DOI:
10.1039/C7RA12360D
(Paper)
RSC Adv., 2018,
8, 5373-5381
Facile complexation reactions for the selective spectrofluorimetric determination of albendazole in oral dosage forms and spiked human plasma
Received
12th November 2017
, Accepted 14th January 2018
First published on 31st January 2018
Abstract
Two simple, sensitive, and rapid spectrofluorimetric methods were developed and validated for the determination of albendazole. The first method (method I) was based on the quenching effect of albendazole on the native fluorescence of erythrosine B. The fluorescence intensity was measured at 554 nm after extraction at 527 nm. In the second method (method II) the drug was reacted with lanthanum(III) ions to form a metal complex, which was measured at 340 nm after excitation at 295 nm. The suitable pH was 3.4 (Teorell–Stenhagen buffer) and pH 5.5 (phosphate buffer solution), for method I and II, respectively. The influence of experimental factors on the fluorescence intensity of the reaction products was investigated and optimized. The linear concentration ranges were 0.2–3.5 and 0.06–0.90 μg mL−1, with detection limits of 0.049 and 0.019 μg mL−1 for method I and II, respectively. ICH guidelines were followed for validation of the developed procedures, and the results were acceptable. The Gibb's free energy change of the reactions was −24.6 and −27.5 kJ mol−1 for method I and II, respectively. These negative values indicated the high feasibility of these reactions at ambient temperature. The proposed procedures were applied successfully for the determination of albendazole in commercial dosage forms and spiked human plasma. The results showed high precision, accuracy and recovery of the reported methods without any significant interference from pharmaceutical excipients or plasma components.
1. Introduction
Albendazole (ABZ, Fig. 1) is an anthelmintic drug belonging to the benzimidazole group which is commonly applied in the treatment of infection caused by a wide range of worms such as pin worms, round worms, hookworms and whipworms.1 ABZ is still an effective treatment for hydrated cysts, echinococcosis, and neurocysticercosis. ABZ as a benzimidazole drug produces its effect through specific binding to tubulin, the microtubule subunit protein. This binding will interrupt the structure and function of the microtubule and hence interfere with the microtubule-mediated transport of secretory vesicles in absorptive tissues of the worm.2 Due to the great clinical importance of ABZ, several analytical methods have been reported for its determination either in pharmaceutical dosage forms or in biological fluids. The reported analytical methods include: spectrophotometry,3–9 spectrofluorimetry,10–13 high-performance liquid chromatography (HPLC)14–17 HPTLC,18 thermogravimetry19 and electrochemical methods.20,21
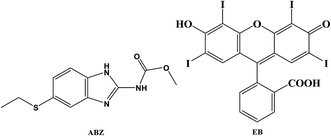 |
| | Fig. 1 Chemical structure of albendazole (ABZ) and erythrosine B (EB). | |
Most of the reported spectrophotometric methods have low sensitivity. In addition, some of these methods were indirect and involved time consuming, multi steps and multi-reagents reactions. This could affect the accuracy and precision of these methods.6 In spite of the suitable sensitivity of the ion-pair complex based methods,7,8 they suffer from the extraction step which involve the use of hazardous organic solvents and the possibility of emulsion formation or incomplete extraction. HPLC consume large volumes of highly pure organic solvent and labor sample pretreatment steps. HPLC, capillary electrophoresis and electrochemical methods require relatively expensive and sophisticated instruments. On the other hand, the spectrofluorimetry apply a relatively simple procedure and easy to handle instrument. The reported spectrofluorimetric methods were not applied in the analysis of human plasma samples.10–13 In addition chloroform was used which is a hazardous organic solvent10 in one method while the charge transfer based method have low sensitivity (LOD = 2.0 μg mL−1).12
Erythrosine B (EB, Fig. 1) as an acidic dye, is a food colorant that has been used for the spectrofluorimetric determination of some basic compounds such as fluoroquinolones22 imipramine23 and proteins.24 As ABZ has a basic centre (secondary amino group), it can form an ion pair associate with the dye in weakly acidic solution. The complex formation process was accompanied by a significant quenching of the native fluorescence of EB. The mechanism of the fluorescence quenching was investigated using Stern–Volmer equation.
Another feature of ABZ molecule is the presence of the benzimidazole moiety which impart a native fluorescence for the drug. However, this fluorescence activity is very weak in aqueous solutions. Fortunately, the secondary amino and carbonyl groups of ABZ, give it the ability to form a stable complex with many metal ions. Lanthanide complexes have been used as fluorescent label for a wide variety of analytical techniques. It was found that, the weak native fluorescence of the drug was greatly enhanced by its metal complex reaction with lanthanum(III) ions.
In the present study, EB dye and La(III) ion were utilized for the development of two spectrofluorimetric methods for the determination of ABZ based on complex formation reactions. The fluorescence quenching in the first method and fluorescence intensity in the second method were directly proportional to the drug concentration. The mole ratios of both reactions were investigated, and the thermodynamic parameters were calculated. Both methods were applied for the determination of the cited drug in oral dosage forms and spiked human plasma.
The simplicity and low detection limits are among the advantages of the proposed methods over the previously reported methods. In addition the applied procedures were less time consuming and utilized: green solvents (water and methanol for method I and II, respectively), inexpensive reagents and relatively low price instrument. Furthermore, the developed methods could determine the cited drug with good recoveries without any interference from frequently encountered excipients or plasma components. Therefore, the proposed methods are suitable for the determination of the ABZ in its oral dosage forms in quality control laboratories.
2. Experimental
2.1. Instrumentation
All spectrofluorimetric measurements were carried out on SCINCO FluoroMate spectrofluorimeter (FS-2, Korea), equipped with 150 W Xe-arc lamp, PMT detector and 1 cm matched quartz cells. Super-mixer (GEMMY industrial CORD, Taiwan, R.O.C), pH-meter, model AD11P (Adwa, Romania) and bath sonicator (SONICOR SC-101TH) were also used. All weighing were performed on an electronic single pan balance (Precisa XB 220A, Switzerland). Distilled water was prepared by water distiller (TYUMEN-MIDI-A0-25 MO, Russia).
2.2. Chemicals and reagents
All chemicals were of analytical grade and reagent solutions were prepared fresh daily. Albendazole powder was kindly supplied by the Egyptian International Pharmaceutical Industries Company (EIPICO, Cairo, Egypt) and was used without further purification. Solution of 5.1 × 10−4 mol L−1 lanthanum ion was freshly prepared by dissolving 12.51 mg lanthanum(III) chloride (Panreac Chemicals, Lyon, France) in 100 mL distilled water. Erythrosine B (Market Harborough Leicestershire, UK) solution was prepared in distilled water as 0.015% g w/v (1.7 × 10−3 M). Cetyl pyridinium chloride (CPC), polyethylene glycol (PEG), sodium dodecyl sulfate (SDS) and Tween-80 were obtained from El-Nasr Chemical Company (Cairo, Egypt). The authors got the permission for using plasma sample of human volunteers from Minia University Hospital, Minia, Egypt, according to institutional guidelines. In all cases, informed written consent was obtained from all participants. Human plasma samples were kept frozen at 20 °C until assay after gentle thawing.
2.2.1. Phosphate buffer. Phosphate buffer solution was prepared by dissolving 8.95 g of disodium hydrogen phosphate and 3.40 g of potassium dihydrogen phosphate in 1 L of distilled water. The pH of the solution was adjusted to the required value using 1 mol L−1 phosphoric acid (El-Nasr Chemical Company, Cairo, Egypt).
2.2.2. Toerell–Stenhagen buffer. Toerell–Stenhagen25 buffer solution was prepared by dissolving 7 g citric acid (United Company for Chem. & Med. Prep., Cairo, Egypt) and 4 g sodium hydroxide (Isochem specifications, Cairo, Egypt) in 300 mL distilled water. Aliquot of 3.5 mL phosphoric acid (El-Nasr chemical company, Cairo, Egypt) was added, and the volume was completed to 1 L with distilled water. An appropriate volume of this stock solution was adjusted to the required pH using 0.1 mol L−1 hydrochloric acid (36%, Lab-Chem Fine Chemicals & Reagents, Cairo, Egypt).
2.3. Pharmaceutical formulations
The following pharmaceutical preparations were analyzed which were present in the local market; Alzental tablets (Egyptian International Pharmaceutical Industries Company, EIPICO, Cairo, Egypt) labeled to contain 200 mg of albendazole per tablet. Alzental suspension was labeled to contain 20 mg of albendazole per 1 mL.
2.4. Preparation of standard solution
An accurately weighed amount (10 mg) of ABZ was dissolved in 100 mL methanol. This gave a stock standard drug solution (0.1 mg mL−1) which was further diluted with the same solvent to obtain working standard drug solution containing concentrations covering the required range.
2.5. General analytical procedure
2.5.1. Procedure for method I. One milliliter of both Toerell–Stenhagen buffer solution (pH 3.4) and erythrosine B solution (1.7 × 10−4 M) were transferred to 10 mL calibrated flask followed by 1.0 mL of the working standard or sample drug solution. The solution was allowed to stand at room temperature for 5 min. The volume was completed with distilled water and the fluorescence intensity of the resulting solution was measured at 554.4 nm after excitation at 527 nm. A reagent blank was prepared and measured in the same manner.
2.5.2. Procedure for method II. Into 10 mL calibrated volumetric flask, the following solutions were added in order; 1.0 mL of working standard or sample solution of the studied drug, 1 mL of phosphate buffer solution (pH 5.5) and 1.4 mL of lanthanum chloride solution (5.1 × 10−4 M). The solution was set aside at room temperature for 15 min. Methanol was added to complete the volume to the mark, and the fluorescence intensity was measured at 340.3 nm after excitation at 295 nm. A blank was carried out in the same time without the addition of the drug solution.
2.6. Construction of calibration curves
Different aliquots of the standard drug solutions (2.0–35 μg mL−1 for method I or 0.7–9.0 μg mL−1 of method I) were transferred into a series of 10 mL calibrated volumetric flasks and the general analytical procedure was applied as previously mentioned. The calibration graph was obtained by plotting the corrected fluorescence intensity versus the final concentration of ABZ, and the corresponding regression equation was estimated.
2.7. Samples preparation of for analysis
2.7.1. Tablets. Ten tablets of ABZ were accurately weighed and finely powdered. An accurately weighed amount of the powder equivalent to 10 mg of the drug was transferred into a 100 mL volumetric flask. The drug was extracted from the powder by sonication with about 50 mL of methanol for about 25 min. After completion of the volume to the mark with the same solvent, the solution was filtered off and the first portion was discarded. The sample solution containing the required drug concentration (0.5 μg mL−1) was prepared by further dilution of the suitable volume of the filtrate. The general analytical procedure was applied on an aliquot of the final solution, and the drug content was calculated using the corresponding regression equation.
2.7.2. Suspension. Into 100 mL volumetric flask, 5.0 mL of the suspension was transferred followed by 50 mL of methanol and the same procedure described for tablets was applied.
2.7.3. Spiked human plasma. Into a heparinized tube, 5.0 mL of drug-free human blood sample was taken from healthy volunteer and centrifuged at 4000 rpm for 5 min. One milliliter of the drug-free plasma (supernatant) was transferred to 10 mL stoppered calibrated tube and spiked with 1.0 mL of the standard drug solution (0.5 μg mL−1). The protein content of the plasma in the mixture was precipitated by the addition of 3 mL acetonitrile and the volume was completed to 10 mL with methanol. The mixture was cooled in ice bath and centrifuged at 4000 rpm for 10 min. A suitable volumes of the supernatant containing ABZ within the recommended concentration range were subjected for drug analysis by applying the general procedure. Blank value was carried out by treating the drug-free blood sample in the same manner without adding the drug.
2.8. Determination of the reaction stoichiometry
Job's method of continuous variation26 was used to find out the molar ratios between the drug and the reagent. Equimolar solutions of both ABZ and the reagents (5.1 × 10−4 mol L−1 in case of lanthanum and 1.7 × 10−4 mol L−1 in case of erythrosine B) were prepared. Series of 1.0 mL solutions were made up comprising different complimentary proportions of the reagent and drug (0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0, 0.1:0.9, …, 0.9:0.1, 1.0:0) in 10 mL volumetric flasks. One milliliter of the buffer solution (Toerell–Stenhagen buffer, pH 3.4 and phosphate buffer solution, pH 5.5) was added to each flask and the general analytical procedure was followed as described previously.
:1.0, 0.1:0.9, …, 0.9:0.1, 1.0:0) in 10 mL volumetric flasks. One milliliter of the buffer solution (Toerell–Stenhagen buffer, pH 3.4 and phosphate buffer solution, pH 5.5) was added to each flask and the general analytical procedure was followed as described previously.
3. Results and discussion
The ability of ABZ to form complex with different substances was utilized for the development of two sensitive spectrofluorimetric methods. The basic nitrogen of the drug could form an ion pair complex with erythrosine, while both the secondary amino group and its neighbor carbonyl group give ABZ the capability to form metal complex.
3.1. Fluorescence spectra
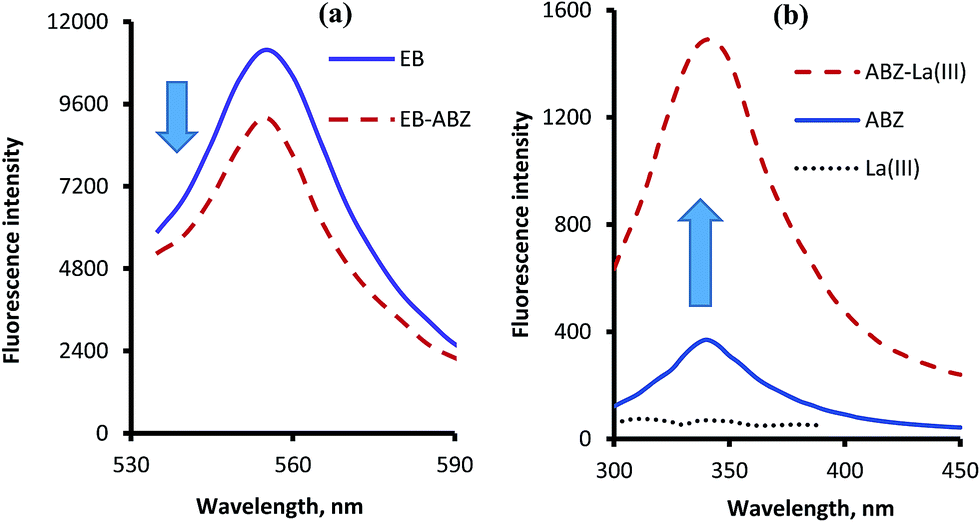
Erythrosine B is an acidic dye which has a native fluorescence appears at 554 nm after excitation at 527 nm. When the solution of ABZ was added to dye solution a significant reduction in the fluorescence intensity of the dye was observed as a result of the ion-pair complex formation. This represent the basis for the first spectrofluorimetric method. However, ABZ itself has a very weak native fluorescence in aqueous solution which could be greatly improved upon the addition of lanthanum ions. The stability of the complex was improved in the presence of phosphate buffer (pH 5.5). Based on this fact, another spectrofluorimetric method was constructed. The excitation and emission spectra of the studied reagents in the presence and absence of ABZ are shown in Fig. 2.
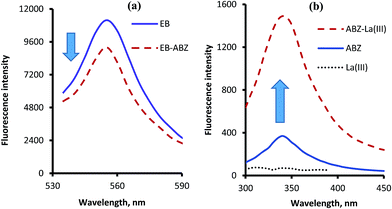 |
| | Fig. 2 Fluorescence emission spectra of the complexes of ABZ (1.0 μg mL−1) with erythrosine B (a) after excitation at 527 nm and with lanthanum(III) ions (b) after excitation at 295 nm. | |
3.2. Optimization of reaction conditions
The best reaction conditions were attained by changing each experimental variable individually and monitoring its effect on the fluorescence intensity. The investigated variables were buffer type, pH and volume, reagents concentration (La(III) or erythrosine B), diluting solvents and reaction time.
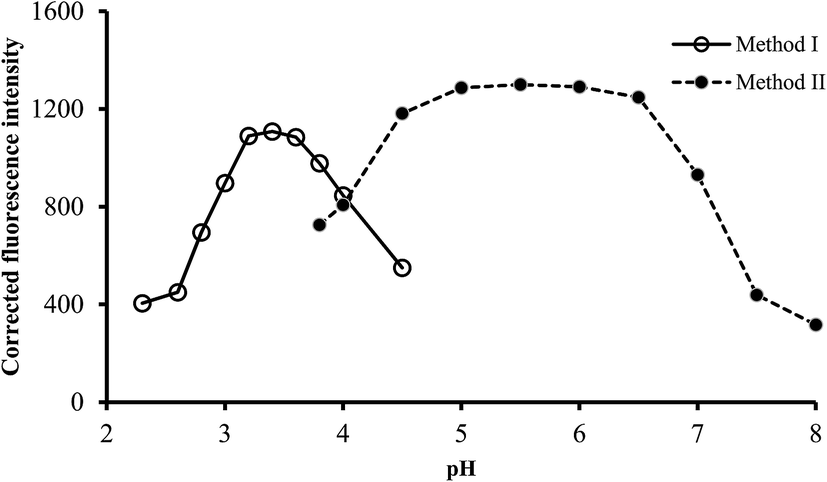
3.2.1. Effect of pH. It is well known that the pH of solution greatly affect the formation and stability of complexes. The effect of pH on the fluorescence intensity of the reaction product was examined using phosphate buffer or Teorell–Stenhagen solution, having pH in the range of 2.0–4.5 and 4.0–8.0 for method I and II, respectively. The highest fluorescence quenching was attained using pH 3.2–3.6 in method I, while in method II, maximum fluorescence intensity was obtained in the pH range of 5.0–6.0. Higher or lower pH values produced a gradually decreased in the obtained values (Fig. 3). As a result, buffer solutions of pH 3.4 and 5.5 were selected for the subsequent work for method I and II, respectively.
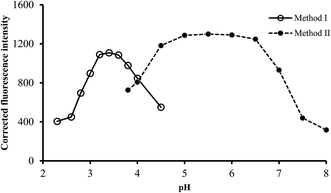 |
| | Fig. 3 Effect of pH on the fluorescence quenching (○) and fluorescence intensity (●) using of 0.5 and 1.0 μg L−1 of ABZ, for method I and II, respectively. | |
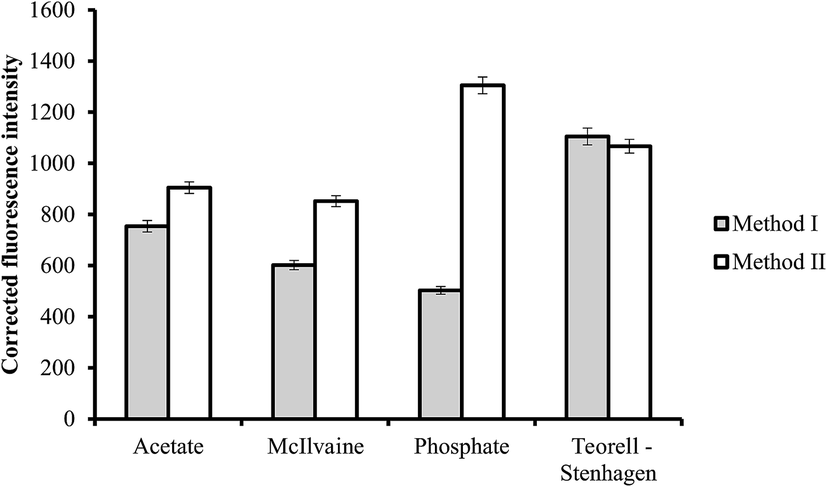
3.2.2. Effect of buffer type. The effect of the chemical composition of the buffer solution on the fluorescence intensity was examined by performing the general analytical procedure using different types of buffers (pH 3.4 for method I and 5.5 for method II). The investigated buffer solutions were acetate, McIlvaine, phosphate and Teorell–Stenhagen buffers (Fig. 4). Maximum fluorescence quenching was achieved using Teorell–Stenhagen buffer in case of method I. While phosphate buffer offered the highest fluorescence intensity, and therefore it was chosen as the optimum buffer in method II (Fig. 4).
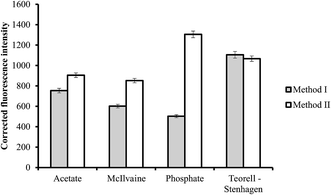 |
| | Fig. 4 Effect of buffer type on the fluorescence quenching (■) and fluorescence intensity (□) using of 0.5 and 1.0 μg L−1 of ABZ, for method I and II, respectively. | |
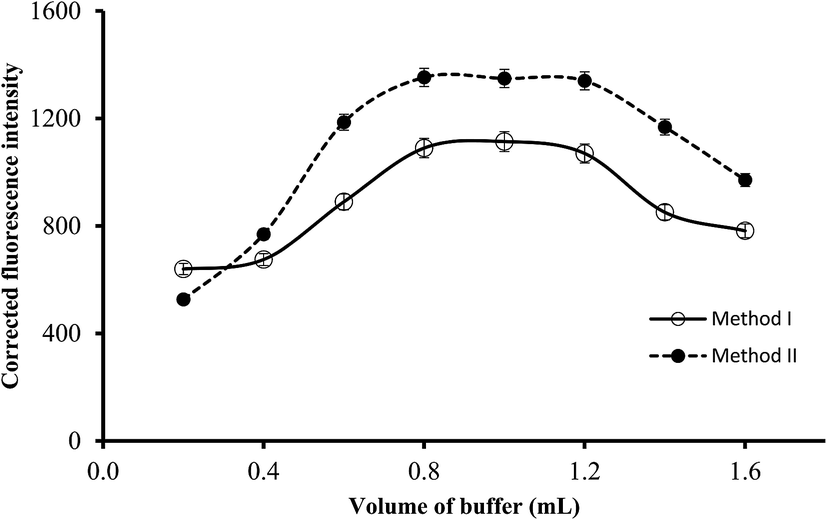
3.2.3. Effect of buffer volume. The volume of the buffer solution of the suitable pH (3.4 or 5.5) was varied in the range of 0.2–1.6 mL and the resulting fluorescence intensity of the blank and reaction product were measured. As shown in Fig. 5, the increase in the buffer volume resulted in a linear increase in the fluorescence intensity (or fluorescence quenching) of the reaction products. Maximum values were obtained using 0.8 mL and remained significantly unchanged up to 1.2 mL. Further increase in the buffer volume produced a slight or negligible decrease in the fluorescence intensity. Therefore, 1 mL of the buffer solution of the suitable pH was selected in both methods.
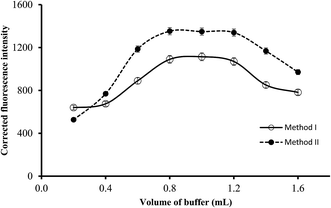 |
| | Fig. 5 Effect of buffer volume on the fluorescence quenching (○) and fluorescence intensity (●) using of 0.5 and 1.0 μg L−1 of ABZ, for method I and II, respectively. | |
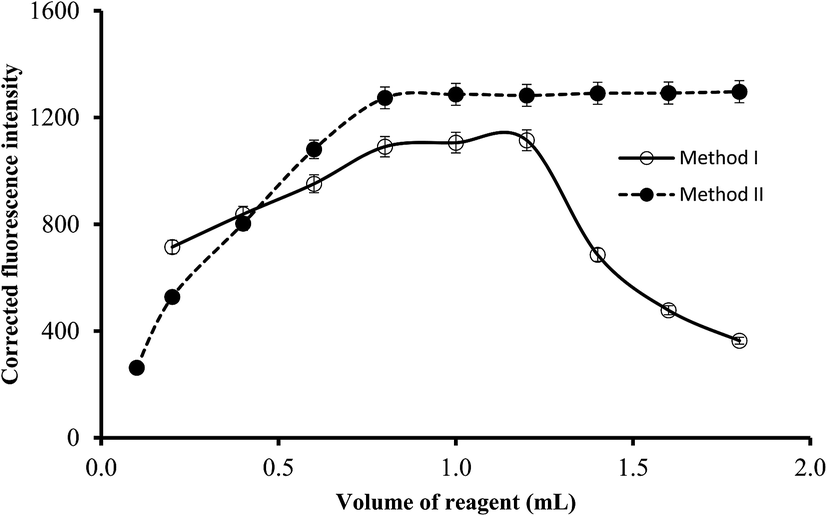
3.2.4. Effect of reagents concentration. The reagent concentrations were varied by using different volumes of the reagents solution (5.1 × 10−4 mol L−1 of La(III) or 1.7 × 10−4 mol L−1 of EB). It was observed that increasing the volume of the reagent resulted in gradual increase in the corresponding fluorescence quenching (or intensity in method II) of the reaction products up to 0.8 mL. Higher reagent volumes have no significant effect on the fluorescence intensity in method II. However in method I, a gradual reduction in the fluorescence quenching was obtained at dye volume higher than 1.2 mL which may be attributed to the formation of EB dimer by self-aggregation (Fig. 6). Thus, it was recommended that 1.0 mL of EB solution or 1.4 mL of La(III) chloride solution to be incorporated in the general analytical procedures.
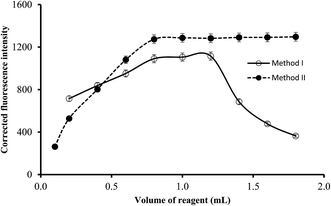 |
| | Fig. 6 The influence of reagent volume (1.7 × 10−4 mol L−1 of EB in method I and 5.1 × 10−4 mol L−1 of lanthanum(III) chloride in method II) on the fluorescence quenching (○) and fluorescence intensity (●) using of 0.5 and 1.0 μg L−1 of ABZ, for method I and II, respectively. | |
3.2.5. Effect of diluting solvents. After completion of the reaction between the reagents and ABZ (1.0 and 0.5 μg mL−1 for method I and II, respectively), the reaction mixture was diluted with different solvents. It was observed that the highest fluorescence quenching was obtained with water (ΔFI = 1309) in method I. Low quenching values were obtained in the presence of diethyl ether (13), dioxane (603) and methanol (276). On the other hand, ethanol (−1329), produced fluorescence enhancement rather than quenching. Therefore, water was the most appropriate solvent for dilution. In method II, it was found that a very low fluorescence intensity was observed in the case of ethanol (66) and dioxin (100). The intensity in the presence of the other of solvents increased in the following order; 2-propanol (715) < acetonitrile (776) < water (918) < DMSO (1269) < methanol (1309). Consequently, methanol was used as the diluting solvent as it gave the highest fluorescence intensity. The selected solvents; (water for method I and methanol for method II), are green solvents although water is more preferable. Both are readily available, cheap and environmentally safe which make the proposed methods superior over the reported spectrofluorimetric method27 which was carried out in DMSO.
3.2.6. Effect of reaction time. The reaction between the drug and the reagents were studied at different time interval after mixing at room temperature. It was observed that, the reaction between ABZ and EB was very fast and completed within 4 min. In the case of the reaction of ABZ with La(III), the fluorescence intensity reached its maximum value within about 12 min. In both reactions the fluorescence intensity remained unchanged for at least 60 min. In order to make sure that the reaction has been completed, the fluorescence intensity was measured after 5 and 15 min for method I and II, respectively.
3.3. Determination of the reactions stoichiometry
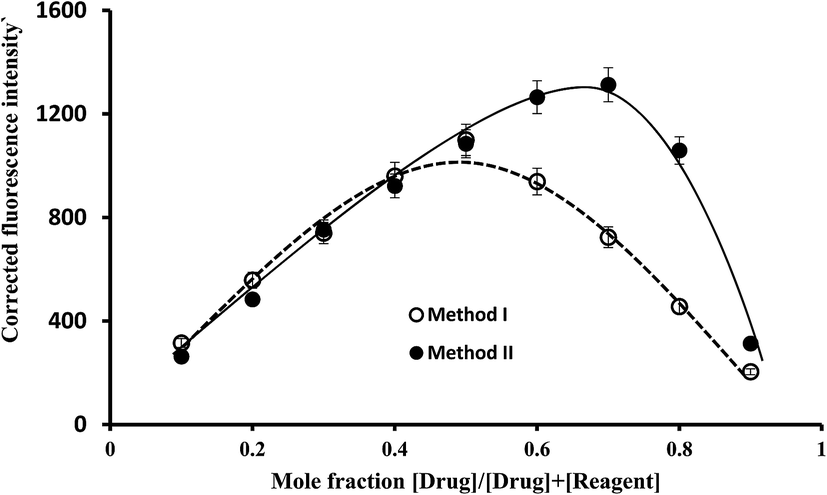
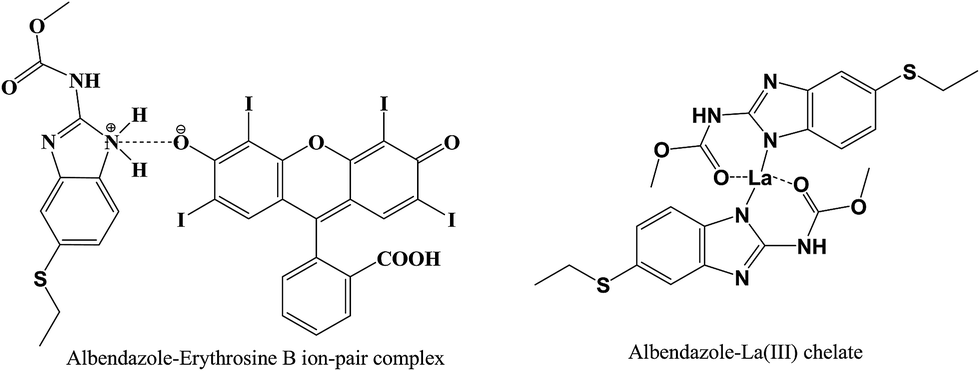
The ratio between ABZ and the reagent was estimated by applying Job's method of continuous-variation. A series of solutions containing various molar ratios of the drug and the reagents (EB or La(III)) were prepared keeping the total moles in all solutions constant. The fluorescence intensities of at the specific wavelengths for each solution was measured. In method I, the observed fluorescence intensity of each mixture at various mole fractions was subtracted from that of the solutions containing the same concentration of the dye alone. While, in method II, the fluorescence intensity of the blank (containing the La(III) ions alone) was subtracted from that of the solution experiment. The difference in the fluorescence intensities was plotted against the mole fraction of the drug, Fig. 7. Curves were obtained in which the position of the maxima indicated that the molar ratio was 1:1 for EB:ABZ and 1:2 for La(III):ABZ. Based on the estimated molar ratios, the chemical structure of the formed complexes were presented in Fig. 8.
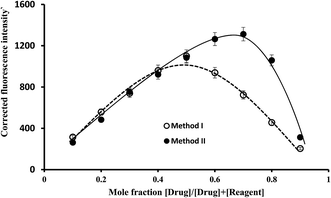 |
| | Fig. 7 Job's plots for molar ratio evaluation for the complex formation reaction using equimolar solutions of the drug and the reagent (1.7 × 10−4 mol L−1 of EB in method I and 5.1 × 10−4 mol L−1 of lanthanum(III) chloride for method II). | |
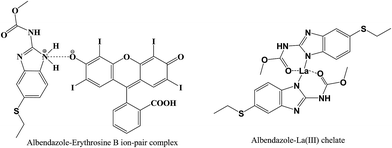 |
| | Fig. 8 The suggested chemical structure of the complexes of ABZ with either erythrosine B or lanthanum(III) ions. | |
In weakly acidic medium (pH 3.4), EB will dissociate to form a mono-valent anion in which the ionization of the hydroxyl group is predominant than carboxylic group of the phenyl ring. This is due to the presence of two strong electron withdrawing groups (iodine atoms) on the xanthene ring adjacent to the hydroxyl. The negatively charged mono-valent anion of EB will interact with the drug cation through both electrostatic interaction and hydrophobic forces to form the ion pair complex.
The mechanism of metal complex formation between ABZ and La(III) was similar to the previously reported complex between ferric(III) and ABZ.28 Accordingly, in the proposed chemical structure of La(III)–ABZ chelate, the drug molecule acts as a neutral bidentate ligand through the carbonyl group of the side chain and NH of five-member ring with octahedral fashion toward La(III) ions.
Furthermore, the quenching mechanism of ABZ on the native fluorescence of EB was studied by applying Stern–Volmer method. Stern–Volmer plot was constructed by plotting the fluorescence intensities in the absence (F0) and presence (F) of the quencher (the drug) against the quencher concentration [Q]. The obtained straight line was fitted to the Stern–Volmer equation.29
The intercept of the straight line is approximately 1 and the slope (2.45 × 104) is called Stern–Volmer constant (KSV) which in turn is the product of the natural radiation lifetime (the lifetime in the absence of quencher), τ0, and the quenching rate constant, Kq, (KSV = Kqτ0). This equation provides an important way for the estimation of the quenching rate constant. Since the reported fluorescence lifetime of erythrosine B is 89 ps,30 hence, the calculated quenching rate constant value is 2.49 × 1014 L mol−1 s−1. This value is significantly higher than the largest reported Kq for collisional quenching (2 × 1010 L mol−1 s−1).31 Consequently, the assumed quenching mechanism for the interaction between ABZ and erythrosine B is considered a single static process.
3.4. The rate constants and free energy changes of the reactions
The reaction rate constant (K) for the reaction between ABZ and EB could be calculated using following equation.32| | |
log((F0 − F)/F) = logK + nlog[D]
| (1) |
where, [D] is the molar concentration of the drug, F0 and F are the fluorescence intensities of the dye in the absence and presence of the drug, respectively. Results for the curve fitting of eqn (1) are summarized in Table 1. The rate constant for the reaction was calculated from the intercept and was found to be 2.07 × 104. The number of mole (n) of ABZ was found to be approximately one (0.96) which give an additional confirmation for the molar ratio (1:1) in the drug–dye ion pair complex.
Table 1 Curve fitting data of eqn (1) and (2) for calculation of the reaction rate constants between ABZ and EB or La(III)
| Parameter |
ABZ–EB |
ABZ–La(III) |
| Equation no. |
(1) |
(2) |
| Slope |
0.9623 |
2.04 × 10−9 |
| Intercept |
4.3166 |
1.32 × 10−4 |
| Correlation coefficient, r2 |
0.9965 |
0.9996 |
| Reaction rate constant, K |
2.07 × 104 |
6.48 × 104 |
| Gibbs free energy, (ΔG°, kJ mol−1) |
−24.63 |
−27.45 |
For the reaction between lanthanum and the drug, the binding constant (K) was calculated using the modified Benesi–Hildebrand equation:33
| | |
1/ΔF = 1/ΔFmax + 1/K[D]ΔFmax
| (2) |
| ΔF = F − F0 and ΔFmax = Fmax − F0 |
where;
F0,
F, and
Fmax are the fluorescence intensities of: the blank, the metal ions in the presence of the drug, and at full saturation, respectively. [D] is the molar drug concentration. Straight line was obtained when 1/Δ
F was plotted
versus 1/[D]. The value of
K was obtained from the values of both the slope and the intercept (
Table 1). The calculated value was 6.48 × 10
4 for lanthanum complexes with ABZ.
Gibb's free energy (ΔG°) of the reactions can be calculated from the obtained reaction rate constants using the formula: ΔG° = −2.303RTlogK, where, R is the gas constant (8.314 J K−1 mol−1), T is the absolute temperature in Kelvin and K is the rate constant. The estimated values for ΔG° were −24.6 and −27.5 kJ mol−1 for EB–ABZ and La–ABZ reactions, respectively. Since the values of the obtained free energy change are largely negative, it is expected that both reactions have high spontaneity and feasibility at room temperature.
3.5. Validation of the developed method
To confirm the feasibility of the developed procedure in routine analysis of ABZ in real samples, the procedure was fully validated according to guidelines of the International Conference on Harmonization (ICH).34 The studied parameters included; linearity and range, accuracy and precision, sensitivity, selectivity and robustness.
3.5.1. Range and linearity. Different standard solutions of ABZ having different concentrations were subjected to analysis using the general analytical procedures. The fluorescence intensities in the absence and presence of the drug were measured. The fluorescence quenching (blank-experiment) in case of method I or the corrected florescence intensity (experiment – blank) in case of method II was plotted versus the corresponding drug concentration. The analytical parameters were calculated by applying linear regression analysis. The fluorescence quenching (or fluorescence intensity for method II) was directly proportional to the drug concentration in the linear range of 0.2–3.5 and 0.06–0.90 μg mL−1 of ABZ for method II and II, respectively. The obtained correlation coefficients were very close to the unit (>0.9996) prove the high linearity of the methods. Other analytical parameters of the proposed methods are summarized in (Table 2).
Table 2 Analytical parameters of the proposed spectrofluorimetric methods for the determination of ABZ with EB and La(III) complexation
| Parameters |
Method I |
Method II |
| Linear range (μg mL−1) |
0.2–3.5 |
0.06–0.90 |
| Intercept (a) |
247.2 |
57.07 |
| Standard deviation of intercept |
12.63 |
7.47 |
| Slop (b) |
850.7 |
1252 |
| Standard deviation of slope |
5.35 |
7.88 |
| Correlation coefficient (r) |
0.9998 |
0.9998 |
| Determination coefficient (r2) |
0.9996 |
0.9996 |
| Limit of detection, LOD (μg mL−1) |
0.049 |
0.019 |
| Limit of quantitation LOQ (μg mL−1) |
0.15 |
0.059 |
3.5.2. Detection and quantitation limits. For evaluation of the sensitivity of the proposed methods, the detection and quantitation limits were calculated using the formula; LOD = 3.3σ/S and LOQ = 10σ/S, respectively, where σ is the standard deviation of intercept and S is the slope of calibration curve. It was found that values of LOD were 0.048 and 0.019 μg mL−1 while LOQ were 0.150 and 0.059 μg mL−1 for method I and II, respectively (Table 2). These values, which are in the nano-gram scale indicated the high sensitivity of both methods. However, metal complexation method was three fold more sensitive than the other method.
3.5.3. Accuracy. Accuracy of the proposed method was checked at three concentration levels within the recommended range (0.5, 1.50, and 2.50 μg mL−1 for method I and 0.10, 0.50, and 0.70 μg mL−1 for method II). Three replicates were measured for each concentration and the results were expressed as % recovery, RSD and percentage relative error (Er%) (Table 3). The high accuracy of the suggested methods was proved from the closeness of the obtained % recovery to 100% and the low value of Er%. This level is satisfactory for the determination of this drug during routine analysis.
Table 3 Accuracy of the proposed spectrofluorimetric methods for the analysis of the studied drug at three concentration levels
| Drug |
Taken (μg mL−1) |
Founda (μg mL−1) |
Recovery ± % SDa |
RSDb% |
Erb% |
| The value is the mean of three replicates measurement, SD is the standard deviation. RSD is the relative standard deviation and Er% is the relative error percentage. |
| Method I |
0.5 |
0.502 |
100.34 ± 0.39 |
0.39 |
0.34 |
| 1.5 |
1.506 |
100.39 ± 0.393 |
0.391 |
0.39 |
| 2.5 |
2.493 |
99.73 ± 0.587 |
0.589 |
−0.27 |
| Method II |
0.1 |
0.1003 |
100.32 ± 0.96 |
0.96 |
0.32 |
| 0.5 |
0.5001 |
100.02 ± 0.88 |
0.88 |
0.02 |
| 0.7 |
0.704 |
100.53 ± 0.92 |
0.91 |
0.53 |
3.5.4. Precision. Two levels of precision were examined for the developed methods; intra-day and inter-day (intermediate) precision. Three independent analyses using three different drug concentrations were performed within the same day to evaluate the intra-day precision and were carried out on three consecutive days in case of inter-day precision. The low value of the calculated relative standard deviation which did not exceed 2.0%, gave an indication about the acceptable level of precision of the methods (Table 4) and its high level of repeatability and reproducibility.
Table 4 Intra-day and inter-day precisions of the proposed spectrofluorimetric methods
| Drug |
Conc. (μg mL−1) |
Intra-day precisions |
Inter-day precisions |
| % recoverya ± SD |
% RSD |
% recoverya ± SD |
% RSD |
| The value is the mean of 3 determinations, SD is the standard deviation, and RSD is relative standard deviation. |
| Method I |
0.5 |
101.86 ± 0.55 |
0.54 |
101.31 ± 0.59 |
0.58 |
| 1.5 |
100.89 ± 0.64 |
0.63 |
100.22 ± 0.38 |
0.37 |
| 2.5 |
100.14 ± 0.38 |
0.38 |
100.48 ± 0.41 |
0.41 |
| Method II |
0.1 |
101.67 ± 0.81 |
0.80 |
101.32 ± 1.48 |
1.46 |
| 0.5 |
100.63 ± 0.30 |
0.30 |
100.40 ± 0.62 |
0.62 |
| 0.7 |
100.20 ± 0.12 |
0.12 |
100.23 ± 0.36 |
0.36 |
3.5.5. Robustness. The effect of small changes in the experimental parameters on the analytical performance of the methods was investigated. In the study of the method's robustness, four factors were altered individually and their effects were examined. These parameters were; buffer pH and volume, reagent concentration (EB or LaCl3) and reaction time. It was found that, neither of these variables had any significant effect on the analytical performance as the relative standard deviation did not exceed 2% (Table 5). This gives an indication about the suitable level of robustness and the reliability of the developed spectrofluorimetric methods.
Table 5 Evaluation of the robustness of the proposed spectrofluorimetric methods for the determination of the studied drug (0.5 μg mL−1)
| Parameter |
Parameter value |
% recoverya ± RSD |
| Method I |
Method II |
| The value is the mean of three determinations and RSD is the relative standard deviation. The recommended reagent volume was 1.0 mL for erythrosine B (1.7 × 10−4 mol L−1) and 1.4 mL for lanthanum chloride (5.1 × 10−5 mol L−1). |
| Buffer pH |
+0.2 |
100.91 ± 1.80 |
99.62 ± 0.40 |
| −0.2 |
99.70 ± 1.93 |
99.99 ± 0.73 |
| Buffer volume |
+0.2 mL |
98.52 ± 0.58 |
99.78 ± 0.88 |
| −0.2 mL |
100.01 ± 1.36 |
99.03 ± 1.00 |
| Reagent volumeb |
+0.2 mL |
101.97 ± 1.35 |
99.94 ± 0.40 |
| −0.2 mL |
99.50 ± 1.78 |
99.51 ± 1.21 |
| Reaction time |
+2 min |
98.41 ± 1.25 |
99.40 ± 0.40 |
| −2 min |
98.60 ± 1.36 |
98.98 ± 1.20 |
3.5.6. Selectivity and effect of interferences. The selectivity of the method was assessed to confirm its ability to determine the drug alone without any interference from the common excipients incorporated into tablets or suspension formulations. The studied excipients included; glucose, lactose, magnesium stearate and maltose. These components were added individually to the standard drug solution in a ratio similar to those in their commercial pharmaceutical formulations. The general analytical procedure was applied to estimate the drug content and the results were expressed in % recovery and standard deviation. As shown in Table 6, neither of the investigated excipients have any significant interference with the results of the proposed method.
Table 6 Determination of the investigated drug (0.5 μg mL−1) in presence of some common excipients using the proposed spectrofluorimetric methods
| Excipient (20 μg mL−1) |
% recoverya ± SD |
| Method I |
Method II |
| Mean value of three determinations, SD standard deviation. |
| Lactose |
100.05 ± 1.11 |
100.10 ± 1.22 |
| Glucose |
100.48 ± 0.83 |
99.56 ± 1.05 |
| Mg-stearate |
99.89 ± 1.37 |
98.98 ± 0.72 |
| Maltose |
100.800 ± 1.53 |
99.94 ± 1.36 |
3.6. Application of the proposed methods
3.6.1. Application to pharmaceutical preparations. The commercially available dosage forms (tablets and oral suspension) of ABZ were analyzed by the proposed methods for their drug contents. In the same time, these dosage forms were also subjected for analysis by the reported methods,5 and the results of the methods were compared with respect to accuracy and precision using the student's t- and F-tests, respectively. It was found that, the calculated t- and F-values did not exceeded the tabulated one (Table 7). This gave an indication about the absence of any significant difference between the proposed and reported methods in respect to accuracy and precision, respectively.
Table 7 Determination of ABZ in its pharmaceutical dosage forms using the proposed spectrofluorimetric and the reported methods
| Dosage forms |
% recoverya ± SD |
| Reported method |
Method I |
Method II |
| The value is the average of five determinations for both the proposed and reported methods. Tabulated values at 95% confidence limit are t = 2.306, F = 6.338. |
| Alzental tablets (200 mg per tablet) |
98.96 ± 1.65 |
100.34 ± 1.37 (t = 1.44, F = 1.46)b |
100.23 ± 1.38 (t = 1.33, F = 2.71)b |
| Alzental suspension (20 mg mL−1) |
99.78 ± 1.36 |
100.54 ± 1.79 (t = 0.76, F = 1.73)b |
100.34 ± 1.33 (t = 0.65, F = 1.84)b |
3.6.2. Application to spiked human plasma. The high sensitivity of the proposed method encouraged the determination of studied drug in human plasma. It was reported that the mean maximum concentration of albendazole in human plasma was 0.33 μg mL−1 after a single oral dose of 400 mg.35 The drug concentrations in the human plasma samples spiked with ABZ were estimated from their corresponding regression equations. The results summarized in Table 8 shows that, the mean recovery values were 96.28–97.41% ± 0.70–2.39. These results confirm the absence of any significant interference encountered from the components of the plasma and prove the suitability of the proposed methods for the analysis of the ABZ in the spiked human plasma.
Table 8 Application of the proposed spectrofluorimetric methods for the determination of ABZ in spiked human plasma
| Method |
Added conc. (μg mL−1) |
Found conc. (μg mL−1) |
% recoverya ± SD |
| Mean value of five determinations, SD standard deviation. |
| Method I |
1.00 |
0.970 |
96.95 ± 0.95 |
| 2.00 |
1.95 |
97.41 ± 0.70 |
| 3.00 |
2.91 |
97.17 ± 0.41 |
| Method II |
0.100 |
0.096 |
96.28 ± 2.39 |
| 0.400 |
0.388 |
96.94 ± 0.79 |
| 0.800 |
0.774 |
96.71 ± 0.87 |
4. Conclusion
In the current article, the formation of complexes of albendazole was applied for the development of two sensitive and simple spectrofluorimetric methods. The methods were validated according to ICH guidelines and successfully applied for the analysis of dosage forms and spiked human plasma containing the drug. The proposed methods have the virtues of simpler and shorter reaction times, more environmentally safe solvents are reagents, easier operation procedures and lower cost than the reported methods. The thermodynamic study reveals the feasibility of the undergoing reactions at room temperature. All these advantages make the method suitable for the use in quality control laboratories for routine work.
Conflicts of interest
There are no conflicts to declare.
References
- K. Parfitt and W. Martindale, The complete drug reference, 1999 Search PubMed.
- E.-M. Bennet and C. Bryant, Mol. Biochem. Parasitol., 1984, 10, 335–346 CrossRef CAS PubMed.
- N. Swamy and K. Basavaiah, Braz. J. Pharm. Sci., 2014, 50, 839–850 CrossRef.
- N. Swamy and K. Basavaiah, ISRN Anal. Chem., 2013, 2013, 1–11 CrossRef.
- A. C. Tella, O. M. Olabemiwo, M. O. Salawu and G. K. Obiyenwa, Int. J. Phys. Sci., 2010, 5, 379–382 CAS.
- C. P. Sastry, V. N. Sarma, U. Prasad and C. R. Lakshmi, Indian J. Pharm. Sci., 1997, 59, 161 CAS.
- C. Sastry, A. Sarma Varahabhatia, U. Prasad and C. Lakshmi, Indian Drugs, 1997, 34, 102–104 CAS.
- M. S. Kamel, B. Barsoum and R. Sayed, J. Appl. Sci. Res., 2008, 4, 1242–1248 CAS.
- S. M. Derayea, H. R. H. Ali, A. A. Hamad and R. Ali, J. Appl. Pharm. Sci., 2017, 7, 076–082 Search PubMed.
- S. Küçükkolbaşı, B. Gündüz and E. Kılıç, Anal. Lett., 2008, 41, 104–118 CrossRef.
- G. Y. Zhao, H. Wu, S. L. Dong and L. M. Du, Chin. Chem. Lett., 2008, 19, 951–954 CrossRef CAS.
- K. A. Attia, A. A. Mohamad and M. S. Emara, Anal. Methods, 2016, 8, 5136–5141 RSC.
- F. Tian, W. Huang, J. Yang and Q. Li, Spectrochim. Acta, Part A, 2014, 126, 135–141 CrossRef CAS PubMed.
- G. Balizs, J. Chromatogr. B: Biomed. Sci. Appl., 1999, 727, 167–177 CrossRef CAS.
- N. Sher, N. Fatima, S. Perveen and F. A. Siddiqui, Instrum. Sci. Technol., 2016, 44, 672–682 CrossRef CAS.
- M. Whelan, B. Kinsella, A. Furey, M. Moloney, H. Cantwell, S. J. Lehotay and M. Danaher, J. Chromatogr. A, 2010, 1217, 4612–4622 CrossRef CAS PubMed.
- A. Wojnicz, T. Cabaleiro-Ocampo, M. Román-Martínez, D. Ochoa-Mazarro, F. Abad-Santos and A. Ruiz-Nuño, Clin. Chim. Acta, 2013, 426, 58–63 CrossRef CAS PubMed.
- J. J. Pandya, M. Sanyal and P. S. Shrivastav, Biomed. Chromatogr., 2017, 31, e3947 CrossRef PubMed.
- J. Moyano, J. Liro, J. Pérez, M. Arias and P. Sánchez-Soto, Microscopy, 2014, 4, 1043–1050 Search PubMed.
- J. I. Gowda, R. B. Kantikar, D. G. Harakuni, K. Y. Jadhav, V. C. Chanagoudar and S. T. Nandibewoor, J. AOAC Int., 2016, 99, 1522–1526 CrossRef CAS PubMed.
- B. C. Lourencao, M. Baccarin, R. A. Medeiros, R. C. Rocha-Filho and O. Fatibello-Filho, J. Electroanal. Chem., 2013, 707, 15–19 CrossRef CAS.
- J. Wang, Z. Liu, J. Liu, S. Liu and W. Shen, Spectrochim. Acta, Part A, 2008, 69, 956–963 CrossRef PubMed.
- T. Pérez-Ruiz, C. Martínez-Lozano, V. Tomás and C. Sidrach, Analyst, 1995, 120, 1103–1106 RSC.
- X. Zhu, J. Sun and Y. Hu, Anal. Chim. Acta, 2007, 596, 298–302 CrossRef CAS PubMed.
- M. Pesez, J. Bartos, Colorimetric and fluorimetric analysis of organic compounds and drugs, Marcel Dekker, New York, 1974 Search PubMed.
- C. Y. Huang, Methods Enzymol., 1982, 87, 509–525 CAS.
- K. Semahat, G. Beniz and K. Esma, Gazi Üniversitesi Gazi Eğitim Fakültesi Dergisi, 2004, 24, 159–174 Search PubMed.
- M. S. Refat and G. A. M. Mersal, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2012, 42, 786–791 CAS.
- J. G. Xu and Z. B. Wang, Fluorimetric Analysis Method, Science Press, Beijing, 3rd edn, 2006 Search PubMed.
- N. Boens, W. Qin, N. Basarić, J. Hofkens, M. Ameloot, J. Pouget, J.-P. Lefèvre, B. Valeur, E. Gratton and M. VandeVen, Anal. Chem., 2007, 79, 2137–2149 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer-Verlag, Berlin, Heidelberg, 3rd edn, 2006 Search PubMed.
- S. Bi, L. Ding, Y. Tian, D. Song, X. Zhou, X. Liu and H. Zhang, J. Mol. Struct., 2004, 703, 37–45 CrossRef CAS.
- I. Ravikumar and P. Ghosh, Inorg. Chem., 2011, 50, 4229–4231 CrossRef CAS PubMed.
- ICH Harmonized Tripartite Guideline, Validation of analytical procedures: text and methodology Q2 (R1), 2005 Search PubMed.
- S. Marriner, D. Morris, B. Dickson and J. Bogan, Eur. J. Clin. Pharmacol., 1986, 30, 705–708 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c
*c