
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: SNHG5 promotes proliferation and induces apoptosis in melanoma by sponging miR-155
Lu Yan *a,
Suihai Wangb,
Yue Lic,
Linda Tognettid,
Rui Tana,
Kang Zenge,
Elisa Pianigianid,
Xiangbin Mia,
Hui Lia,
Michele Fimianid and
Pietro Rubegni*d
*a,
Suihai Wangb,
Yue Lic,
Linda Tognettid,
Rui Tana,
Kang Zenge,
Elisa Pianigianid,
Xiangbin Mia,
Hui Lia,
Michele Fimianid and
Pietro Rubegni*d
aDepartment of Dermatology, Zhujiang Hospital of Southern Medical University, Guangzhou, 510000, China. E-mail: michelley8051@126.com; Tel: +86-020-61642833
bSchool of Biotechnology, Southern Medical University, Guangzhou, 510000, China
cDepartment of Laboratory, Nanfang Hospital, Southern Medical University, Guangzhou, 510000, China
dDepartment of Medical, Surgical and Neuro Sciences, Section of Dermatology, University of Siena, Policlinico Le Scotte Viale Bracci, Siena, 53100, Italy. E-mail: rubegni@unisi.it; Tel: +39-0577-40190
eDepartment of Dermatology, Nanfang Hospital, Southern Medical University, Guangzhou, 510000, China
First published on 7th February 2018
Abstract
Background: Melanoma is the most common malignancy of skin cancer. Small nucleolar RNA host gene 5 (SNHG5), a long non-coding RNA (lncRNA), has been demonstrated to be abnormally expressed in multiple malignances. However, the roles and molecular mechanisms of SNHG5 in melanoma progression have not been well identified. Methods: RT-qPCR assays were used to detect the expression patterns of SNHG5 and microRNA-155 (miR-155). Cell proliferation was assessed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and colony formation assays. Cell apoptosis rate was measured by flow cytometry via double-staining of fluorescein isothiocyanate (FITC)-labeled annexin V (Annexin V-FITC) and propidium iodide (PI). The interaction between SNHG5 and miR-155 was validated using bioinformatics analysis, subcellular fraction assay, luciferase assay and RNA immunoprecipitation (RIP) assay. A mouse model of melanoma was established to further verify the effect of SNHG5 on tumor growth in vivo. Results: SNHG5 expression was upregulated in melanoma tumor tissues and cell lines. Moreover, higher SNHG5 expression was associated with advanced pathogenic status and poor prognosis. Functional analysis showed that SNHG5 knockdown suppressed proliferation and facilitated apoptosis in melanoma cells. Mechanical exploration revealed that SNHG5 acted as a molecular sponge of miR-155 in melanoma cells. Restoration experiments delineated that miR-155 down-regulation partly abrogated SNHG5-knockdown-mediated anti-proliferation and pro-apoptosis effect in melanoma cells. In vivo assays further demonstrated that SNHG5 depletion hindered tumor growth through up-regulating miR-155 expression. Conclusion: SNHG5 promoted the development of melanoma by sponging miR-155 in vitro and in vivo, implying the important implication of lncRNAs in melanoma progression and providing a potential therapeutic target for melanoma.
1. Introduction
Melanoma is the most common malignancy of skin cancer with an estimated 76![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 380 new cases and 10130 death in 2016 in the United States, accounting for 91% of new skin cancer cases and 74% of skin cancer death.1 The incidence rate of melanoma has been rising by about 1.4% each year over the past 10 years.2 Moreover, patients with stage IV metastatic melanoma have a median survival time of less than 1 year.3 Although a variety of treatment strategies (such as targeted therapies, immune-based approaches, and chemoprevention) have been applied to melanomas, the prognosis of patients with advanced melanoma is still far from being satisfied.4,5 For searching more effective treatment options to prevent melanoma developing, the better understanding of the molecular mechanism underlying melanoma progression is imperative.
380 new cases and 10130 death in 2016 in the United States, accounting for 91% of new skin cancer cases and 74% of skin cancer death.1 The incidence rate of melanoma has been rising by about 1.4% each year over the past 10 years.2 Moreover, patients with stage IV metastatic melanoma have a median survival time of less than 1 year.3 Although a variety of treatment strategies (such as targeted therapies, immune-based approaches, and chemoprevention) have been applied to melanomas, the prognosis of patients with advanced melanoma is still far from being satisfied.4,5 For searching more effective treatment options to prevent melanoma developing, the better understanding of the molecular mechanism underlying melanoma progression is imperative.
Long non-coding RNAs (lncRNAs), a class of transcripts longer than 200 nucleotides (nt), are associated with the development of cancers by regulating multiple cellular processes such as proliferation, metastasis and apoptosis.6 Moreover, some lncRNAs have been identified as critical regulators in the development of melanoma. For example, SPRY4 intronic transcript 1 (SPRY4-IT1) was highly expressed in melanoma, and SPRY4-IT1 knockdown resulted in the inhibition of proliferation, differentiation and invasion and the enhancement of apoptosis in melanoma cells.7 HOX transcript antisense RNA (HOTAIR) was overexpressed in metastatic melanoma tissues and higher HOTAIR expression promoted melanoma cell motility and invasion.8 Small nucleolar RNA (SnoRNA) host gene family has been reported to be important participants in determining cell fate and oncogenesis.9 For instance, growth arrest-specific 5 (GAS5), a member of snoRNA host gene family, has been identified as a tumor suppressor in various malignant tumors, such as breast cancer,10 gastric cancer,11 prostate cancer12 and melanoma.13 SnoRNA host gene 5 (SNHG5), a snoRNA-U50-associated lncRNA, belongs to snoRNA host gene family and 5′-terminal oligopyrimidine (5′-TOP) family.14 Abnormal expression of SNHG5 was also found to be involved in multiple malignant diseases, including chronic myeloid leukemia,15 gastric cancer16,17 and colorectal cancer,18 via different pathways. Moreover, Ichigozaki et al. showed that SNHG5 expression was markedly up-regulated in serum of malignant melanoma patients compared with healthy volunteers, and SNHG5 serum level was strikingly reduced after surgery,19 indicating SNHG5 was associated with a poor prognosis of melanoma patients. However, the roles and molecular mechanism of SNHG5 in melanoma progression has not been absolutely elucidated.
In the present study, we first demonstrated that SNHG5 was highly expressed in melanoma tumor tissues and cells. Moreover, higher SNHG5 level was positively associated with advanced pathological stage and poor prognosis. Hence, the roles and pathogenesis of SNHG5 in the progression of melanoma were further investigated.
2. Materials and methods
2.1. Clinical specimens and cell culture
Melanoma tumor tissues and adjacent normal tissues were collected from 22 melanomas patients (60% male, 40% female, mean age of 50 years old) in different pathological stages at our hospital. The clinical stage of melanoma patients was differentiated according to the standard of American Joint Committee on Cancer (AJCC). Resected tumors were fixed with formalin and then embedded using paraffin, followed by preserved at −80 °C refrigerator. Our study was conducted with the approval of Medical Ethics Committee of Zhujiang Hospital, Southern Medical University and complied with the Declaration of Helsinki. Every patient signed the consent form prior to participating in this project.Melanoma cell lines (HBL, A375, SK-MEL-2, CHL-1 and VMM917) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in DMEM medium (Invitrogen, Carlsbad, CA, USA) containing 10% FBS (Invitrogen). Normal human epidermal melanocytes (NHEM) were obtained from Lonza Co., Ltd (Basel, Switzerland) and cultured in medium 154 (Invitrogen) supplemented with human melanocyte growth supplement (Invitrogen).
2.2. Reagents and cell transfection
Small interfering RNAs (SiRNAs) targeting SNHG5 (si-SNHG5#1 and si-SNHG5#2) and one scramble control siRNA (si-con), miR-155 mimic and its scramble control (miR-con), miR-155 inhibitor (anti-miR-155) and its scramble control (miR-con) were purchased from GenePharma Co. Ltd (Suzhou, China). Also, full length cDNA sequences of SNHG5 were cloned into pcDNA3.1 vector (Invitrogen) to produce pcDNA3.1-SNHG5 overexpression plasmid (SNHG5). All these substances were transfected into melanoma cells using lipofectamine 2000 reagent (Invitrogen) referring to the instructions of manufacturer.2.3. RT-qPCR assays
Total RNA in melanoma tissues and cells was extracted using mirVana miRNA Isolation Kit (Ambion, Austin, TX, USA) and RNA concentration was determined by Nanodrop 2000 instrument (Thermo Fisher Scientific, Rockford, IL, USA). Then the same amount (1 μg) of RNA was employed to synthesize the cDNA first strand by PrimeScript™ 1st Strand cDNA Synthesis Kit (Takara, Dalian, China). The cDNAs of SNHG5 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were synthesized using random primer, and cDNAs of miR-155 and U6 small nuclear RNA (snRNA) were synthesized using specific reverse transcription (RT) primers. Finally, the expression analyses of SNHG5, GAPDH, miR-155 and U6 snRNA were performed using SYBR® Premix Ex Taq™ reagent (Takara) and quantitative primers. U6 snRNA and GAPDH were used for normalization of miR-155 and SNHG5 levels, respectively. RT primers sequences for miR-155 and U6 snRNA were listed as follows: miR-155, 5′-GTC GTA TCC AGT GCA GGG TCC GAG GTA TTC GCA CTG GAT ACG ACC CCC TA-3′. U6 snRNA, 5′-AAC GCT TCA CGA ATTTGC GT-3′. Quantitative primers sequences were also listed as below: miR-155, 5′-CTC GTG GT AAT GCT AAT TGT GA-3′ (forward) and 5′-GTG CAG GGT CCG AGG T-3′ (reverse); U6 snRNA, 5′-CTC GCT TCG GCA GCA CA-3′ (forward) and 5′-AAC GCT TCA CGA ATT TGC GT-3′ (reverse); GAPDH, 5′-CCA CCC ATG GCA AAT TCC ATG GCA-3′ (forward) and 5′-TCT AGA CGG CAG GTC AGG TCC ACC-3′ (reverse); SNHG5, 5′-TAG AGA TGC AAA GAT ACA CGA AA (forward) and 5′-CAC ACT CAG AAC GCT GTT CAC-3′ (reverse).2.4. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays
Transfected melanoma cells were plated into 96-well plates, followed by the detection of cell proliferation capacity by MTT assays. At the indicated time point (0, 24, 48, 72 h) after transfection, 10 μl MTT solution (5 mg ml−1, Sigma-Aldrich, St. Louis, MO, USA) was inoculated into each well and the plates were maintained for an additional 4 h at 37 °C. Then 150 μl DMSO (Sigma-Aldrich) was added into each well to dissolve formed MTT formazan crystals. Finally, cell absorbance was measured at the wavelength of 490 nm.2.5. Colony formation assays
Transfected A375 and SK-MEL-2 cells were plated in 60 mm culture dishes and cultured in DMEM medium supplemented with 10% FBS for 2 weeks. Next, cells were fixed with absolute ethylalcohol and stained using 0.1% crystal violet solution (Sigma-Aldrich) for 10 min. Finally, dishes were used to photographed and scanned for colony counts.2.6. Apoptosis assays
Melanoma cells were collected 48 h after transfection, followed by the detection of apoptosis rate using FITC Annexin V apoptosis detection kit I (BD Biosciences, San Jose, CA, USA). Briefly, collected cells were resuspended in 1 × binding buffer (100 μl) and then stained using 5 μl FITC-Annexin V and 5 μl PI solutions for 15 min at room temperature in the dark. At last, apoptotic rates were measured by flow cytometry (BD Biosciences) following the addition of 400 μl 1 × binding buffer.2.7. Luciferase activity assays
Partial fragments of SNHG5 encompassing putative miR-155 binding sites were amplified by PCR and cloned into psiCHECK-2 vector (Promega, Madison, WI, USA) to construct wide type SNHG5 reporter (SNHG5-WT). Fast Site-Directed Mutagenesis Kit (Tiangen Biotech, Beijing, China) was used to generate mutant type SNHG5 reporter (SNHG5-MUT) containing mutant miR-155 binding sites. Then constructed SNHG5-WT or SNHG5-MUT reporter was co-transfected with miR-con, miR-155, anti-miR-con, or anti-miR-155 into melanoma cells. At last, luciferase activities in cells were determined using a dual luciferase reporter assay kit (Promega) at 48 h posttransfection.2.8. Subcellular fraction assays
RNA in nuclear and cytoplasm of A375 and SK-MEL-2 cells was extracted by SurePrep™ Nuclear or Cytoplasmic RNA Purification Kit (Thermo Fisher Scientific, Rockford, IL, USA) according to the protocols of manufacturer. Then the expression patterns of GAPDH, U6 snRNA and SNHG5 in nuclear and cytoplasm fractions were detected by RT-qPCR assays. GAPDH acted as an endogenous cytoplasm control and U6 snRNA served as an endogenous nuclear control.2.9. RNA immunoprecipitation (RIP) assays
RIP assays were carried out in A375 and SK-MEL-2 cells using Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, Bedford, MA, USA) according to the protocols of manufacturer. Generally, A375 and SK-MEL-2 cells were lysed using RIP lysis buffer. Then cell extraction was subjected to the treatment of protein A/G magnetic beads conjugated with IgG (Millipore) or Ago2 (Millipore) antibody, followed by the purification of RNA. At last, purified RNA was used to detect the enrichment profiles of SNHG5 and miR-155 using RT-qPCR assays.2.10. Lentivirus production and infection
Designed SNHG5 short hairpin RNA (shRNA) fragments were subcloned into pLKO.1 vector (Addgene, Cambridge, Massachusetts, USA) to construct sh-SNHG5 lentivirus plasmid. Next, sh-SNHG5 lentivirus plasmid or pLKO.1 empty plasmid was introduced into HEK293T cells along with psPAX2 (Addgene) and pMD2.G (Addgene) plasmids to generate sh-SNHG5 or sh-con lentivirus. Then, A375 cells were infected with sh-SNHG5 or sh-con lentivirus, followed by the screening using puromycin (Sigma-Aldrich) for at least 1 week to construct sh-SNHG5 or sh-con stably-transfected A375 cell line.2.11. In vivo assays
Fifteen adult athymic nude mice (6–8 weeks old) were purchased from Slaccas Co. Ltd (Shanghai, China). All animal procedures were performed following the National Institute of Health Guide for the Care and Use of Laboratory Animals. Also, mice experiments got the approval of Institutional Committee for Animal Research. Mice were randomly distributed into 3 groups (sh-con, sh-SNHG5, and sh-SNHG5 + miR-155) with 5 mice in each group. Then, an accurately measured amount of 5 × 106 A375 cells transfected with sh-con or sh-SNHG5 were inoculated into the right flanks of mice. Seven days after injection, miR-155 mimic was injected every 7 days into visible xenograft tumors of mice in sh-SNHG5 + miR-155 group. Tumor volume was measured using a caliper every 7 days for 42 days after injection. In the end, xenograft tumors were excised, photographed and weighted. Also, the expression patterns of miR-155 in resected tumors were detected using RT-qPCR assays.2.12. Statistical analysis
SPSS software (IBM, Armonk, NY, USA) was used to perform all statistical analyses. One-way variance analysis or Student's t-test was used for difference comparisons in different groups. A value of P < 0.5 was regarded as statistically significant.3. Results
3.1. SNHG5 expression was up-regulated in melanoma tumor tissues and cells
Firstly, RT-qPCR analysis was carried out to detect SNHG5 expression levels in 22 pairs of melanoma tumor tissues and adjacent normal tissues. Results showed that SNHG5 expression was markedly up-regulated in melanoma tumor tissues compared with adjacent normal tissues (Fig. 1A). Further analysis indicated that SNHG5 levels were higher in melanoma patients with advanced pathological stage (III + IV) than that in earlier stage (I + II) (Fig. 1B). To further explore the association between SNHG5 and clinical prognosis, melanoma patients were divided into SNHG5 high expression group and SNHG5 low expression group using the median value of SNHG5 expression as a cutoff point. Subsequent Kaplan–Meier analysis and log-rank test suggested that higher SNHG5 level was associated with decreased overall survival rate and shortened overall survival time (Fig. 1C). Moreover, SNHG5 expression was detected in several melanoma cell lines (HBL, A375, SK-MEL-2, CHL-1 and VMM917) and normal human epidermal melanocytes (NHEM). As expected, a notable increase of SNHG5 levels was observed in melanoma cell lines compared with NHEM (Fig. 1D). In a word, these data outlined that SNHG5 might be linked to the progression and poor prognosis of melanoma. | ||
| Fig. 1 SNHG5 expression was up-regulated in tumor tissues and cells of melanoma. Expression detection of SNHG5 in melanoma tumor tissues (n = 22) and adjacent normal tissues (n = 22) (A), different clinical pathological stages (B), n(I + II) = 13, n(III + IV) = 9, melanoma cell lines (HBL, A375, SK-MEL-2, CHL-1 and VMM917) and normal human epidermal melanocytes (NHEM) (D). (C) Kaplan–Meier analysis of overall survival for melanoma patients according to the difference of SNHG5 expression. *P < 0.05. | ||
3.2. Knockdown of SNHG5 suppressed proliferation and facilitated apoptosis in melanoma cells
To further explore biological significance of SNHG5 in the progression of melanoma, two different SNHG5-specific siRNAs (si-SNHG5#1 and si-SNHG5#2) and scramble control siRNA (si-con) were synthesized, followed by the detection of knockdown efficiency in human melanoma cells. Results showed that SNHG5-specific siRNAs markedly reduced the endogenous expression of SNHG5 in A375 (Fig. 2A) and SK-MEL-2 (Fig. 2B) cells compared with mock control, indicating that si-SNHG5#1 and si-SNHG5#2 could be used for the following loss-of-function assays. Subsequent MTT assays showed that depletion of SNHG5 by si-SNHG5#1 and si-SNHG5#2 strikingly blocked the growth ability of A375 (Fig. 2C) and SK-MEL-2 (Fig. 2D) cells. Colony formation assays further demonstrated that deficiency of SNHG5 suppressed melanoma cells proliferation, revealed by the decrease of colonies number in A375 and SK-MEL-2 cells transfected with si-SNHG5#1 or si-SNHG5#2 with respect to that in si-con-transfected cells (Fig. 2E). Next, the effect of SNHG5 knockdown on apoptosis of melanoma cells was assessed by flow cytometry. Results showed that down-regulation of SNHG5 resulted in an obvious increase of apoptosis rate in A375 and SK-MEL-2 cells (Fig. 2F). In summary, these results indicated that knockdown of SNHG5 hindered proliferation and induced apoptosis in melanoma cells. | ||
| Fig. 2 Knockdown of SNHG5 hindered proliferation and induced apoptosis in human melanoma cells. A375 and SK-MEL-2 cells were transfected with two specific siRNAs targeting SNHG5 and one scramble control siRNA. RT-qPCR assays were performed to measure the efficiency of SNHG5 knockdown in A375 (A) and SK-MEL-2 (B) cells 48 h after siRNA treatment. Cell viability was assessed by MTT assays in A375 (C) and SK-MEL-2 (D) cells at the indicated time points (0, 24, 48, 72 h) after siRNA transfection. (E) Colonies numbers were counted by colony formation assays in A375 and SK-MEL-2 cells at 2 weeks post siRNA treatment. (F) Cell apoptosis rate was analyzed by flow cytometry in A375 and SK-MEL-2 cells at 48 h upon siRNA treatment. *P < 0.05. | ||
3.3. SNHG5 acted as a molecular sponge of miR-155 in melanoma cells
To further investigate the molecular basic of SNHG5 in melanoma progression, miRcode online website was used to search for the potential miRNAs that could interact with SNHG5. In several candidate miRNAs, we focused on miR-155 for further study since miR-155 has been previously reported to be a tumor suppressor in melanoma (Fig. 3A).20 Subsequent subcellular fraction assays showed that SNHG5 was mainly distributed in the cytoplasm of A375 (Fig. 3B) and SK-MEL-2 cells (Fig. 3C), indicating that SNHG5 had a chance to interact with miR-155 in space. In order to further confirm the interaction between SNHG5 and miR-155, dual luciferase reporter assays were carried out in melanoma cells. As shown in Fig. 3D and E, the luciferase activity of SNHG5 wide-type reporter was significantly decreased in miR-155-overexpressed A375 cells, but was strikingly increased in SK-MEL-2 cells with lowered miR-155 expression. However, the introduction of miR-155 mimic and inhibitor displayed no effect on the luciferase activity of SNHG5 mutant-type reporter in melanoma cells, indicating that the interplay between SNHG5 and miR-155 was mediated by the putative binding sites. Argonaute2 (Ago2), a critical component of RNA-induced silencing complex (RISC), is involved in the regulation of miRNA silencing signaling.21 Hence, RIP assays were performed to test whether SNHG5 and miR-155 were co-existed in RISC complex. Results manifested that SNHG5 and miR-155 expressions were substantially enriched by Ago2 antibody in A375 (Fig. 3F) and SK-MEL-2 (Fig. 3G) cells compared with IgG control antibody, indicating direct binding between SNHG5 and miR-155. Next, the effects of SNHG5 up-regulation and down-regulation on miR-155 expression were detected by RT-qPCR assays in A375 and SK-MEL-2 cells. Results showed that miR-155 expression was strikingly increased in A375 and SK-MEL-2 cells following the depletion of SNHG5 (Fig. 3H), but was significantly reduced in SNHG5-overexpressed melanoma cells (Fig. 3I). In other words, SNHG5 acted as a molecular sponge of miR-155 to inhibit miR-155 expression in melanoma cells. Additionally, we further demonstrated that miR-155 expression was down-regulated in melanoma tumor tissues (n = 22) compared with adjacent normal tissues (n = 22) (Fig. 3J). Moreover, miR-155 expression was inversely correlated with SNHG5 expression in 22 cases of melanoma tumor tissues (Fig. 3K). All these outcomes suggested that SNHG5/miR-155 axis might be involved in the regulation of melanoma progression. | ||
| Fig. 3 SNHG5 acted as a molecular sponge of miR-155 in melanoma cells. (A) Graphical representation of the putative binding sites between SNHG5 and miR-155, as well as the mutant sequences in SNHG5-MUT reporter. (B and C) Subcellular fraction assays were performed to explore the location of SNHG5 in A375 and SK-MEL-2 cells with GAPDH as a cytoplasm control and U6 as a nucleus control. (D and E) The effects of miR-155 overexpression and knockdown on the luciferase activity of SNHG5-WT and SNHG5-MUT reporters were assessed by dual luciferase reporter assays. (F and G) RIP assays were performed using Ago2 antibody or IgG control antibody to detect the enrichment degrees of SNHG5 and miR-155 in A375 and SK-MEL-2 cells. (H and I) RT-qPCR assays were conducted to measure the effect of SNHG5 knockdown and overexpression on miR-155 expression in A375 and SK-MEL-2 cells. (J) miR-155 expression in 22 pairs of melanoma tumor tissues and adjacent normal tissues. (K) Correlation analysis between SNHG5 and miR-155 in 22 cases of melanoma tumor tissues. *P < 0.05. | ||
3.4. Down-regulation of miR-155 partly abrogated SNHG5-knockdown-mediated anti-proliferation and pro-apoptosis effect in melanoma cells
Considering the negative correlation between SNHG5 and miR-155, we further discussed whether SNHG5 exerted its cancerigenic effect via regulating miR-155 in melanoma cells. Si-SNHG5#2-transfected A375 and SK-MEL-2 cells were further transfected with anti-miR-155, followed by the analyses of cell proliferation and apoptosis. RT-qPCR assays revealed that si-SNHG5#2-mediated elevation of miR-155 expression was pronouncedly reduced by miR-155 inhibitor in A375 and SK-MEL-2 cells (Fig. 4A). Moreover, miR-155 inhibition reversed the suppression effect of SNHG2 deficiency on cell proliferation (Fig. 4B) and colony formation (Fig. 4C) in A375 and SK-MEL-2 cells. Additionally, miR-155 down-regulation dramatically abated si-SNHG5#2-induced apoptosis in A375 and SK-MEL-2 cells (Fig. 4D). Collectively, these data demonstrated that SNHG5 down-regulation blocked proliferation and accelerated apoptosis of melanoma cells through regulating miR-155 expression. | ||
| Fig. 4 miR-155 downregulation partly abrogated SNHG5-knockdown-mediated anti-proliferation and pro-apoptosis effect in melanoma cells. A375 and SK-MEL-2 cells were transfected with si-con, si-SNHG5#2, and si-SNHG5#2 + anti-miR-155, followed by the measurement of miR-155 expression (A), cell viability (B), colony formation capability (C) and apoptosis rate (D). *P < 0.05. | ||
3.5. SNHG5 depletion hindered tumor growth through enhancing miR-155 expression in vivo
To further test the roles and molecular mechanisms of SNHG5 in tumorigenesis of melanoma in vivo, nude mice were injected with A375 cells stably transfected with sh-con or sh-SNHG5. Introtumorous injection of miR-155 was performed at 7 days after cell inoculation. As shown in Fig. 5A and B, SNHG5 knockdown suppressed tumor growth in mice xenograft model of melanoma, embodied by smaller tumor volume and lower tumor weight, while this effect was partly abolished by miR-155 down-regulation. Further analysis revealed that miR-155 expression was elevated in tumors derived from SNHG5-knockdown melanoma cells, whereas suppression of miR-155 markedly undermined sh-SNHG5-mediated promotion effect on miR-155 expression (Fig. 5C). Taken together, these data suggested that knockdown of SNHG5 hindered tumor growth through facilitating miR-155 expression in vivo.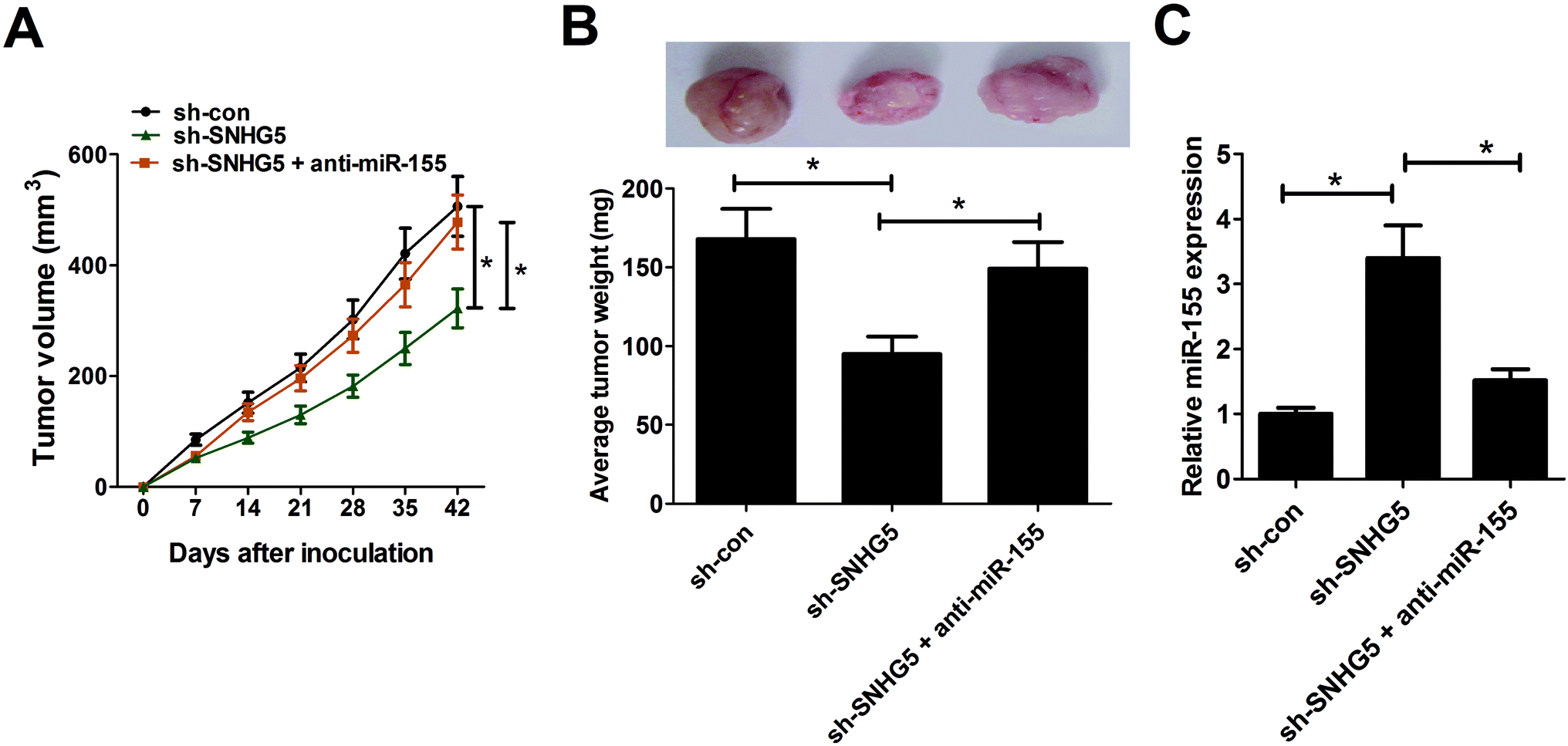 | ||
| Fig. 5 Depletion of SNHG5 hindered tumor growth through facilitating miR-155 expression in vivo. Nude mice were injected with A375 cells stably transfected with sh-con or sh-SNHG5 to generate mice xenograft model of melanoma. After 7 days, mice inoculated with sh-SNHG5-transfected A375 cells were divided into two groups with one group mice was injected with miR-155 mimic every 7 days. (A) Tumor volume was detected using a caliper every 7 days. (B) Mice were sacrificed at 42 days after cell inoculation, and xenograft tumors were resected, photographed and weighted. (C) The expression patterns of miR-155 in excised tumor tissues. *P < 0.05. | ||
4. Discussion
Non-coding RNAs including miRNAs and lncRNAs have been identified as vital regulators in the pathology, diagnosis, and management in melanoma.22,23 LncRNA SNHG5, as a tumor suppressor or an oncogene, has been reported to be abnormally expressed in some cancers. For example, SNHG5 expression was depressed in gastric cancer tissues and SNHG5 overexpression inhibited proliferation and metastasis of gastric cancer cells by regulating miR-32/Kruppel-like factor 4 (KLF-4) or metastasis-associated protein 2 (MTA2).16,17 However, SNHG5 was highly expressed in colorectal cancer and SNHG5 knockdown triggered cell cycle arrest in vitro and suppressed tumor growth in vivo.18 In the present study, we demonstrated that SNHG5 expression was up-regulated in melanoma tumor tissues and cell lines. Moreover, higher SNHG5 expression was related with progressively pathogenic status and poor prognosis in melanoma clinical samples. These results were in accordance with a previous report discovering that SNHG5 was highly expressed in melanoma patient serum and SNHG5 serum levels were strikingly reduced after surgery.19 Hence, the roles of SNHG5 in the development of melanoma were further investigated by loss-of-function assays. Results showed that SNHG5 knockdown markedly suppressed proliferation and induced apoptosis of melanoma cells, indicating that SNHG5 acted as an oncogenic factor in melanoma.A substantial amount of evidence points out that lncRNAs can act as competing endogenous RNAs (ceRNAs) of miRNAs to regulate miRNAs expression.24 To elucidate the action mechanism of SNHG5 in contributing to melanoma progression, miRcode online website was used to search for potential miRNAs that might interact with SNHG5. Among candidate miRNAs, miR-155 was chosen for further exploration due to the its vital roles in multiple physiological and pathological processes such as immunity, inflammation and cancer.25 miR-155 has been described as an oncogenic factor with a high-level expression in some solid tumors such as breast cancer,26 cervical cancer27 and clear-cell renal cell carcinoma.28 However, in melanoma, the expression of miR-155 was controversial. Levati et al. showed that miR-155 expression was reduced in melanoma cells compared with melanocytes, and miR-155 overexpression suppressed proliferation and induced apoptosis of melanoma cells.20,29 Moreover, Segura et al. manifested that higher miR-155 level in metastatic melanoma tissues was associated with longer survival,30 indicating that miR-155 might act as a tumor suppressor in vivo. However, Grignol and Philippidou et al. pointed out miR-155 expression was up-regulated in melanoma samples.31,32
Our study further demonstrated that SNHG5 could interact with miR-155 by subcellular fraction, dual luciferase reporter and RIP assays. Moreover, SNHG5 knockdown promoted miR-155 expression and SNHG5 up-regulation suppressed miR-155 expression, indicating SNHG5 can act as a molecular sponge of miR-155 in melanoma. Furthermore, miR-155 expression was decreased in melanoma tumor tissues, and miR-155 expression was inversely associated with SNHG5 expression in melanoma tumor tissues. All these data raised the possibility that SNHG5 exerted tumor-suppressive role via regulating miR-155 in melanoma. Subsequent restoration experiments further demonstrated that miR-155 down-regulation could reverse SNHG-knockdown-mediated anti-proliferation and pro-apoptosis effect in melanoma cells. Finally, xenotransplanted tumor model of melanoma in nude mice validated that down-regulation of SNHG resulted in a blockage of tumor growth in vivo via enhancing miR-155 expression.
5. Conclusions
In summary, our present study demonstrated that SNHG knockdown hampered melanoma progression by facilitating miR-155 expression in vitro and in vivo, providing a novel potential target for the treatment of melanoma.Authors' contribution
Lu Yan, Linda Tognetti, Kang Zeng and Xiangbin Mi designed the study. Suihai Wang, Michele Fimiani and Pietro Rubegni performed the experiments and analyzed the data. Lu Yan, Elisa Pianigiani and Yue Li prepared the manuscript. Rui Tan and Hui Li supervised the study and reviewed the manuscript.Conflicts of interest
There is no conflict of interest regarding the publication of this paper.References
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 7–30 CrossRef PubMed.
- S. S. Agarwala, Oncology, 2016, 30, 436 Search PubMed.
- C. Garbe, T. K. Eigentler, U. Keilholz, A. Hauschild, J. M. Kirkwood, C. Garbe, T. K. Eigentler, U. Keilholz, A. Hauschild and J. M. Kirkwood, Oncologist, 2011, 16, 5–24 CrossRef CAS PubMed.
- G. Chhabra, M. A. Ndiaye, L. M. Garcia-Peterson and N. Ahmad, Photochem. Photobiol., 2017, 93, 975–989 CrossRef CAS PubMed.
- A. Guennoun, H. Sidahmed, C. Maccalli, B. Seliger, F. M. Marincola and D. Bedognetti, Expert Rev. Clin. Immunol., 2016, 12, 879–893 CrossRef CAS PubMed.
- E. A. Gibb, C. J. Brown and W. L. Lam, Mol. Cancer, 2011, 10, 38 CrossRef CAS PubMed.
- D. Khaitan, M. E. Dinger, J. Mazar, J. Crawford, M. A. Smith, J. S. Mattick and R. J. Perera, Cancer Res., 2011, 71, 3852–3862 CrossRef CAS PubMed.
- L. Tang, W. Zhang, B. Su and B. Yu, BioMed Res. Int., 2013, 2013, 251098 Search PubMed.
- G. T. Williams and F. Farzaneh, Nat. Rev. Cancer, 2012, 12, 84–88 CrossRef CAS PubMed.
- M. Mourtadamaarabouni, M. R. Pickard, V. L. Hedge, F. Farzaneh and G. T. Williams, Oncogene, 2009, 28, 195–208 CrossRef CAS PubMed.
- M. Sun, F. Y. Jin, R. Xia, R. Kong, J. H. Li, T. P. Xu, Y. W. Liu, E. B. Zhang, X. H. Liu and W. De, BMC Cancer, 2014, 14, 319 CrossRef PubMed.
- M. R. Pickard, M. Mourtada-Maarabouni and G. T. Williams, Biochim. Biophys. Acta, 2013, 1832, 1613–1623 CrossRef CAS PubMed.
- L. Chen, H. Yang, Y. Xiao, X. Tang, Y. Li, Q. Han, J. Fu, Y. Yang and Y. Zhu, Int. J. Oncol., 2016, 48, 1509–1518 CrossRef CAS PubMed.
- Y. Nakamura, N. Takahashi, E. Kakegawa, K. Yoshida, Y. Ito, H. Kayano, N. Niitsu, I. Jinnai and M. Bessho, Cancer Genet. Cytogenet., 2008, 182, 144–149 CrossRef CAS PubMed.
- B. He, Y. Bai, W. Kang, X. Zhang and X. Jiang, Am. J. Cancer Res., 2017, 7, 1704–1713 Search PubMed.
- L. Zhao, T. Han, Y. Li, J. Sun, S. Zhang, Y. Liu, B. Shan, D. Zheng and J. Shi, FASEB J., 2016, 31, 893–903 CrossRef PubMed.
- L. Zhao, H. Guo, B. Zhou, J. Feng, Y. Li, T. Han, L. Liu, L. Li, S. Zhang and Y. Liu, Oncogene, 2016, 35, 5770–5780 CrossRef CAS PubMed.
- N. D. Damas, M. Marcatti, C. Côme, L. L. Christensen, M. M. Nielsen, R. Baumgartner, H. M. Gylling, G. Maglieri, C. F. Rundsten and S. E. Seemann, Nat. Commun., 2016, 7, 13875 CrossRef CAS PubMed.
- Y. Ichigozaki, S. Fukushima, M. Jinnin, A. Miyashita, S. Nakahara, A. Tokuzumi, J. Yamashita, I. Kajihara, J. Aoi and S. Masuguchi, Exp. Dermatol., 2016, 25, 67–69 CrossRef PubMed.
- L. Levati, E. Pagani, S. Romani, D. Castiglia, E. Piccinni, C. Covaciu, P. Caporaso, S. Bondanza, F. R. Antonetti and E. Bonmassar, Pigm. Cell Melanoma Res., 2011, 24, 538–550 CrossRef CAS PubMed.
- T. Kawamata, M. Yoda and Y. Tomari, EMBO Rep., 2011, 12, 944–949 CrossRef CAS PubMed.
- M. N. Aftab, M. E. Dinger and R. J. Perera, Arch. Biochem. Biophys., 2014, 563, 60–70 CrossRef CAS PubMed.
- G. Richtig, B. Ehall, E. Richtig, A. Aigelsreiter, T. Gutschner and M. Pichler, Int. J. Mol. Sci., 2017, 18, 715 CrossRef PubMed.
- L. Salmena, L. Poliseno, Y. Tay, L. Kats and P. P. Pandolfi, Cell, 2011, 146, 353–358 CrossRef CAS PubMed.
- I. Faraoni, F. R. Antonetti, J. Cardone and E. Bonmassar, Biochim. Biophys. Acta, 2009, 1792, 497–505 CrossRef CAS PubMed.
- S. Mattiske, R. J. Suetani, P. M. Neilsen and D. F. Callen, Cancer Epidemiol., Biomarkers Prev., 2012, 21, 1236–1243 CrossRef CAS PubMed.
- G. Lao, P. Liu, Q. Wu, W. Zhang, Y. Liu, L. Yang and C. Ma, Tumor Biol., 2014, 35, 11933–11938 CrossRef CAS PubMed.
- H. Ji, D. Tian, B. Zhang, Y. Zhang, D. Yan and S. Wu, Exp. Ther. Med., 2017, 13, 2286–2292 CrossRef PubMed.
- L. Levati, E. Alvino, E. Pagani, D. Arcelli, P. Caporaso, S. Bondanza, L. G. Di, M. Ferracin, S. Volinia and E. Bonmassar, Int. J. Oncol., 2009, 35, 393–400 CAS.
- M. F. Segura, I. Belitskayalévy, A. E. Rose, J. Zakrzewski, A. Gaziel, D. Hanniford, F. Darvishian, R. S. Berman, R. L. Shapiro and A. C. Pavlick, Clin. Cancer Res., 2010, 16, 1577–1586 CrossRef CAS PubMed.
- V. Grignol, E. T. Fairchild, J. M. Zimmerer, G. B. Lesinski, M. J. Walker, C. M. Magro, J. E. Kacher, V. I. Karpa, J. Clark and G. Nuovo, Br. J. Cancer, 2011, 105, 1023–1029 CrossRef CAS PubMed.
- D. Philippidou, M. Schmitt, D. Moser, C. Margue, P. V. Nazarov, A. Muller, L. Vallar, D. Nashan, I. Behrmann and S. Kreis, Cancer Res., 2010, 70, 4163–4173 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |