
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot two-step synthesis of 3-iodo-4-aryloxy coumarins and their Pd/C-catalyzed annulation to coumestans†
Niranjan Panda * and
Irshad Mattan
* and
Irshad Mattan
Department of Chemistry, National Institute of Technology, Rourkela, Odisha-769008, India. E-mail: npanda@nitrkl.ac.in; niranjanpanda@gmail.com
First published on 16th February 2018
Abstract
An efficient protocol for the synthesis of various coumestans from the intramolecular annulation of 3-iodo-4-aryloxy coumarins through C–H activation has been developed. When 3-iodo-4-aryloxy coumarins were treated with 10% Pd/C (0.3 mol% Pd) in the presence of sodium acetate, the corresponding coumestans were produced in good to excellent yield. Reusability of the palladium catalyst was investigated in up to three consecutive cycles and it was found that the yield of the reaction was almost unaltered. Sequential iodination and O-arylation of 4-hydroxy coumarins leading to the 3-iodo-4-aryloxy coumarins were also achieved in a one-pot two-step process starting from aryl iodides in high yield. Pivalic acid was revealed to be the most effective additive for the later method to produce 3-iodo-4-phenoxy coumarins. Different functional groups bearing electron-donating as well as withdrawing groups are also tolerant to the reaction conditions.
Introduction
Benzofuran-fused coumarins, commonly called coumestans, constitute a class of plant products (secondary metabolites) found abundantly in split peas, pinto beans, lima beans, alfalfa and clover sprouts. Coumestans including coumestrol, wedelolactone, isoglycyrol, psoralidin, erosnin, medicagol, and many others exhibit diverse biological properties such as phytoestrogenic, antioxidant, antihepatotoxic, antifungal and antitumor activities (Fig. 1).1 Further, these compounds serve as chemo-preventive agents for breast and prostate cancers.2 Considering their physiological and pharmacological potential, it is desirable to develop more efficient synthetic strategies to achieve newer coumestans for pharmaceutical research.3 Among the reported synthetic protocols, transition metal mediated assembly of furan nucleus of the fused tetracyclic scaffold is emerged to be the most promising. Annulation of 3-aryl-4-hydroxyl coumarins through oxidative C–O bond formation (Scheme 1, path a) is a classical pathway to achieve coumestans.4 Zhao and co-workers developed a FeCl3-mediated dehydrogenative C–O cross-coupling protocol to achieve coumestans.5 However, limited substrate scope, multistep sequence to access starting material and use of stoichiometric excess of catalyst which in-turn is wasteful, often vitiates its large scale use.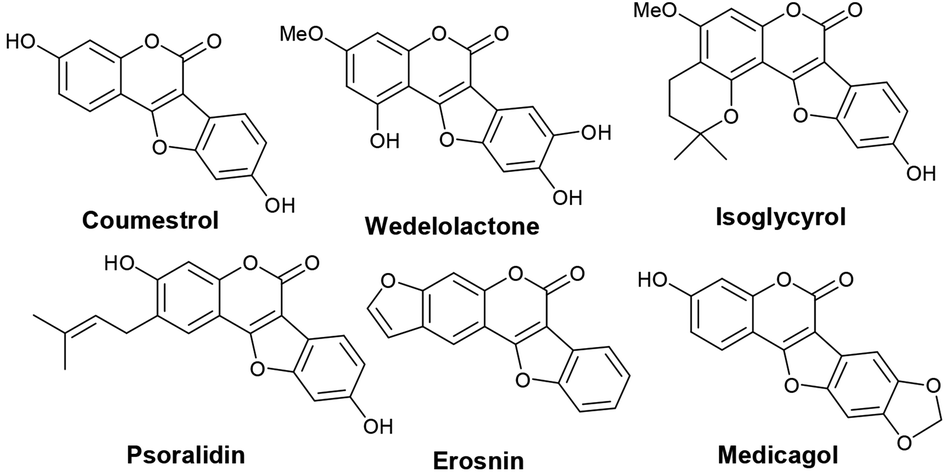 | ||
| Fig. 1 Examples of biologically potent naturally occurring coumestans. | ||
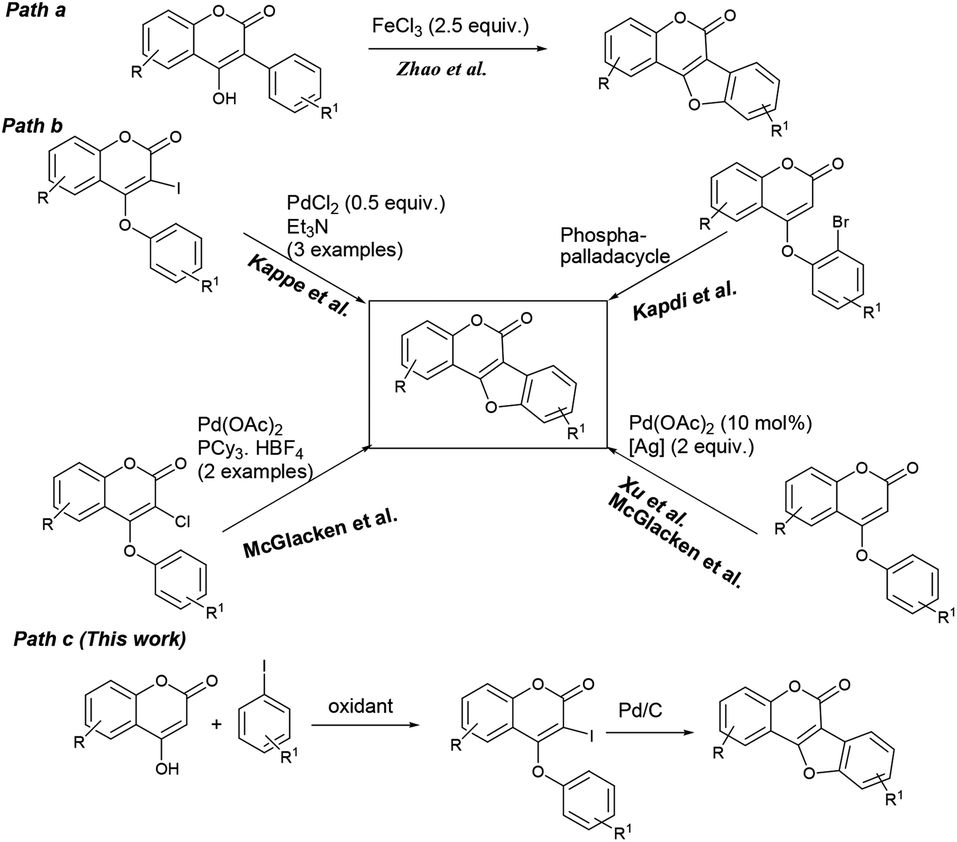 | ||
| Scheme 1 Transition metal catalyzed routes to coumestans. | ||
On the other hand, ring closure of preformed aryl-coumarin ether through C–C bond formation has been employed successfully (Scheme 1, path b) to construct the coumestan skeletal framework. In 1990, Laschober and Kappe6 first reported the PdCl2 (0.5 equiv.) mediated synthesis of coumestans (3 examples) from 4-aryloxy-3-iodocoumarins in triethyl amine. Later, in 2013, Kapdi and co-workers7 employed palladacycles to construct the furan ring through C–H functionalization of substituted 4-(2-bromophenoxy)-coumarins. Recently, Cheng et al. developed a Pd(OAc)2-catalyzed one-pot, two-step cyclization reaction of iodobenzene with coumarin to produce the corresponding coumestan.8 McGlacken9 developed an intramolecular direct arylation approach using pre-halogen functionalized 4-aryloxy coumarins as substrates. Very recently, McGlacken10a and Xu10b independently reported Pd(OAc)2-catalyzed cross-dehydrogenative coupling reaction of 4-phenoxy coumarins to access coumestans in the presence of excess amount of expensive silver salt. Despite largely improved efficiency, these methodologies generally requires the use of ligands, precious transition metal catalyst such as Pd(OAc)2, PdCl2, AgOAc, Ag2O and multi-step strategy to the starting materials (e.g. 4-aryloxy coumarin and 3-halo 4-aryloxy coumarin). In this context, development of a stable and less expensive catalytic system to achieve these biologically potent tetracycles under ligand-free conditions is admirable from the synthetic point of view. In continuation of our recent interest in the synthesis of heterocycles,11 herein, we report a more convenient ligand-free and less expensive catalytic method using Pd supported on charcoal (i.e. Pd/C) as a heterogeneous catalyst for the successful synthesis tetracyclic coumestans (path c) through C–H activation reaction. It is noteworthy that the precursor for coumestans i.e. o-iodocoumarin-aryl ethers were also achieved in a more simplified process directly from the reaction of coumarins and aryl iodides in one-pot.
Results and discussion
We initiated our work with the development of an amicable protocol for the synthesis of 3-iodo-4-aryloxy coumarin (3aa) from 4-hydroxy coumarin (2a) using in situ generated hypervalent iodine for intramolecular annulation. Note worthy that hypervalent diaryliodonium salts are typically used for selective O-arylation reactions.12 However, use of diaryliodonium salt causes the loss of an equimolar amount of expensive iodobenzene impedes its large-scale utility. On the other hand, hypervalent iodine reagent such as phenyl iododiacetate (PIDA) could be employed for sequential ortho-iodination and O-arylation reactions.13 Notably, iodonium salts are produced in such methods; that subsequently undergo phenyl migration leading to ortho-iodo-O-phenyl arene. Despite of remarkable success of the reaction with PIDA, similar reactions with other substituted hypervalent iodine derivatives always end with a poor result. Indeed, preparation of such substituted aryl iododiacetates for tandem iodination/arylation is often associated with certain limitations. Not only stoichiometric excess (10 equiv. in most cases) of oxidants were employed for the synthesis of aryl iododiacetates, tedious purification procedure resulting in poor yield of the reagent. Therefore, we motivated to access o-iododiaryl ethers (e.g. 3aa) directly in a one-pot two-step method using readily available aryl iodide for both arylating as well as iodinating agent. Notably, aryliodide-catalyzed oxidative C–C and C–N bond formation reactions leading to heterocyclic carbazoles, benzimidazoles, indoles and phenanthridinones were reported in the literature.14 It has been proposed that aryl iodide oxidized by the terminal oxidant (e.g. mCPBA, K2S2O8, Oxone®, etc.) in hexaflouro isopropanol (HFIP) to hypervalent iodine(III) species, which served as the catalyst for the said oxidative transformations under metal-free conditions. Contrastingly, use of in situ generated hypervalent iodine species for tandem iodination and O-arylation is not much literature precedent.Our optimization studies commenced with 4-hydroxy coumarin 2a and iodobenzene (1a) by the screening of tandem O-arylation and ortho-iodination promoters in one-pot (Table 1). Firstly, iodobenzene was treated with oxidant at 50 °C over a period of 5 h for the formation of hypervalent iodine reagent, and then 2a was added and refluxed for overnight for sequential iodination and O-arylation reaction. We began our study with the use of an oxidants such as Oxone®, K2S2O8, NaIO4, TBHP and NaBO3·H2O for in situ oxidation of iodobenzene and subsequent transformation to 3-iodo 4-aryloxy coumarin (3aa). Unfortunately, in all such cases the desired product 3aa was not observed (entries 1–5). Interestingly, when the reaction was conducted in the presence of mCPBA (3 equiv.) in polar solvent like 1,4-dioxane, 3aa was obtained in 38% yield (entry 6). When the reaction was carried out in the non-polar solvent such as toluene under reflux, 3aa was obtained in 45% yield (entry 7).
|
|||||
|---|---|---|---|---|---|
| Entry | Oxidant (equiv.) | PhI (equiv.) | Additive (3 equiv.) | Solvent | % yieldb |
| a Reaction conditions: a mixture of oxidant and iodobenzene was heated at 50 °C for 5 h in a sealed tube. Then, 2a was added and the reaction mixture was refluxed.b GC yield. | |||||
| 1 | Oxone (3) | 3 | — | 1,4-Dioxane | n.d. |
| 2 | NaIO4 (3) | 3 | — | 1,4-Dioxane | n.d. |
| 3 | K2S2O8 (3) | 3 | — | 1,4-Dioxane | n.d. |
| 4 | TBHP (3) | 3 | — | 1,4-Dioxane | n.d. |
| 5 | NaBO3·H2O (3) | 3 | — | 1,4-Dioxane | n.d. |
| 6 | mCPBA (3) | 3 | — | 1,4-Dioxane | 38 |
| 7 | mCPBA (3) | 3 | — | Toluene | 45 |
| 8 | mCPBA (3) | 3 | AcOH | 1,4-Dioxane | 50 |
| 9 | mCPBA (3) | 3 | AcOH | Toluene | 70 |
| 10 | mCPBA (3) | 3 | TsOH | Toluene | n.r. |
| 11 | mCPBA (3) | 3 | TFA | Toluene | 60 |
| 12 | mCPBA (3) | 3 | K2CO3 | Toluene | n.r. |
| 13 | mCPBA (3) | 3 | PivOH | Toluene | 90 |
| 14 | mCPBA (3) | 3 | PivOH | CH3CN | 16 |
| 15 | mCPBA (3) | 3 | PivOH | MeOH | 37 |
| 16 | mCPBA (3) | 3 | PivOH | DCE | 48 |
| 17 | mCPBA (3) | 3 | PivOH | DMF | n.d. |
| 18 | mCPBA (3) | 3 | PivOH | DMSO | n.d. |
| 19 | mCPBA (1) | 1 | PivOH | Toluene | 45 |
| 20 | mCPBA (1.5) | 1.5 | PivOH | Toluene | 68 |
| 21 | mCPBA (2) | 1.5 | PivOH | Toluene | 88 |
| 22 | mCPBA (2) | 1.5 | AcOH | Toluene | 70 |
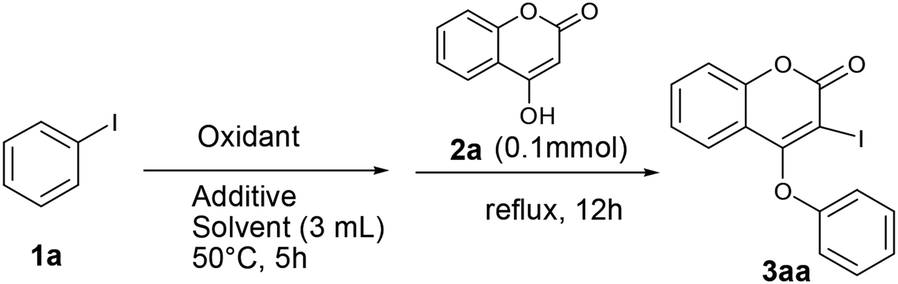
Gratifyingly, the yield of the desired product was further enhanced up to 70% by conducting the reaction in the presence of acetic acid as an additive (entries 8 and 9). Effect of other additives in the above reaction was also examined (entries 10–13). In the presence of the base such as Na2CO3, no desired product was achieved. Significantly, when the reaction was performed in the presence of 3 equiv. of pivalic acid (as an additive) and mCPBA (3 equiv.) in toluene, 3aa was produced in 90% yield (entry 13). To realize the solvent effect on reaction yield, other solvents (such as DMF, DMSO, CH3CN and DCE) were screened (entries 14–18) and none of the cases the better result was produced. While attempting to reduce the concentrations of iodobenzene and oxidant towards the development of a cost-effective method, we noticed that 1 equiv. of iodobenzene reacts with 4-hydroxy coumarin (1 equiv.) in the presence of 1 equiv. of mCPBA and pivalic acid (3 equiv.) in toluene affords 3aa in 45% yield (entry 19). The increase in the concentration of iodobenzene and mCPBA to 1.5 equiv. each, produces 3aa in 68% yield (entry 20). Further raising the oxidant concentration to 2 equiv., 3aa was achieved in 88% yield (entry 21). Additionally, the effect of inert atmosphere for the reaction was not realised as the yield of the reaction was unchanged both in the presence of air as well as nitrogen atmosphere.
Towards the mechanistic insight, we speculated that mCPBA could oxidize the iodobenzene to iodosobenzene (PhIO, A), which might be one of the key intermediates14a,b (Scheme 2). According to the previous report of Lei et al.,15 iodosobenzene could react with pivalic acid to form B, which further gives rise to phenyl iododipivalate (PhI(OPiv)2) C in the presence of PivOH. Indeed, the formation of C was evident from 1H NMR of the crude reaction mixture. For instance, the appearance of signals at δ 8.13–8.10 (m, 2H), 7.66–7.62 (m, 1H), 7.58–7.54 (m, 2H) and 1.02 (s, 18H) in 1H NMR spectrum along with the signals for PhIO and PhIO2 confirms the formation of C. The reaction of C with 2a might produce the intermediate D. Subsequent intramolecular aromatic nucleophilic attack of the oxygen atom causes the intramolecular migration of phenyl group from iodo to oxygen to produce 3aa with the release of PivOH. Indeed, when PhIO, prepared from the decomposition of PIDA with NaOH, was treated with 2a in the absence of pivalic acid, 3aa was formed fairly in low yield (40%), whereas similar reaction in the presence of pivalic acid, affords 3aa in excellent yield (90%). This observation suggests that pivalic acid serves as a better ligating agent in the overall transformation. Additionally, when over oxidized iodine(V) derivative i.e. PhIO2, produced from the disproportionation of PhIO, was treated with 2a both in the presence of pivalic acid, 3aa was achieved in 42% whereas similar reaction in the absence of the later results in 43% of 3aa. Thus, the in situ generated PhIO from PhI is the active intermediate for sequential iodination and O-arylation of coumarins and not the PhIO2.
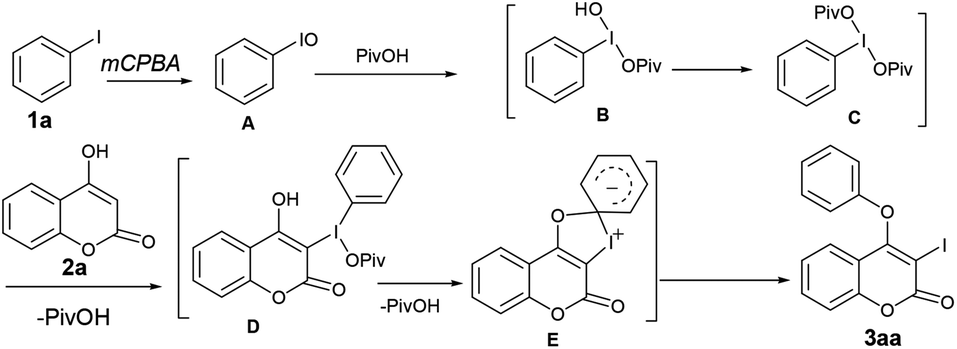 | ||
| Scheme 2 Plausible mechanism. | ||
With the optimum reaction condition in hand, we next explored the scope and generality of the tandem iodination and O-arylation of coumarins (Scheme 3). Thus, the one-pot two-step reaction of coumarins with different substitutions such as –Me, –OMe, –Cl, –OBn, –NO2 etc. and PhI in the presence of mCPBA produced 4-phenoxy 3-iodo coumarins in moderate to good yield. Furthermore, aryl iodides bearing electron-donating as well as withdrawing substituents (–Me, –Cl, –OMe, –OBn, –NO2) coupled with 4-hydroxy coumarin to produce 3 in good yield (63–82%) though the yield of reaction slightly diminished in the case of aryl halides containing an electron withdrawing group over a period of 12 h, while prolonged heating for 24 h did not improve the yield of the reaction.
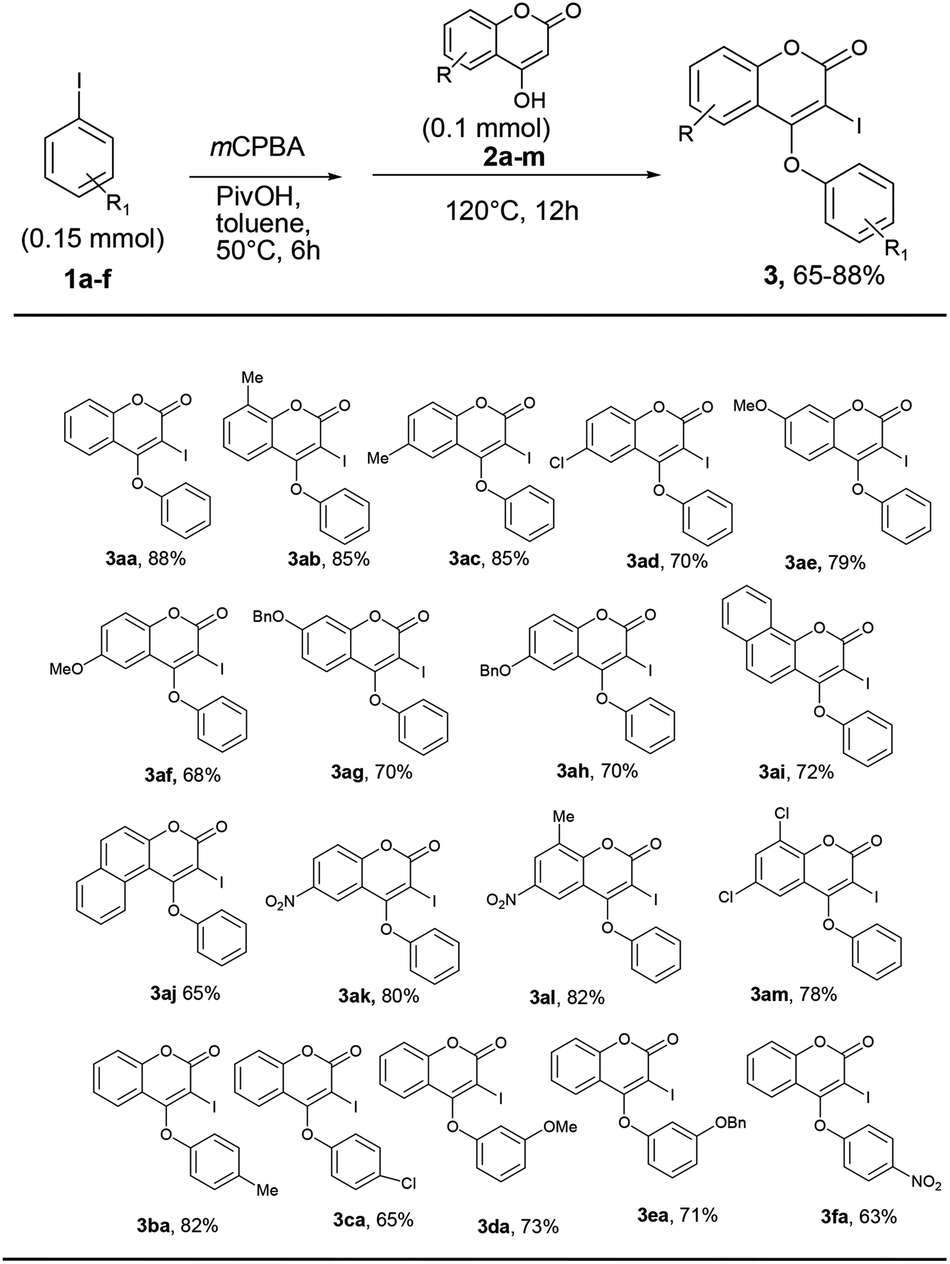 | ||
| Scheme 3 Synthesis of 4-aryloxy 3-iodo isocoumarins. aReaction conditions: a mixture of 1 and mCPBA (2 equiv.) in toluene was heated at 50 °C for 5 h in a sealed tube. Then, 2 was added and the reaction mixture was heated at 120 °C for 12 h. bIsolated yield. | ||
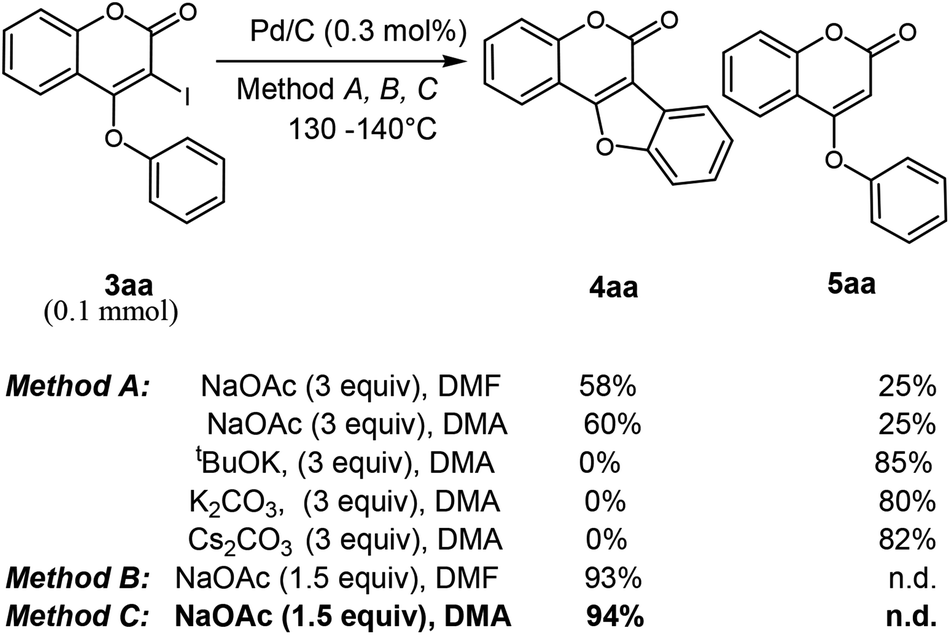
Next, we turn our focus towards intramolecular annulation of 4-aryloxy 3-iodocoumarins to coumestans using less expensive reusable Pd-catalyst. Notably, earlier reports on coumestan synthesis from 4-aryloxy 3-halocoumarins rely on the use of PdCl2, Pd(OAc)2 and phospha-palladacycles in the presence of expensive phosphorous-ligands. As a side note, to the best of our knowledge there is no report for the cross-coupling reaction of coumarin using heterogeneous Pd-catalyst. The quest for commercially viable, highly active, recoverable, and reusable palladium catalysts for the cross-coupling reaction allows us to start our investigation with Pd on carbon (i.e. Pd/C) catalyst for intramolecular C–C bond formation. Despite the low-cost, stability and reusability, the catalytic activity of Pd/C was underdeveloped except in catalytic hydrogenation. Moreover, scattered precedents of Pd/C-catalyzed C–C coupling reactions16 gratify its catalytic potential. Thus, when 3-iodo-4-phenoxy coumarin was reacted with NaOAc (3 equiv.), in the presence of Pd/C catalyst in DMA at 130–140 °C, tetracyclic coumestan 4aa was produced along with the dehalogenated product, 5aa. Lowering of temperature to 110 °C did not produce the product 4aa over a prolonged period of 24 h; rather the dehalogenated product was produced in 35% yield with the recovery of remaining starting material. To suppress the undesired dehalogenated product, alternative additives were tested for the intramolecular annulation (Table 2). These experiments revealed that other bases such as tBuOK, K2CO3, Cs2CO3 afford 5aa only (80% yield). Notably, after several experimentations, it has been realized that 1.5 equiv. of NaOAc in DMF at 130–140 °C produces 4aa predominantly in 93% yield. Furthermore, replacing the solvent from DMF to DMA, 4aa was also isolated in 94% yield without detection of any trace of dehalogenated product 5aa.
|
|||
|---|---|---|---|
| Entry | Substrate (3) | Coumestan (4) | % yielda |
| a Reaction conditions: [method C] a mixture of 3 (0.1 mmol), Pd/C (10 wt%) (3 mg, 0.3 mol%), NaOAc (1.5 equiv.) in 3 mL of DMA at 140 °C for 16 h. | |||
| 1 | 3aa | 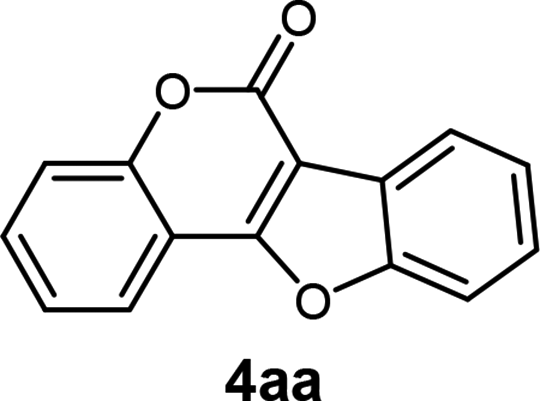 |
94 |
| 2 | 3ab | 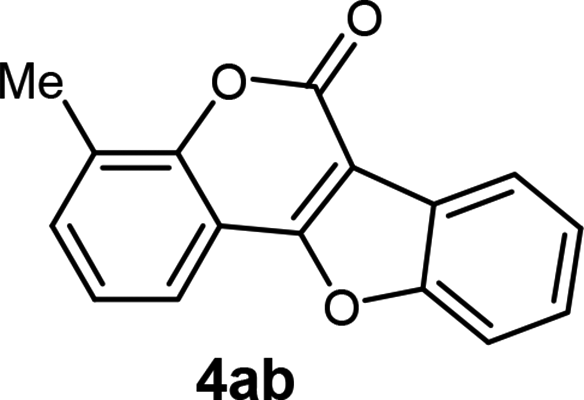 |
94 |
| 3 | 3ac | 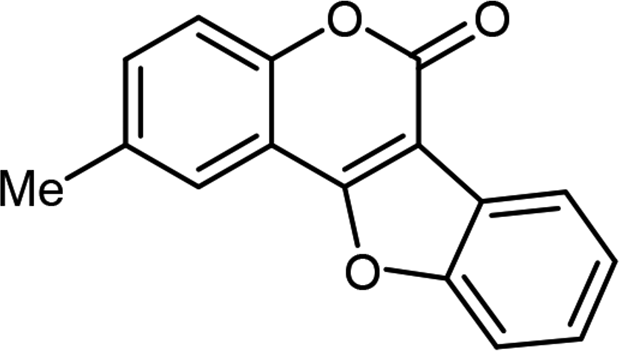 |
85 |
| 4 | 3ad | 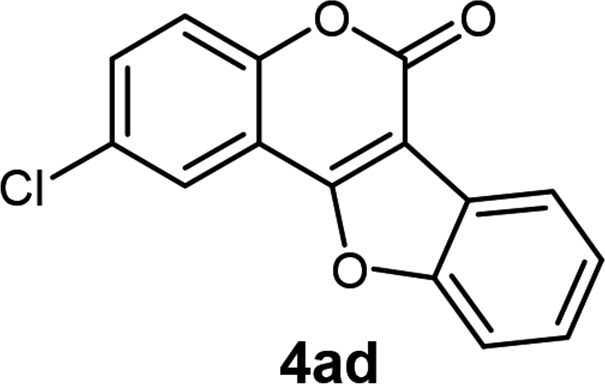 |
90 |
| 5 | 3ae |  |
79 |
| 6 | 3af | 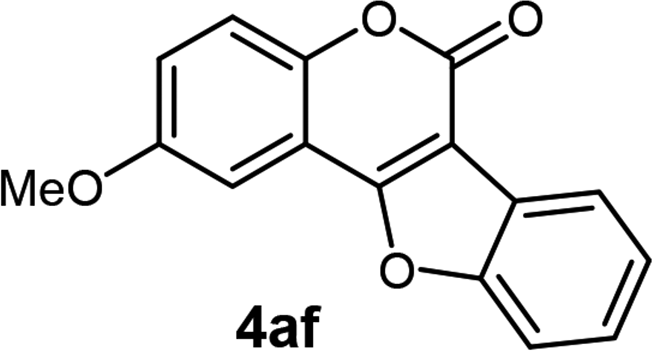 |
78 |
| 7 | 3ag | 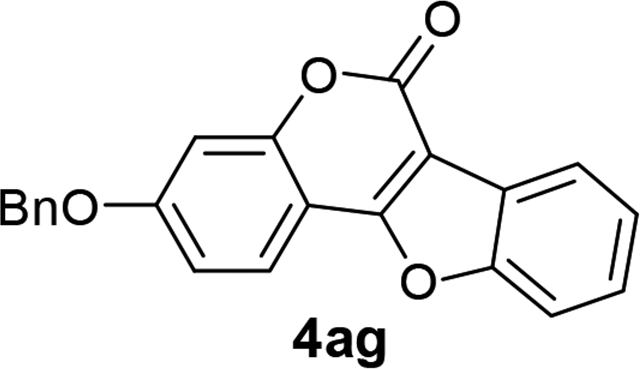 |
96 |
| 8 | 3ah | 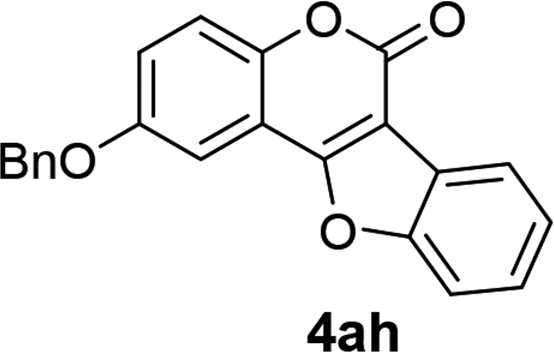 |
89 |
| 9 | 3ai | 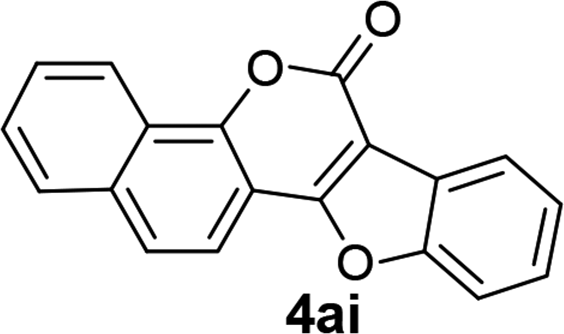 |
89 |
| 10 | 3aj | 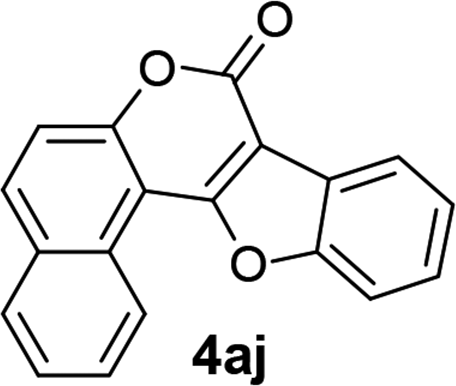 |
91 |
| 11 | 3ba | 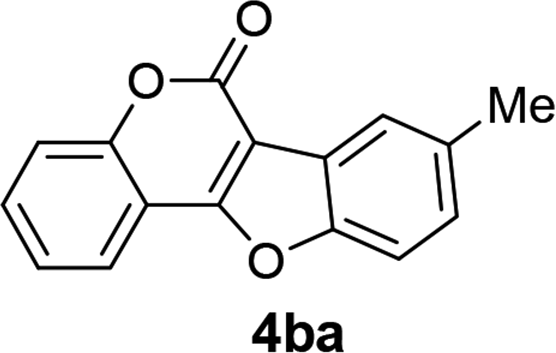 |
93 |
| 12 | 3ca | 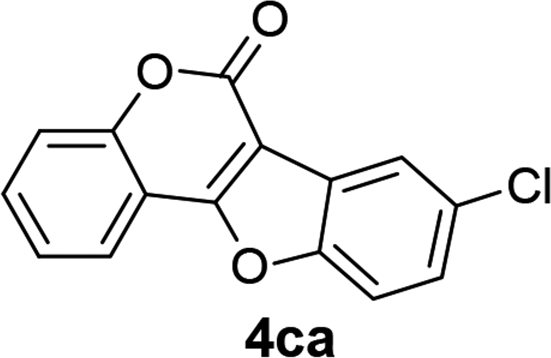 |
75 |
| 13 | 3da | 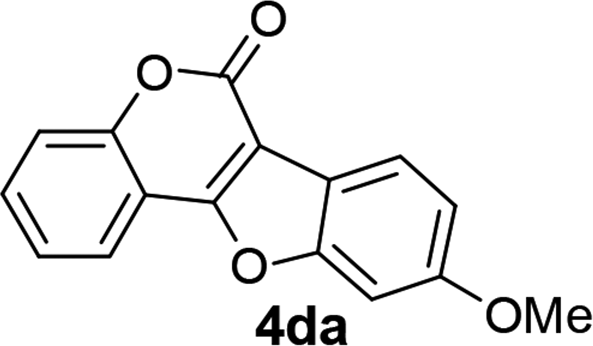 |
81 |
| 14 | 3ea | 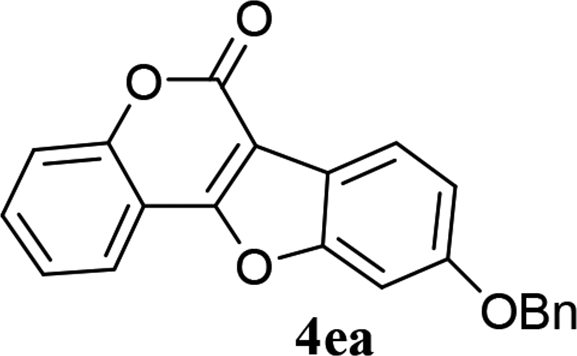 |
73 |
In the course of optimization study as we are isolating some amount of dehalogenated product, 5aa, it may be envisaged that the annulation might be occurring through Pd/C-catalyzed C–C bond formation through double dehydrogenative cross-coupling in the presence of atmospheric oxygen. However, when 5aa was treated with Pd/C following method C in the presence of O2 atmosphere, no conversion occurred. With this optimum reaction condition, we attempted the cyclization of synthesized 3-iodo-4-aryloxy coumarins 3 to coumestans 4. To our delight, in all cases, coumestans are produced in good to excellent yield.
Recycle and reusability is a prime concern in heterogeneous catalysis. Leaching and deactivation of catalyst often encountered by resulting in the decrease in the rate of reaction and hence yield of the isolated product. To investigate the reusability and catalytic efficiency of the catalyst in annulation process (3aa → 4aa), the workup process was modified slightly. After completion of the reaction, yield was calculated by GC. Then, to the reaction mixture another equiv. (0.1 mmol) of 3aa and 1.5 equiv. of the base were added for the 2nd catalytic cycle. Our effort to study the catalytic activity of Pd/C for three consecutive cycles reveals that yield of the annulation process is almost unaltered (cycle 1, 94%; cycle 2, 93%; and cycle 3, 92% GC yield) though the rate of the reactions becomes sluggish. Additionally, in another batch of reaction, after 7 h, the catalyst was filtered out under hot condition (61% yield from GC analysis) and the filtrate was heated at 140 °C for the completion of the reaction. Notably, after 30 h the yield (60%) of the reaction was not increased any more. This confirms that the leaching of Pd from the catalyst (Pd/C) is not significant for the completion of the reaction. It may be assumed that the annulation reaction proceeds through the usual oxidative addition to C–I bond, ortho C–H insertion and finally reductive elimination cycle11c to produce the coumestans 4. As leaching of Pd(0) is not occurring significantly to the solution, probably the overall transformation is occurring on the surface of the catalyst. Alternatively, after 1st catalytic cycle (after 16 h) the catalyst was filtered and washed with methanol and dried in an oven for 3 h at 140 °C (along with the filter paper). Next, the 2nd cycle was performed using the catalyst obtained from the first cycle (along with the filter paper) in the presence of 1.5 equiv. of NaOAc at 140 °C. After 24 h, the yield of the reaction was found to be 90%. For cycle 3 and 4 similar procedure was repeated, and yield of the annulation process was monitored after 30 h (88% yield) and 36 h (88% yield) respectively. This confirms that Pd/C is truly a heterogeneous catalyst and can be reusable for C–C coupling reactions to afford coumestans. Notably, the slight lowering of obtained yield for the catalytic cycles from the GC yield might be due to the handling loss, as the reaction was performed in small scale.
Conclusions
In summary, an efficient one-pot two step strategy for the synthesis of 3-iodo-4-aryloxy coumarin from the reaction of 4-hydroxy coumarin and aryl iodide was developed. Both electron-donating and withdrawing groups are observed to be inert towards sequential ortho-iodination and O-arylation reaction. Intramolecular annulation leading to a series of coumestans through C–C bond formation was achieved using commercially viable Pd on charcoal (Pd/C) catalyst under ligand free conditions. Simple workup procedure and utilization of low-cost reusable catalyst in the present method show promising synthetic potential in exploring biologically potent compounds of the coumestan family. Application of this method for the total synthesis of psoralidin, medicagol and isoglycyrol is going on in our laboratory.Experimental
General information
Unless otherwise noted, the reagents (chemicals) were purchased from commercial sources and used without further purification. The reactions were monitored by TLC, and the residue was chromatographed on silica gel (Ranchem, India), using an ethyl acetate–petroleum ether (60–80 °C) mixture as eluent. All NMR spectra were recorded on a 400 MHz (for 1H NMR) and 100 MHz (for 13C NMR) NMR spectrometer, and chemical shifts were expressed in δ units relative to the TMS signal as an internal reference in CDCl3. Multiplicities are indicated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet, when multiplicity is complex) for 1H NMR. Coupling constants, J were reported in Hz. High-resolution mass spectrometry (ESI-HRMS) (Agilent 6520 Q-TOF) was used to determine the elemental composition.General procedure for synthesis of 4-hydroxy coumarins
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) to give the desired nitro compounds (2k and 2l) as white crystalline solid.
:3) to give the desired nitro compounds (2k and 2l) as white crystalline solid.General procedure for the synthesis of 3-iodo-4-aryloxy coumarins
In a dry sealed tube, a mixture of aryl iodide (1.5 equiv.), m-CPBA (2 equiv.), and pivalic acid (3 equiv.) in 3 mL of toluene was added and stirred at 50 °C, during which yellowish white solid was formed. After 5 h, 4-hydroxy coumarin (0.1 mmol) was added to the reaction mixture and heated at 120 °C for 12 h. Then, it was cooled to room temperature and 20 mL of water was added and extracted with dichloromethane (3 × 15 mL). The combined organic layer was washed with dilute sodium bicarbonate (10%) solution (2 × 10 mL). Then, the solvent was removed under reduced pressure, and the crude reaction mixture was purified by column chromatography over silica gel (60–120 mesh) using mixture of ethyl acetate and petroleum ether as eluent to give 3-iodo-4-aryloxy coumarins.General procedure for the Pd/C-catalyzed synthesis of coumestans
To a mixture of 3-iodo-4-aryloxy coumarins (0.1 mmol) and NaOAc (0.15 mmol, 1.5 equiv.) in DMA (3 mL) was added 3 mg (0.3 mol%) of Pd/C (10 wt%) and the reaction mixture was heated at 130–140 °C for 16 h. After completion of the reaction (monitored by TLC), it was cooled to room temperature and poured into water (10 mL). Then, the product was extracted with ether (3 × 15 mL), and solvent was removed by rotary evaporator. The crude product was purified by column chromatography over silica gel (60–120 mesh) using a mixture of ethyl acetate and petroleum ether as eluent to afford the coumestans.Reusability study
To a mixture of 3aa (0.5 g, 1.37 mmol) and NaOAc (168.5 mg, 2.05 mmol) in DMA (7 mL) was added 41 mg of Pd/C (10 wt%) and the reaction mixture was heated at 140 °C for 16 h. After completion of the reaction (monitored by TLC), it was cooled to room temperature and sampling was made for GC analysis, which shows 95% yield of the product. For the 2nd cycle, to the original reaction mixture additional amount of 3aa (0.5 g, 1.37 mmol) and NaOAc (168.5 mg, 2.05 mmol) was added and heated at the same temperature for 20 h. Then, the reaction mixture was cooled to room temperature and yield of the reaction was calculated to be 93%. For 3rd cycle additional 0.5 g (1.37 mmol) of 3aa and NaOAc (168.5 mg, 2.05 mmol) was added and heated at 140 °C. After 30 h, the yield of the reaction was calculated to be 92% (by GC analysis). For the isolation of the product, the reaction mixture was poured into water (50 mL) and extracted with ether (3 × 25 mL). The combined organic layer was dried over anhydrous sodium sulphate and solvent was removed under reduced pressure. The crude product was purified by column chromatography over silica gel (60–120 mesh) using a mixture of ethyl acetate and petroleum ether as eluent to afford the coumestan 4aa (894 mg from 1.5 g of 3aa in 92% over all yield from three consecutive batches). Following general procedure, the isolated yield for the 1st cycle is 306 mg from 0.5 g of 3aa (94.4% yield) which is agreeable with the GC yield (95%).Analytical data
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors like to thank SERB (Ref. No. SB/S1/OC-21/2014) and CSIR (Ref. No. 02(0278)/16/EMR-II), Government of India for financial support.Notes and references
- (a) P. M. Dewick, in The Flavonoids – Advances in Research Since 1986, ed. J. B. Harborne, Chapman and Hall, London, 1994, p. 117 Search PubMed; (b) T. Nehybova, J. Smarda and P. Benes, Anti-Cancer Agents Med. Chem., 2014, 14, 1351–1362 CrossRef CAS; (c) V. A. Tuskaev, Pharm. Chem. J., 2013, 47, 1 CrossRef CAS; (d) M. Y. Xu and Y. S. Kim, Food Chem. Toxicol., 2014, 74, 311 CrossRef CAS PubMed; (e) T. Nehybova, J. Smarda, L. Daniel, J. Brezovsky and P. Benes, J. Steroid Biochem. Mol. Biol., 2015, 152, 76 CrossRef CAS PubMed; (f) N. Tsutsumi, Biol. Pharm. Bull., 1995, 18, 1012 CrossRef CAS PubMed; (g) A. J. M. Da Silva, P. A. Melo, N. M. V. Silva, F. V. Brito, C. D. Buarque, D. V. de Souza, V. P. Rodrigues, E. S. C. Pocas, F. Noel, E. X. Albuquerque and P. R. R. Costa, Bioorg. Med. Chem. Lett., 2001, 11, 283 CrossRef CAS PubMed; (h) W. Wang, Y. Y. Zhao, H. Liang, Q. Jia and H. B. Chen, J. Nat. Prod., 2006, 69, 876 CrossRef CAS PubMed; (i) P. H. Nguyen, T. N. A. Nguyen, T. T. Dao, H. W. Kang, D. T. Ndinteh, J. T. Mbafor and W. K. Oh, J. Nat. Prod., 2010, 73, 598 CrossRef CAS PubMed; (j) P. L. Whitten and F. Naftolin, J. Clin. Endocrinol. Metab., 1998, 12, 667 CAS; (k) N. Tsutsumi, Biol. Pharm. Bull., 1995, 18, 1012 CrossRef CAS PubMed.
- S. Kolukula and R. J. Anderson, J. Cancer Sci. Ther., 2011, S1 Search PubMed.
- For selected syntheses of coumestans, see: (a) L. Jurd, Tetrahedron Lett., 1963, 4, 1151 CrossRef; (b) T. Kappe and A. Z. Brandner, Z. Naturforsch., B: Chem. Sci., 1974, 29, 292 CAS; (c) R. Laschober and T. Kappe, Synthesis, 1990, 387 CrossRef CAS; (d) K. Hiroya, N. Suzuki, A. Yasuhara, Y. Egawa, A. Kasano and T. Sakamoto, J. Chem. Soc., Perkin Trans. 1, 2000, 4339 RSC; (e) G. A. Kraus and N. Zhang, J. Org. Chem., 2000, 65, 5644 CrossRef CAS PubMed; (f) C. C. Li, Z. X. Xie, Y. D. Zhang, J. H. Chen and Z. Yang, J. Org. Chem., 2003, 68, 8500 CrossRef CAS PubMed; (g) N. Takeda, O. Miyata and T. Naito, Eur. J. Org. Chem., 2007, 1491 CrossRef CAS; (h) T. Tricotet, P. Fleming, J. Cotter, A.-M. L. Hogan, C. Strohmann, V. H. Gessner and D. F. O'Shea, J. Am. Chem. Soc., 2009, 131, 3142 CrossRef CAS PubMed; (i) T. Yao, D. Yue and R. C. Larock, J. Org. Chem., 2005, 70, 9985 CrossRef CAS PubMed; (j) K. Neog, A. Borah and P. Gogoi, J. Org. Chem., 2016, 81, 11971 CrossRef CAS PubMed; (k) J. Sheng, T. Xu, E. Zhang, X. Zhang, W. Wei and Y. Zou, J. Nat. Prod., 2016, 79, 2749 CrossRef CAS PubMed.
- O. N. Emerson and E. M. Bickoff, J. Am. Chem. Soc., 1958, 80, 4381 CrossRef CAS.
- L. Tang, Y. Pang, Q. Yan, L. Shi, J. Huang, Y. Du and K. Zhao, J. Org. Chem., 2011, 76, 2744 CrossRef CAS PubMed.
- R. Laschober and T. Kappe, Synthesis, 1990, 387 CrossRef CAS.
- A. R. Kapdi, A. Karbelkar, M. Naik, S. Pednekar, C. Fischer, C. Schulzke and M. Tromp, RSC Adv., 2013, 3, 20905 RSC.
- F. Hong, Y. Chen, B. Lu and J. Cheng, Adv. Synth. Catal., 2016, 358, 353 CrossRef CAS.
- M.-T. Nolan, L. M. Pardo, A. M. Prendergast and G. P. McGlacken, J. Org. Chem., 2015, 80, 10904 CrossRef CAS PubMed.
- (a) K. Mackey, L. M. Pardo, A. M. Prendergast, M.-T. Nolan, L. M. Bateman and G. P. McGlacken, Org. Lett., 2016, 18, 2540 CrossRef CAS PubMed; (b) C. Cheng, W.-W. Chen, B. Xua and M.-H. Xu, Org. Chem. Front., 2016, 3, 1111 RSC.
- (a) N. Panda and A. K. Jena, J. Org. Chem., 2012, 77, 9401 CrossRef CAS PubMed; (b) N. Panda and R. Muthkuri, New J. Chem., 2014, 38, 5727 RSC; (c) N. Panda and I. Mattan, J. Org. Chem., 2015, 80, 6590 CrossRef CAS PubMed; (d) N. Panda, P. Mishra and I. Mattan, J. Org. Chem., 2016, 81, 1047 CrossRef CAS PubMed; (e) N. Panda and S. A. Yadav, Tetrahedron, 2018 DOI:10.1016/j.tet.2018.02.012; (f) N. Panda and D. K. Nayak, Monatsh. Chem., 2018 DOI:10.1007/s00706-018-2152-8.
- N. Jalalian, E. E. Ishikawa, L. F. Jr Silva and B. Olofsson, Org. Lett., 2011, 13, 1552 CrossRef CAS PubMed.
- (a) G. Wells, A. Seaton and M. F. G. Stevens, J. Med. Chem., 2000, 43, 1550 CrossRef CAS PubMed; (b) M. Takaku, Y. Hayashi and H. Nozaki, Tetrahedron, 1970, 26, 1243 CrossRef CAS; (c) R. M. Moriarty, J. Org. Chem., 2005, 70, 2893 CrossRef CAS PubMed; (d) E. G. Bakalbassis, S. Spyroudis and E. Tsiotra, J. Org. Chem., 2006, 71, 7060 CrossRef CAS PubMed; (e) O. Prakash, D. Kumar, R. K. Saini and S. P. Singh, Tetrahedron Lett., 1994, 35, 4211 CrossRef CAS; (f) G. F. Koeer, Hypervalent Halogen Compounds, in The Chemistry of Functional Groups, Supplement, ed. D. S. Patai and Z. Rappoport, Wiley, Chichester, 1983, p. 774 Search PubMed.
- (a) D. Liang, W. Yu, N. Nguyen, J. R. Deschamps, G. H. Imler, Y. Li, A. D. MacKerell Jr, C. Jiang and F. Xue, J. Org. Chem., 2017, 82, 3589 CrossRef CAS PubMed; (b) M. Ochiai and K. Miyamoto, Eur. J. Org. Chem., 2008, 4229 CrossRef CAS; (c) F. Berthiol, Synthesis, 2015, 47, 587 CrossRef CAS; (d) S. Maiti, T. K. Achar and P. Mal, Org. Lett., 2017, 19, 2006 CrossRef CAS PubMed; (e) R. D. Richardson and T. Wirth, Angew. Chem., Int. Ed., 2006, 45, 4402 CrossRef CAS PubMed; (f) M. Ochiai, Chem. Rec., 2007, 7, 12 CrossRef CAS PubMed; (g) T. Dohi, A. Maruyama, M. Yoshimura, K. Morimoto, H. Tohma and Y. Kita, Angew. Chem., Int. Ed., 2005, 44, 6193 CrossRef CAS PubMed; (h) T. Dohi, A. Maruyama, Y. Minamitsuji, N. Takenaga and Y. Kita, Chem. Commun., 2007, 1224 RSC; (i) T. Yakura and T. Konishi, Synlett, 2007, 765 CrossRef CAS; (j) T. Dohi, Y. Minamitsuji, A. Maruyama, S. Hirose and Y. Kita, Org. Lett., 2008, 10, 3559 CrossRef CAS PubMed; (k) H. Liu and C.-H. Tan, Tetrahedron Lett., 2007, 48, 8220 CrossRef CAS; (l) A. Moroda and H. Togo, Synthesis, 2008, 1257 CAS; (m) K. Miyamoto, Y. Sei, K. Yamaguchi and M. Ochiai, J. Am. Chem. Soc., 2009, 131, 1382 CrossRef CAS PubMed.
- Y. Zhao, H. Yan, H. Lu, Z. Huang and A. Lei, Chem. Commun., 2016, 52, 11366 RSC.
- For Pd/C-catalyzed C-C bond forming reactions see: (a) G. Marck, A. Villiger and R. Buchecker, Tetrahedron Lett., 1994, 35, 3277 CrossRef CAS; (b) D. Gala, A. Stamford, J. Jenkins and M. Kugelman, Org. Process Res. Dev., 1997, 1, 163 CrossRef CAS; (c) D. S. Ennis, J. McManus, W. WoodKaczmar, J. Richardson, G. E. Smith and A. Carstairs, Org. Process Res. Dev., 1999, 3, 248 CrossRef CAS; (d) R. R. LeBlond, A. T. Andrews, Y. Sun and J. R. Jr. Sowa, Org. Lett., 2001, 3, 1555 CrossRef; (e) B. Schmidt, M. Riemer and M. Karras, J. Org. Chem., 2013, 78, 8680 CrossRef CAS PubMed; (f) F.-X. Felpin, T. Ayad and S. Mitra, Eur. J. Org. Chem., 2006, 2679 CrossRef CAS; (g) F.-X. Felpin, E. Fouquet and C. Zakri, Adv. Synth. Catal., 2008, 350, 2559 CrossRef CAS; (h) F.-X. Felpin, O. Ibarguren, L. Nassar-Hardy and E. Fouquet, J. Org. Chem., 2009, 74, 1349 CrossRef CAS PubMed; (i) F.-X. Felpin, E. Fouquet and C. Zakri, Adv. Synth. Catal., 2009, 351, 649 CrossRef CAS; (j) F.-X. Felpin and E. Fouquet, Chem.–Eur. J., 2010, 16, 12440 CrossRef CAS PubMed; (k) C. Rossy, E. Fouquet and F.-X. Felpin, Synthesis, 2012, 44, 37 CrossRef CAS; (l) H. Sakurai, T. Tsukuda and T. Hirao, J. Org. Chem., 2002, 67, 2721 CrossRef CAS PubMed.
- P. K. Ambre, R. R. S. Pissurlenkar, R. D. Wavhale, M. S. Shaikh, V. M. Khedkar, B. Wan, S. G. Franzblau and E. C. Coutinho, Med. Chem. Res., 2014, 23, 2564 CrossRef CAS.
- J.-C. Jung, Y.-J. Jung and O.-S. Park, Synth. Commun., 2001, 31, 1195 CrossRef CAS.
- C.-N. Huang, P.-Y. Kuo, C.-H. Lin and D.-Y. Yang, Tetrahedron, 2007, 63, 10025 CrossRef CAS.
- D.-H. Gong, C.-Z. Li and C.-Y. Yuan, Chin. J. Chem. Phys., 2001, 19, 522 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12419h |
| This journal is © The Royal Society of Chemistry 2018 |