. Rev., 2007, 6, 143–165 CrossRef.
- N.-H. Tan and J. Zhou, Chem. Rev., 2006, 106, 840–895 CrossRef PubMed.
- M. A. Beg, U. V. S. Teotia and S. Farooq, J. Med. Plants Stud., 16, 4, 230–233 Search PubMed.
- M. Goyal, B. P. Nagori and D. Sasmal, Spatula DD, 2012, 2, 107–116 CrossRef.
- S. Suksamrarn, P. Panseeta, S. Kunchanawatta, T. Distaporn, S. Ruktasing and A. Suksamrarn, Chem. Pharm. Bull., 2006, 54, 535–537 CrossRef PubMed.
- P. Panseeta, K. Lomchoey, S. Prabpai, P. Kongsaeree, A. Suksamrarn, S. Ruchirawat and S. Suksamrarn, Phytochemistry, 2011, 72, 909–915 CrossRef PubMed.
- S. Suksamrarn, N. Suwannapoch, N. Aunchai, M. Kuno, P. Ratananukul, R. Haritakun, C. Jansakul and S. Ruchirawat, Tetrahedron, 2005, 61, 1175–1180 CrossRef.
- K. Tangjitman, C. Wongsawad, K. Kamwong, T. Sukkho and C. Trisonthi, J. Ethnobiol. Ethnomed., 2015, 11, 1–13 CrossRef PubMed.
- S. Hout, A. Chea, S.-S. Bun, R. Elias, M. Gasquet, P. Timon-David, G. Balansard and N. Azas, J. Ethnopharmacol., 2006, 107, 12–18 CrossRef PubMed.
- X. Li, T. Ohtsuki, S. Shindo, S. Sato, T. Koyano, S. Preeprame, T. Kowithayakorn and M. Ishibashi, Planta Med., 2007, 73, 1195–1196 CrossRef PubMed.
- M. A. Arai, C. Tateno, T. Hosoya, T. Koyano, T. Kowithayakorn and M. Ishibashi, Bioorg. Med.Chem., 2008, 16, 9420–9424 CrossRef PubMed.
- B. H. Han, M. H. Park and Y. N. Han, Phytochemistry, 1990, 29, 3315–3319 CrossRef.
- M. Abu-Zarga, S. Sabri and A. Al-Aboudi, J. Nat. Prod., 1995, 58, 504–511 CrossRef.
- S. D. Zhao, L. Shen, D. Q. Luo and H. J. Zhu, Curr. Org. Chem., 2011, 15, 1843–1862 CrossRef.
- X.-C. Li, D. Ferreira and Y. Ding, Curr. Org. Chem., 2010, 14, 1678–1697 CrossRef PubMed.
- C. Diedrich and S. Grimme, J. Phys. Chem. A, 2003, 107, 2524–2539 CrossRef.
- P. J. Stephens, D. M. McCann, F. J. Devlin, J. R. Cheeseman and M. J. Frisch, J. Am. Chem. Soc., 2004, 126, 7514–7521 CrossRef PubMed.
- D. M. McCann and P. J. Stephens, J. Org. Chem., 2006, 71, 6074–6098 CrossRef PubMed.
- G. Bringmann, T. Bruhn, K. Maksimenka and Y. Hemberger, Eur. J. Org. Chem., 2009, 2009, 2717–2727 CrossRef.
- J. Frelek, A. Fryszkowska, M. Kwit and R. Ostaszewski, Tetrahedron: Asymmetry, 2006, 17, 2469–2478 CrossRef.
- B.-D. Zhou, J. Ren, X.-C. Liu and H.-J. Zhu, Tetrahedron, 2013, 69, 1189–1194 CrossRef.
- A. E. Nugroho and H. Morita, J. Nat. Med., 2014, 68, 1–10 CrossRef PubMed.
- C.-Y. Mang, C.-P. Liu, G.-M. Liu, B. Jiang, H. Lan, K.-C. Wub, Y. Yan, H.-F. Li, M.-H. Yang and Y. Zhao, Spectrochim. Acta A., 2015, 136, 1401–1408 CrossRef PubMed.
- B. Komjáti, Á. Urai, S. Hosztafi, J. Kökösi, B. Kováts, J. Nagy and P. Horváth, Spectrochim. Acta A., 2016, 155, 95–102 CrossRef PubMed.
- D. C. Gournelis, G. G. Laskaris and R. Verpoorte, in Progress in the Chemistry of Organic Natural Products, ed. W. Herz, H. Falk, G. W. Kirby, R. E. Moore and C. H. Tamm, Springer-Verlag/Wein, New York, 1998, 75, pp. 1–179 Search PubMed.
- R. Tschesche, M. Elgamal and G. Eckhardt, Chem. Ber., 1977, 110, 2649–2655 CrossRef.
- A. H. Shah, V. B. Pandey, G. Eckhard and R. Tschesche, Phytochemistry, 1985, 24, 2765–2767 CrossRef.
- H. Lin, C.-H. Chen, B.-J. You, K. C. S. Chen Liu and S.-S. Lee, J. Nat. Prod., 2000, 63, 1338–1343 CrossRef.
- A. F. Morel, G. Maldaner, V. Ilha, F. Missau, U. F. Silva and I. I. Dalcol, Phytochemistry, 2005, 66, 2571–2576 CrossRef PubMed.
- G. Maldaner, P. Marangon, V. Ilha, M. S. B. Caro, R. A. Burrow, I. I. Dalcol and A. F. Morel, Phytochemistry, 2011, 72, 804–809 CrossRef PubMed.
- J. Han, C.-J. Ji, W.-J. He, Y. Shen, Y. Leng, W.-Y. Xu, J.-T. Fan, G.-Z. Zeng, L.-D. Kong and N.-H. Tan, J. Nat. Prod., 2011, 74, 2571–2575 CrossRef PubMed.
- M. A. Mostardeiro, V. Ilha, J. Dahmer, M. S. B. Caro, I. I. Dalcol, U. F. da Silva and A. F. Morel, J. Nat. Prod., 2013, 76, 1343–1350 CrossRef PubMed.
- R. Tschesche, J. Rheingans and H.-W. Fehhaber, Chem. Ber., 1967, 100, 3924–3936 CrossRef PubMed.
- R. Tschesche, H. Last and H.-W. Fehhaber, Chem. Ber., 1967, 100, 3937–3943 CrossRef PubMed.
- B. H. Han, M. H. Park and Y. N. Han, Pure Appl. Chem., 1989, 61, 443–448 CrossRef.
- D. Xiao, S. P. East and M. M. Joullie, Tetrahedron Lett., 1998, 39, 9631–9632 CrossRef.
- P. L. de Oliveira, C. M. A. Tanaka, L. Kato, C. C. da Silva, R. P. Medina, A. P. Moraes, J. R. Sabino and C. M. A. de Oliveira, J. Nat. Prod., 2009, 72, 1195–1197 CrossRef PubMed.
- W. Trager and J. B. Jensen, Science, 1976, 193, 673–675 Search PubMed.
- R. E. Desjardins, C. J. Canfield, J. D. Haynes and J. D. Chulay, Antimicrob. Agents Chemother., 1979, 17, 710–718 CrossRef.
- L. Collins and S. G. Franzblau, Antimicrob. Agents Chemother., 1997, 41, 1004–1009 Search PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenny and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef PubMed.
- E. Tuenter, V. Exarchou, A. Baldé, P. Cos, L. Maes, S. Apers and L. Pieters, J. Nat. Prod., 2016, 79, 1746–1751 CrossRef PubMed.
- M. Toumi, V. Rincheval, A. Young, D. Gergeres, E. Turos, F. Couty, B. Mignotte and G. Evano, Eur. J. Org. Chem., 2009, 3368–3386 CrossRef.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Cryst., 1999, 32, 115–119 CrossRef.
- G. M. Sheldrick, Acta Cryst., 2008, A64, 112–122 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Rev B01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- L. Hunt, M. Jordan, M. De Jesus and F. M. Wurm, Biotechnol. Bioeng., 1999, 65, 201–205 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1469415. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra13050c |
| This journal is © The Royal Society of Chemistry 2018 |