DOI:
10.1039/C7RA13006F
(Paper)
RSC Adv., 2018,
8, 8747-8754
The surface chemical composition effect of a polyacrylic acid/polyvinyl alcohol nanofiber/quartz crystal microbalance sensor on ammonia sensing behavior
Received
3rd December 2017
, Accepted 12th February 2018
First published on 27th February 2018
Abstract
Polyacrylic acid (PAA)/polyvinyl alcohol (PVA)-based quartz crystal microbalance (QCM) ammonia sensors were fabricated by depositing composite PAA/PVA nanofibrous substrates onto QCM gold electrodes. Morphological analysis of the PAA/PVA substrates revealed a homogenous smooth surface and similar specific surface areas. X-ray photoelectron spectroscopy results indicated their distinct chemical properties with different carboxyl group contents on the surface. The ammonia sensing tests demonstrated the significant effect of the surface chemical characteristics on ammonia-sensing sensitivity, and the sensing process was proven to be derive from a monolayer adsorption mechanism. Because of the inherent high specific surface areas (SSAs) and three-dimensional porous architecture, the prepared PAA/PVA nanofiber-based QCM sensors exhibited ultrahigh sensitivity (100 ppb) and rapid response (several seconds) with good selectivity and repeatability in terms of ammonia detection.
1. Introduction
As an industrial refrigerant and raw material in the production of fertilizers and nitrogenous chemicals, ammonia plays an important role in agricultural and industrial areas over the past decades.1 However, it is toxic and hazardous to human beings and ecosystems. For example, under long-term exposure to trace ammonia at the parts per million level, humans could face harm to their skin, eyes, and respiratory tract.2,3 Hence, the development of sensors to detect accidental ammonia leakage in the environment has become important for ensuring human safety, and various types of ammonia sensors based on electrical-, optical-, mass-sensitivity have been fabricated in recent years.4–6
Electrochemical sensors are most widely studied because of their cost effectiveness and high sensitivity.7 Their sensing mechanism involves the detection of the conductivity variation resulting from the redox reaction of ammonia and the sensing material on the surface of the sensitive membrane.8 Metal oxides such as SnO2, WO3 and TiO2 are widely used as sensitive materials for ammonia detection, but poor sensing selectivity and high operation temperature limit their application.9,10 Conducting polymers, such as polyaniline,11 polythiophene,12 and polypyrrole,7 have also been proposed as sensing materials because of their exceptional advantages such as high conductivity, easy preparation, and capability to operate at room temperature, in comparison to metal oxide sensors. However, the reaction of ammonia with these polymers is irreversible, leading to the decrease in sensitivity during long-term detection.13 In addition, many efforts have been made to develop optical gas sensors because of their ability to detect ammonia gas with high sensitivity and fast response rate dependent on the reversible interaction of ammonia molecules with optical sensing materials.14 However, most optical sensors are complex and expensive, and not suitable for miniaturization and integration.15
Recently, mass-sensitive gas sensors based on quartz crystal microbalance (QCM) technology have been frequently mentioned because of the prominent features of QCM such as fast response, easy operation, high stability, and potential integration.16 Nanostructured materials such as nanowires,17 nanotubes,18 nanoparticles,16 and nanofibers,19 are widely chosen as sensing materials for QCM sensors to detect harmful gases, in order to take advantage of their large specific surface areas (SSAs). Compared with other fabrication technologies, electrospinning is generally regarded as an effective, facile, and low cost approach for the manufacture of high-SSA nanofibers and three dimensional (3D) structures, which makes them promising for the fabrication of high sensitivity gas sensors.20–24 In 2005, Ding et al. first integrated electrospun nanofibers with QCM to establish nanofiber/QCM sensing systems for ammonia detection.25 The prepared electrospun polyacrylic acid (PAA) nanofiber/QCM ammonia sensors exhibited quick response, good selectivity, as well as relatively low detection limit (130 ppb). Subsequently, Horzum et al. broadened the detection range for nanofiber/QCM gas sensing systems, and reported that trace amounts of volatile organic compounds (VOC) such as benzene, ethanol, and methylene chloride could be effectively detected using metal oxides as raw materials to fabricate inorganic nanofibers.26
In general, sensitivity has been the focus of researchers in terms of harmful gases detection. The SSA of sensing substrate is well accepted as the critical factor to tailor the sensitivity of QCM sensors.27 Therefore, to enhance the sensitivity of nanofiber/QCM gas sensors, Wang et al. developed two-dimensional nanofiber/nets (NFNs) as sensing substrates.28 The unique NFN membrane comprises interlinked spider-web-like nano-nets with diameters of 10–30 nm to provide a large SSA (31.23 m2 g−1), and as a result, the formaldehyde detection limit was as low as 50 ppb. Graphene oxide, a 2D material with ultrahigh SSA (∼2630 m2 g−1), was also blended into polystyrene nanofibers serving as the sensing substrate, and the fabricated sensor exhibited a high SSA of 24.64 m2 g−1 and a low ammonia detection limit of 1 ppm.29 Thus, increasing the SSA of sensing substrates is widely regarded as an effective approach to improve the sensitivity of nanofiber/QCM gas sensors for research, but there is limited knowledge of the effect of other factors on sensitivity.
The above results for nanofiber/QCM gas sensors have spurred our interest in exploring other factors that can affect the ammonia sensing sensitivity, which may provide more information to understand the ammonia adsorption behavior on the surfaces of nanofibers. Therefore, in the present study, PAA was blended with polyvinyl alcohol (PVA) in different ratios to fabricate PAA/PVA composite nanofibers as sensing materials. By optimizing the electrospinning process, the prepared PAA/PVA nanofibers exhibited homogenous morphology and similar SSA, whereas there were different ratios of carboxyl groups on the surface of PAA/PVA nanofibers. The sensitivity of ammonia detection for PAA/PVA nanofibers/QCM sensors was explored in order to study the effect of the distinct interfaces in the chemical composition of sensing materials on the ammonia detection sensitivity. In addition, the selectivity and repeatability of PAA/PVA nanofibers/QCM ammonia sensors were also investigated to verify the practicability of the sensors. To the best of our knowledge, this work is the first study to discuss the effect of the surface chemical characteristic of sensing materials on the sensitivity for nanofiber/QCM gas sensors.
2. Experimental
2.1 Materials
Poly(acrylic acid) (PAA) aqueous solution of 25 wt% (Mw = 240 kDa) was purchased from J&K Scientific Ltd. in Shanghai (China). Poly(vinyl alcohol) (PVA-1788) with an alcoholysis degree of 87–89% was obtained from Sigma-Aldrich Corporation. Distilled water with a resistance of 18.2 MΩ was used as the solvent. Volatile organic compounds (VOCs), including analytical grade acetone, ethanol, dichloromethane, formaldehyde, and tetrahydrofuran, were obtained from Sinopharm Chemical Regent Corporation in Shanghai (China). Ammonia (analytical grade, degree of purity: 99.999%) and the interfering gases (NO, NO2, H2S, CO, CO2 and SO2, degrees of purity: 99.9%) were supplied by Gaonengda Chemical Industry Co. Ltd. (Zhaoqing, China).
2.2 Preparation of spinning solutions
A 10 wt% PVA aqueous solution was obtained at 60 °C with vigorous stirring for 10 h. To fabricate the PAA/PVA composite nanofibers with distinct interfaces in chemical composition, different volume ratios of PAA solution (25 wt%) and PVA solution (10 wt%) were blended together according to the ingredient listing in Table 1, and the corresponding samples were named as F0–F10. The obtained mixed solutions were stirred for 20 min to fully disperse the PAA/PVA polymers.
Table 1 The operation conditions (feeding rate and voltage) and BET surface data corresponding to different polymer solutions with various volume ratios
| Sample |
F0 |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
| The volume ratio of PAA/PVA |
0/10 |
1/9 |
2/8 |
3/7 |
4/6 |
5/5 |
6/4 |
7/3 |
8/2 |
9/1 |
10/0 |
| Feeding rate (mL h−1) |
1 |
0.5 |
0.5 |
0.5 |
0.5 |
0.5 |
0.5 |
1 |
0.5 |
0.5 |
0.5 |
| Applied voltage (kV) |
15 |
15 |
15 |
15 |
15 |
15 |
15 |
15 |
15 |
15 |
10 |
| Mean diameter ± SD (nm) |
218 ± 40 |
301 ± 30 |
327 ± 28 |
313 ± 35 |
328 ± 29 |
336 ± 30 |
295 ± 23 |
346 ± 51 |
380 ± 40 |
309 ± 41 |
213 ± 36 |
| Specific surface area (m2 g−1) |
5.55 |
4.14 |
4.29 |
5.63 |
5.43 |
5.35 |
5.03 |
5.08 |
5.02 |
4.28 |
5.60 |
2.3 Deposition of PAA/PVA membranes on QCM electrode
Fig. 1a illustrates the process by which the fibrous PAA/PVA membranes were deposited on the electrode of the QCM via electrospinning. A syringe loaded with the PAA/PVA mixed solution was fixed on a peristaltic pump (Model: LSP02-1B, Longer Pump® Co., China), and a size-seven needle was connected to the positive electrode of a high voltage power supply (TXR1020, TeslaMan Co., China). The distance from the syringe to the grounded electrode of the QCM (5 MHz, AT-cut quartz crystal with Au electrodes) was adjusted to 10 cm. The electrospinning operational details of the feeding rate and the applied voltage are listed in Table 1. As shown in Fig. 1a, the PAA/PVA fibers were continuously deposited on the QCM electrode, and the coating load of the PAA/PVA fibers on the surface of QCM was regulated to be around 1500 Hz, and was measured using a frequency counter (details are given in Sec. 2.4). Subsequently, fibrous PAA/PVA membrane coated QCM electrode was dried at 30 °C in vacuum for 1 h prior to the ammonia sensing test.
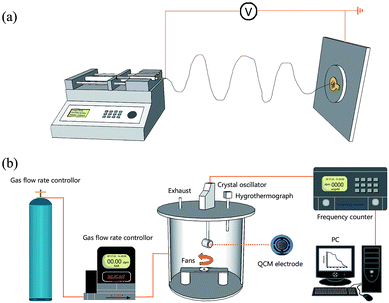 |
| | Fig. 1 Schematic diagram of the deposition of sensing PAA/PVA membranes on QCM electrode surface (a); and the ammonia-sensing system (b). | |
2.4 Apparatus for gas sensing
The schematic diagram of the gas sensing system is shown in Fig. 1b. In brief, the fibrous PAA/PVA membrane coated QCM electrode was installed in a testing chamber (10 L), in which temperature and relative humidity (RH) were monitored in real time through a Thermo-Hygrometer (Testo605-H1, Testo Ltd., Germany), and was kept at 25 °C and 55 ± 2%, respectively. During the sensing test, ultra-high purity nitrogen gas was used as the carrier gas to remove the residual test gas and regulate the humidity in the testing chamber, and the concentration of the test gas was adjusted by a flow gas meter (MCS-Series, Alicat Scientific Inc., USA) that was injected into the chamber. The resonance frequencies were monitored by a QCM digital controller (QCM-1000, Nanosensing Research System) and the data were recorded by a computer. The entire gas detection process was carried out in a constant temperature and humidity laboratory.
2.5 Equilibrium isotherm study
Sample F10 (PAA membrane) was chosen for the adsorption isotherm test because of its high ammonia sensitivity. Various ammonia concentrations, ranging from 0.1 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm, were successively injected into the testing chamber at room temperature. The frequency shifts were recorded, and the corresponding mass variations were calculated by the Sauerbrey equation,30 as follows:
000 ppm, were successively injected into the testing chamber at room temperature. The frequency shifts were recorded, and the corresponding mass variations were calculated by the Sauerbrey equation,30 as follows:| | |
Δf = −2f02Δm/A(μρ)1/2
| (1) |
here, Δf is the frequency shift of the QCM sensor (Hz), f0 is the intrinsic frequency of a blank QCM platform (5 MHz), Δm is the mass variation per unit area (ng), A is the surface area of the QCM electrode (1 cm−2), μ is the shear modulus of the quartz crystal (2.947 × 1011 dyne per cm2), and ρ is the density of quartz (2.648 g cm−3). As a result, the relationship between the frequency shift and mass variation can be described as:
2.6 Characterization
The morphology of the as-prepared fibrous PAA/PVA membranes was observed by field emission scanning electron microscopy (FE-SEM, NoVaTM Nano SEM 430, FEI Co., USA). The mean diameter of the fibers in the acquired FE-SEM images was calculated using an image-analytical software (Image-Pro Plus 6.0, Media Cybernetics, USA). Sixty fibers in each image were arbitrarily chosen for the calculation. Brunauer–Emmett–Teller (BET) analyses were performed to investigate the SSAs of the PAA/PVA membranes using a surface area analyzer (ASAP2020, Micromeritics Co., USA). The chemical composition of the fibrous PAA/PVA membranes was characterized by Fourier transform infrared spectroscopy (FT-IR, VERTEX 70, Bruker Optics, Germany). Elemental analyses were carried out using X-ray photoelectron spectrometry (XPS, ESCALAB 250Xi, Thermo, USA) to determine the carboxyl group ratio on the interface of each sample.
3. Results and discussion
3.1 Characterization of PAA/PVA nanofibers
Fig. 2 displays the FE-SEM images of PAA/PVA composite nanofibers. A continuous fibrous structure with a smooth surface (inset images on the top right corner) is clearly seen for each sample. The mean diameters and SSAs results are listed in Table 1, revealing similar average diameters ranging from 213 ± 36 nm to 380 ± 40 nm and analogical BET surface areas at a range of 4.14–5.63 m2 g−1 for F0–F10. Thus, all PAA/PVA nanofibers present a homogenous feature in the physical morphology through the optimization of the electrospinning process, and the 3D structures of these samples lead to easy accessibility to the gases, rendering the PAA/PVA nanofibrous membrane an optimal candidate for the application in gas sensing tests.29,31–33
 |
| | Fig. 2 FE-SEM images of PAA/PVA nanofibers (F0–F10, a–k). | |
The FT-IR spectra of the PAA/PVA composite nanofibrous membranes are shown in Fig. 3. The spectrum of pristine PVA fibers (F0) exhibits a characteristic peaks at 3417 cm−1 (–OH stretching), 2920 cm−1 (C–H asymmetric stretching), and 2852 cm−1 (C–H symmetric stretching). The PAA fibers (F10) characteristic peaks appear at 3434 cm−1 (–OH stretching), 2921 cm−1 (C–H asymmetric stretching), 2853 cm−1 (C–H symmetric stretching), and 1714 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching). In the PAA/PVA composite fibers (F1–F9) spectra, the representative peaks of PAA and PVA both appear in each sample, indicating the excellent compatibility of PAA and PVA polymers. In addition, with the increasing PAA ratio in PAA/PVA mixed solution from F1 to F10, the –CO characteristic peaks of PAA at 1714 cm−1 strengthen continuously.
O stretching). In the PAA/PVA composite fibers (F1–F9) spectra, the representative peaks of PAA and PVA both appear in each sample, indicating the excellent compatibility of PAA and PVA polymers. In addition, with the increasing PAA ratio in PAA/PVA mixed solution from F1 to F10, the –CO characteristic peaks of PAA at 1714 cm−1 strengthen continuously.
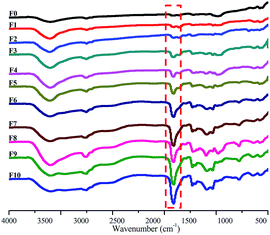 |
| | Fig. 3 FT-IR images of PAA/PVA nanofibers (F0–F10, from top to bottom). | |
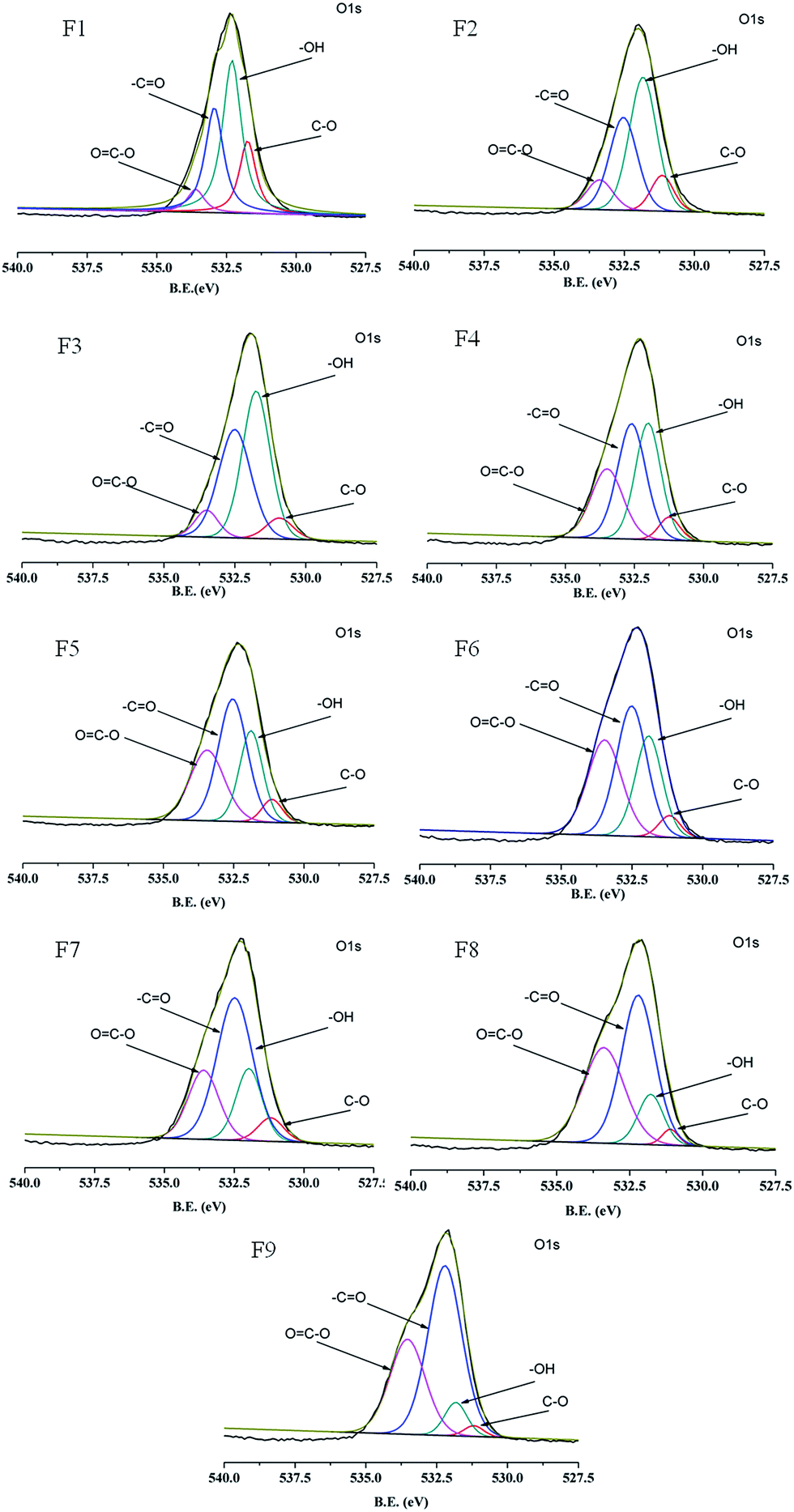
To analyze the surface chemical composition of the PAA/PVA composite nanofibers, XPS measurement is performed, and images of O 1s region recorded for samples with various PAA/PVA weight ratios are shown in Fig. 4. All the spectra present a sharp peak at a binding energy (BE) range from 527.5 to 540.0 eV, together with the relevant four fitted peaks representing four different kinds of surface chemical groups for PAA/PVA composite nanofibers. As listed in Table 2, the fitted peaks are well in agreement with those reported in the literature,34 commonly assigned to oxygen in the C–O bond at ∼531.1 eV, hydroxyl group at ∼531.9 eV, –CO bond at ∼532.8 eV, and carboxyl group around 533.3 eV, respectively. The content of the surface carboxyl group for the composite samples are calculated on the basis of the intensity percentage of the OC–O group in the total sample O 1s area, increasing from 6.1% to 37.6% for F1–F9 in according to the increase in the PAA weight ratio in the spinning solution (Table 2).
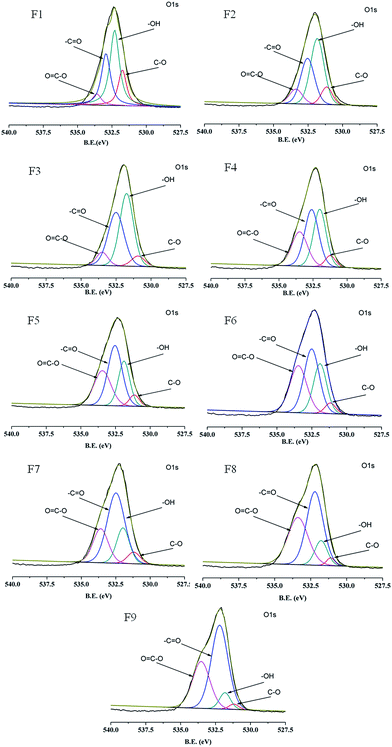 |
| | Fig. 4 XPS of the O 1s regions in the PAA/PVA nanofibers. | |
Table 2 XPS characteristics of the O 1s regions in the PAA/PVA nanofibers with different PAA/PVA feeding weight ratios
| PAA/PVA weight ratio |
Binding energy (eV) |
Peak intensityb (%) |
| C–O |
–OH |
–CO |
OC–O |
OC–O |
| Number in parentheses refer to FWHM in eV. Intensity of the peak in percentage of the total sample O 1s area. |
| F1 (1/9) |
531.4 (0.7)a |
532.2 (0.8) |
532.8 (0.8) |
533.5 (0.7) |
6.1 |
| F2 (2/8) |
531.1 (1.0) |
531.8 (1.2) |
532.5 (1.2) |
533.4 (1.2) |
10.0 |
| F3 (3/7) |
531.1 (1.2) |
531.8 (1.0) |
532.4 (1.1) |
533.1 (1.2) |
18.6 |
| F4 (4/6) |
531.2 (1.1) |
531.9 (1.1) |
532.5 (1.5) |
533.6 (1.3) |
22.0 |
| F5 (5/5) |
531.2 (0.9) |
532.0 (1.1) |
532.6 (1.3) |
533.5 (1.4) |
27.1 |
| F6 (6/4) |
531.1 (0.9) |
531.9 (1.0) |
532.5 (1.4) |
533.4 (1.4) |
28.4 |
| F7 (7/3) |
531.2 (0.9) |
531.9 (1.2) |
532.5 (1.3) |
533.4 (1.5) |
31.5 |
| F8 (8/2) |
531.2 (0.8) |
531.9 (1.0) |
532.2 (1.4) |
533.5 (1.5) |
32.9 |
| F9 (9/1) |
531.1 (0.7) |
531.8 (1.0) |
532.2 (1.4) |
533.4 (1.7) |
37.6 |
3.2 Ammonia-sensing sensitivity and adsorption isotherms
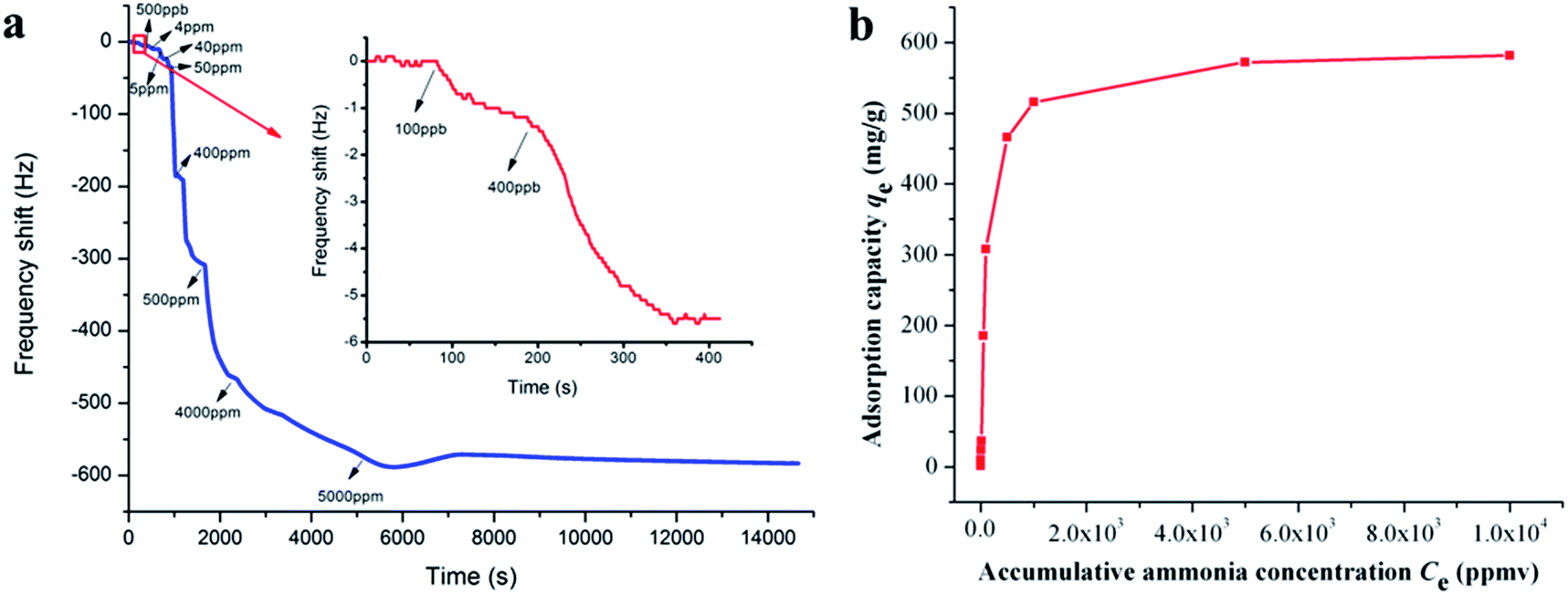
Ammonia adsorption isotherm over PAA membranes (F10) was measured to gain a deeper insight into the adsorption mechanism. As shown in Fig. 5a, when the ammonia gases ranging from 0.1 to 5000 ppm were introduced into the test chamber, the frequency shifts of QCM detector decreased dramatically within several seconds and maintained balance or only showed slight fluctuations after several minutes. The detailed frequency shifts of the PAA membranes exposed to 0.1 and 0.4 ppm of ammonia were 1.0 and 4.3 Hz (Fig. 5a, inset top right), thus demonstrating the surprisingly high sensitivity (100 ppb) and quick response (several seconds) of the PAA nanofiber membranes/QCM sensor system to ammonia. The data of 0.5, 4, 5, 40, 50, 400, 500, 4000, and 5000 ppm were 4.7, 13.8, 12.5, 148.7, 123.0, 158.0, 50.0, 56.0, and 10.0 Hz, respectively. It can be concluded that the response amplitude of the sensor gradually increased with ammonia gas concentration, but high concentrations of last two injections in less variation of the responses due to the saturated adsorption of ammonia molecules.29 The total frequency shift at saturation should be cumulative sum of the frequency shifts of each ammonia injection, and the saturated ammonia content was calculated by the eqn (2) as 32.9 ng.
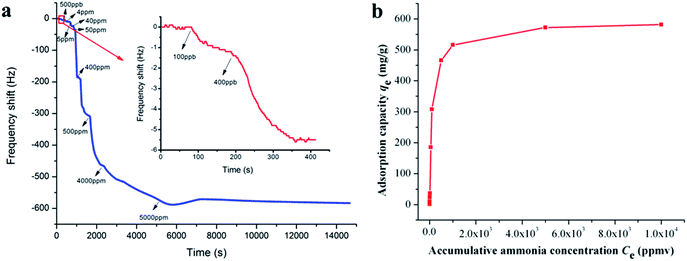 |
| | Fig. 5 Frequency shifts of QCM sensors coated with PAA nanofibers (F10) upon exposure to increasing ammonia concentrations from 0.1 ppm to 5000 ppm (the inset shows the sensing limit starting from 0.1 ppm) (a); and the corresponding adsorption isotherm curve exposed to various accumulative ammonia concentrations (Ce) from 0.1 to 10000 ppm (b). | |
The adsorption isotherm curve was evaluated by exploring ammonia adsorption capacity (qe) exposed to various accumulative ammonia concentrations (Ce) from 0.1 to 10000 ppm, as shown in Fig. 5b. qe was the ratio of adsorption ammonia mass to PAA/PVA substrates mass that was calculated according to Sauerbrey equation. The classical Langmuir, Freundlich, Tempkin and Toth models were chosen to fit the adsorption isotherm, and their corresponding calculated parameters are represented in Table 3. Assuming a free surface on the adsorbent with constant adsorption energy, the Langmuir model is suitable for a monolayer homogeneous adsorption process. While the Freundlich model is a multilayer adsorption process on the heterogeneous surface with distinct adsorption heat and affinities.35,36 Tempkin considered the effects of some indirect adsorbate/adsorbent interactions and suggested that because of these interactions the heat of adsorption of all the molecules in the layer would decrease linearly with coverage. Toth model was derived from potential theory and was applicable to heterogeneous adsorption. A comparison of the correlation coefficient (R2) clearly showed that the monolayer adsorption models fit the adsorption isotherm data better than other models. This result suggested that a monolayer adsorption mechanism controls the adsorption process of the interface between ammonia and the PAA nanofiber membranes.
Table 3 Isotherm models for ammonia adsorption by PAA nanofiber membranes
| Isotherms model |
Liner form |
q∞ (mg g−1) |
KL (L mg−1) |
R2 |
| Langmuir |
1/qe = 1/q∞ + 1/(KL × q∞ × Ce) |
3.8 |
0.0467 |
0.9991 |
| |
|
n |
KF |
R2 |
| Freundlich |
log(qe) = 1/nlog(Ce) + log(KF) |
1.8 |
160.88 |
0.9088 |
| |
|
f |
A |
R2 |
| Tempkin |
qe = 1/f × ln(A × Ce) |
0.83 |
2.75 |
0.9843 |
| |
|
KT |
B |
β |
R2 |
| Toth |
qe = KT × Ce/(1 + B × Ceβ)1/β |
2.31 |
1.26 |
0.65 |
0.9415 |
3.3. Effect of carboxyl group content on sensitivity
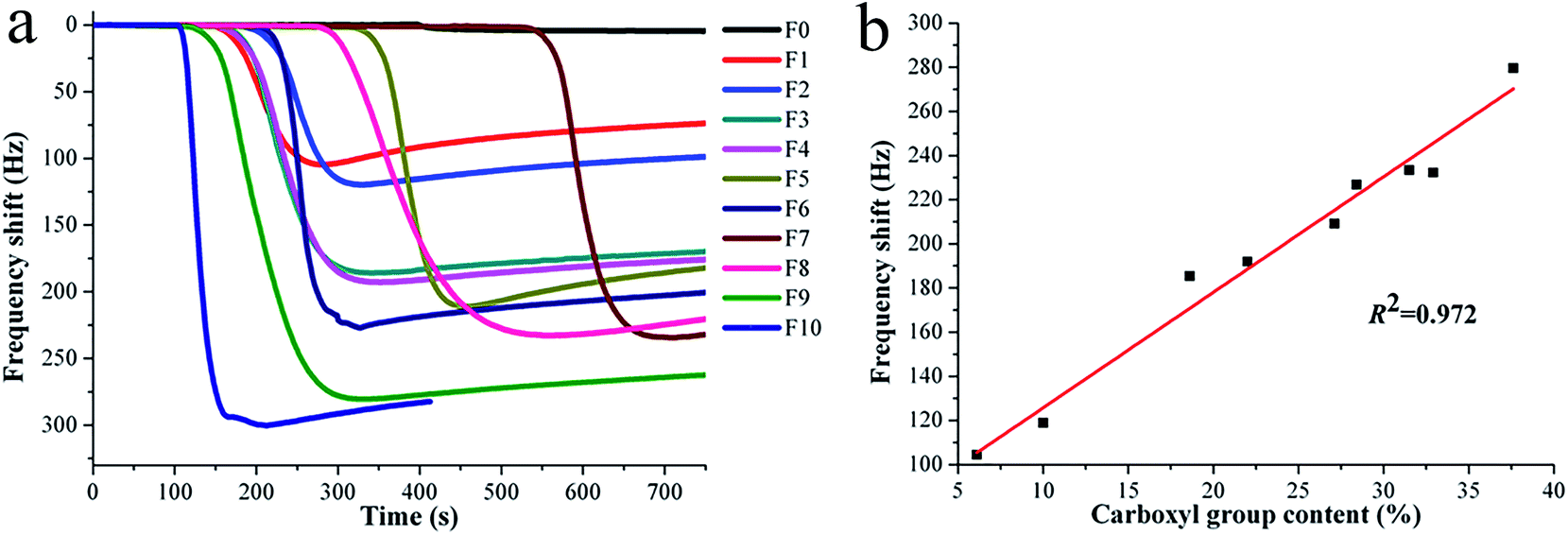
It is well known that SSA will have a significant effect on the gas sensor sensitivity.27,28 Researchers have focused their attention on exploring the relationship between SSA and sensing sensitivity in the past years. Apart from the sensitivity, the gas adsorption behavior is also a research focus for gas sensors. Based on the above facts, the SSAs of sensing substrates were tailored to have similar values by adjusting the electrospinning conditions in this work, and the ammonia sensing sensitivity was investigated in the view of a new perspective, i.e., the content of sensitive group on the surface of sensing substrate. Fig. 6a illustrates the dynamic responses of QCM sensors for the composite nanofibrous PAA/PVA substrates toward ammonia gas at 50 ppm. All samples except F0 (PVA substrate) present a sharp drop, and the detailed frequency shifts for F0–F10 are 4.6, 104.5, 119.0, 185.5, 192.0, 209.2, 226.9, 233.5, 232.3, 279.6, and 300.0, respectively. This result indicates that the sensing substrates with different PAA contents produced significantly different ammonia responses. As shown in Fig. 6b, the specific relationship between the frequency shift (sensing sensitivity) and carboxyl group content (sensing substrates with different PAA contents) exhibits a direct proportional linear correlation (R2 = 0.972) for samples F1–F9. This result strongly supported the above monolayer adsorption hypothesis, with the increasing of carboxyl groups on the surface of sensitive substrates, the adsorbed ammonia mass (frequency shift) increased in linearity.
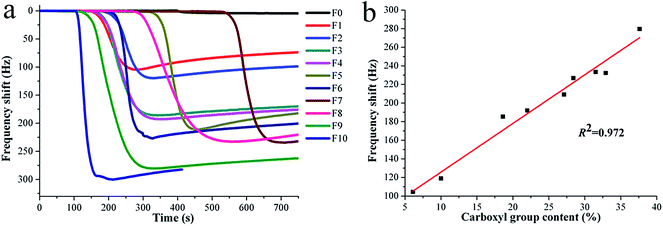 |
| | Fig. 6 Frequency shifts of PAA/PVA nanofiber/QCM sensors (F0–F10) upon exposure to certain ammonia concentration at 50 ppm (a); and the correlation curve between frequency shifts and carboxyl group contents for samples F1–F9 (b). | |
3.4 Selectivity and repeatability of ammonia sensor
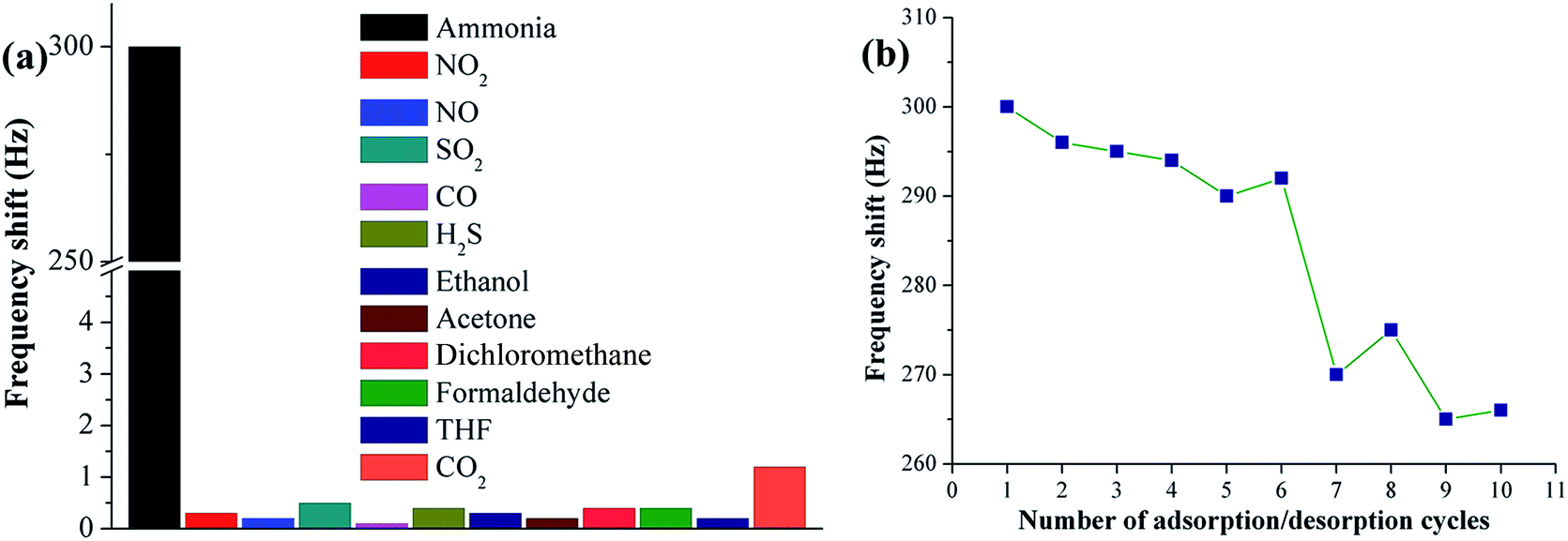
Apart from sensitivity, selectivity and repeatability are two other vital parameters for evaluating the performance of gas sensors for practical use. Sample F10 (PAA substrate) was chosen for ammonia sensing selectivity and repeatability assays because of its highest sensitivity among all samples. The selectivity test was performed in a constant temperature and humidity atmosphere (25 °C and 55 ± 2% RH) for common interfering gases, such as NO2, NO, SO2, CO, H2S, ethanol, acetone, dichloromethane, formaldehyde, tetrahydrofuran and CO2. Fig. 7a shows the responses (frequency shift) of the PAA sensor exposed to various gases at fixed concentrations of 50 ppm. The observed response for ammonia is 300 Hz, and those for NO2, NO, SO2, CO, H2S, ethanol, acetone, dichloromethane, formaldehyde, tetrahydrofuran and CO2 are 0.3, 0.2, 0.5, 0.1, 0.4, 0.3, 0.2, 0.4, 0.4, 0.2 and 1.2 Hz, respectively. The frequency variation towards ammonia is much higher than that towards other gases, which means that the sensor is hardly influenced by other common interfering gases.
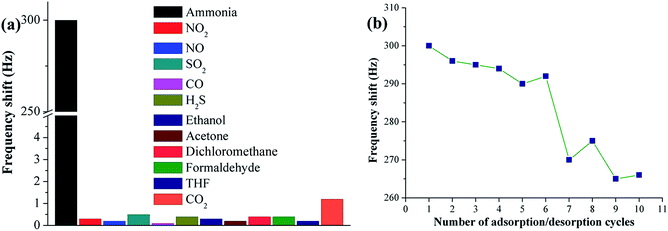 |
| | Fig. 7 Selectivity testing for PAA nanofiber (F10) sensor upon exposure to different gases at a fixed concentration of 50 ppm (a); repeatability testing for F10 sensor upon repetitive exposure to ammonia at 50 ppm (b). | |
To investigate the repeatability of the PAA sensor, the sensor was exposed to ammonia at a fixed concentration of 50 ppm. When the QCM frequency reached equilibrium after each ammonia injection, the PAA-coated electrode was purged with high nitrogen flow until complete ammonia desorption. Fig. 7b shows the sensing repeatability by exposing the sensor to repeated ammonia adsorption/desorption cycles. The sensor was cycled 10 times while QCM frequency showed similar shifts during the initial six rounds, indicating its outstanding repeatability.
4. Conclusions
In summary, a series of PAA/PVA-based QCM sensors with different carboxyl group contents on the surface of the sensing substrates were successfully prepared via adjusting the PAA/PVA blend ratios. The tailored PAA/PVA nanofibrous substrates exhibited homogenous surface and similar SSA, but distinct ammonia-sensing sensitivity, which could be effectively enhanced by increasing the carboxyl group content. Importantly, the monolayer adsorption mechanism was validated to control the specific adsorption process for ammonia gas on the surface of the PAA/PVA substrates. Moreover, the PAA sensor demonstrated ultrahigh sensitivity as low as 100 ppb with excellent selectivity and repeatability as well, together suggesting its practical application in trace ammonia detection.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Foundation of Ministry of Industry and Information Technology ([2016]-562), and the Foundation of Higher Education of Guangdong, China (No. 2016KTSCX146).
Notes and references
- T. Cai, S. Y. Park and Y. Li, Renewable Sustainable Energy Rev., 2013, 19, 360–369 CrossRef CAS.
- R. Nkoa, Agron. Sustainable Dev., 2014, 34, 473–492 CrossRef.
- Z. Y. Pang, J. P. Fu, L. Luo, F. L. Huang and Q. F. Wei, Colloids Surf., A, 2014, 461, 113–118 CrossRef CAS.
- L. Q. Li, P. Gao, M. Baumgarten, K. Müllen, N. Lu, H. Fuchs and L. F. Chi, Adv. Mater., 2013, 25, 3419–3425 CrossRef CAS PubMed.
- F. Tavoli and N. Alizadeh, Sens. Actuators, B, 2013, 176, 761–767 CrossRef CAS.
- Z. Q. Wu, X. D. Chen, S. B. Zhu, Z. W. Zhou, Y. Yao, W. Quan and B. Liu, Sens. Actuators, B, 2013, 178, 485–493 CrossRef CAS.
- O. S. Kwon, S. J. Park, H. Yoon and J. Jang, Chem. Commun., 2012, 48, 10526–10528 RSC.
- B. H. King, A. Gramada, J. R. Link and M. J. Sailor, Adv. Mater., 2007, 19, 4044–4048 CrossRef CAS.
- B. Timmer, W. Olthuis and A. Van den Berg, Sens. Actuators, B, 2005, 107, 666–677 CrossRef CAS.
- G. K. Mani and J. B. B. Rayappan, Sens. Actuators, B, 2013, 183, 459–466 CrossRef CAS.
- Y. X. Zhang, J. J. Kim, D. Chen, H. L. Tuller and G. C. Rutledge, Adv. Funct. Mater., 2014, 24, 4005–4014 CrossRef CAS.
- M. Z. Dai, Y. H. Chen, M. Y. Chuang, H. W. Zan and H. F. Meng, Sensors, 2014, 14, 16287–16295 CrossRef PubMed.
- Y. L. Shang, X. B. Wang, E. C. Xu, C. L. Tong and J. M. Wu, Anal. Chim. Acta, 2011, 685, 58–64 CrossRef CAS PubMed.
- A. Airoudj, D. Debarnot, B. Bêche and F. Poncin-Epaillard, Anal. Chem., 2008, 80, 9188–9194 CrossRef CAS PubMed.
- J. Kim, S. H. Lim, Y. Yoon, T. D. Thangadurai and S. A. Yoon, Tetrahedron Lett., 2011, 52, 2645–2648 CrossRef CAS.
- Y. Hwang, H. Sohn, A. Phan, Q. M. Yaghi and R. N. Candler, Nano Lett., 2013, 13, 5271–5276 CrossRef CAS PubMed.
- X. P. Chen, C. K. Y. Wong, C. A. Yuan and G. Q. Zhang, Sens. Actuators, B, 2013, 177, 178–195 CrossRef CAS.
- A. Ndiaye, P. Bonnet, A. Pauly, M. Dubois, J. Brunet, C. Varenne, K. Guerin and B. Lauron, J. Phys. Chem. C, 2013, 117, 20217–20228 CAS.
- X. F. Wang, J. L. Wang, B. Ding, J. Y. Yu, G. Sun, W. J. Luo and G. Zheng, Nanoscale, 2012, 4, 7585–7592 RSC.
- S. Chen and G. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 6473–6477 CAS.
- S. Xiao, M. Shen, R. Guo, S. Wang and X. Shi, J. Phys. Chem. C, 2009, 113, 18062–18068 CAS.
- S. Xiao, M. Shen, H. Ma, R. Guo, M. Zhu, S. Wang and X. Shi, J. Appl.
Polym. Sci., 2010, 116, 2409–2417 CAS.
- S. Xiao, M. Shen, R. Guo, Q. Huang, S. Wang and X. Shi, J. Mater. Chem., 2010, 20, 5700–5708 RSC.
- S. Xiao, H. Ma, M. Shen, S. Wang, Q. Huang and X. Shi, Colloids Surf., A, 2011, 381, 48–54 CrossRef CAS.
- B. Ding, M. Yamazaki and S. Shiratori, Sens. Actuators, B, 2005, 106, 477–483 CrossRef CAS.
- N. Horzum, D. Tascioqlu, C. Özbek, S. Okur and M. M. Demir, New J. Chem., 2014, 38, 5761–5768 RSC.
- B. Ding, X. F. Wang, J. Y. Yu and W. R. Wang, J. Mater. Chem., 2011, 21, 12784–12792 RSC.
- X. F. Wang, B. Ding, J. Y. Yu, Y. Si, S. B. Yang and G. Sun, Nanoscale, 2011, 3, 911–915 RSC.
- Y. T. Jia, L. Z. Chen, H. Yu, Y. M. Zhang and F. C. Dong, RSC Adv., 2015, 5, 40620–40627 RSC.
- G. Sauerbrey, Z. Phys., 1959, 155, 206–222 CrossRef CAS.
- S. Xiao, M. Shen, H. Ma, X. Fang, Q. Huang, W. Weber and X. Shi, J. Nanosci. Nanotechnol., 2011, 11, 5089–5097 CrossRef CAS PubMed.
- H. Ma, Y. Huang, M. Shen, D. Hu, H. Yang, M. Zhu, S. Yang and X. Shi, RSC Adv., 2013, 3, 6455–6465 RSC.
- H. Ma, Y. Huang, M. Shen, R. Guo, X. Cao and X. Shi, J. Hazard. Mater., 2012, 211–212, 349–356 CrossRef CAS PubMed.
- F. Li, L. H. Zhang, D. G. Evans and X. Duan, Colloids Surf., A, 2004, 244, 169–177 CrossRef CAS.
- L. W. Drahushuk and M. S. Strano, Langmuir, 2012, 28, 16671–16678 CrossRef CAS PubMed.
- Y. D. Li, H. H. Yi, X. L. Tang, F. R. Li and Q. Yuan, Chem. Eng. J., 2013, 229, 50–56 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a