
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic reactivity of an iridium complex with a proton responsive N-donor ligand in CO2 hydrogenation to formate†
Gunniya Hariyanandam Gunasekar‡
 a,
Yeahsel Yoon‡a,
Il-hyun Baekb and
Sungho Yoon*a
a,
Yeahsel Yoon‡a,
Il-hyun Baekb and
Sungho Yoon*a
aDepartment of Applied Chemistry, Kookmin University, 77 Jeongneung-ro, Seongbuk-gu, Seoul, 136-702, Republic of Korea. E-mail: yoona@kookmin.ac.kr
bKorea Institute of Energy Research, 152 Gajeong-ro, Yuseong-gu, Daejeon 305-343, Republic of Korea
First published on 3rd January 2018
Abstract
Catalytic hydrogenation of CO2 into formic acid/formate is an attractive conversion in the utilization of CO2. Although various catalysts with high catalytic efficiency are reported, a very few studies have been carried out to correlate/understand the efficacy and stability of the hydrogenation catalysts, which could be helpful to direct the future design strategy of corresponding catalysts. Herein, a half-sandwich iridium complex containing bibenzimidazole as a proton responsive N-donor ligand, [Cp*Ir(BiBzImH2)Cl]Cl, has been synthesized and fully characterized. The generation of an N− anion by the deprotonation of a bibenzimidazole group resulted in a significant enhancement of activity. The Ir complex showed about 20 times higher catalytic efficiency in the hydrogenation of CO2 into formate than that of its bipyridine counterpart [Cp*Ir(Bpy)Cl]Cl. The time dependent catalytic activity studies revealed that the initial excellent activity of [Cp*Ir(BiBzImH2)Cl]Cl was reduced when catalytic cycle proceeds; which was found to be the structural instability of the catalyst caused by steric hindrance between the bibenzimidazole and Cp* ligands.
Introduction
Hydrogenation of greenhouse CO2 gas into formic acid has been a subject of great interest,1,2 although it suffers from both thermodynamic and kinetic limitations.3 The high thermodynamic barrier (eqn (1)), however, can be lowered upon usage of a base (eqn (2)) and/or an aqueous medium (eqn (3)).3 A relatively high kinetic barrier that exists due to the dissociation of H–H bonds and formation of a C–H bond can be overcome by using a suitable catalyst. Various kinds of catalysts operating in organic solvents and supercritical CO2 solutions have been studied over the past few decades.4,5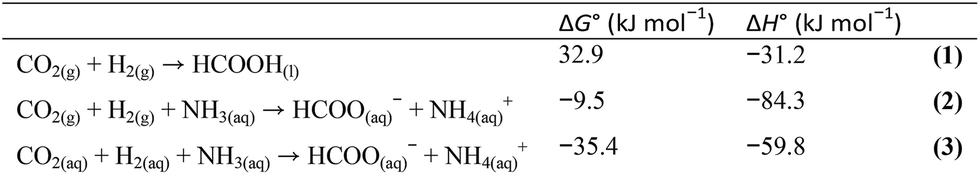
The most remarkable progress in recent years has been the development of bipyridine-based homogeneous catalysts.6 Particularly, complexes containing hydroxyl functional groups such as [Cp*Ir(DHBP)H2O]2+ and [Cp*Ir(DHPT)Cl]2+ (DHBP = 4,4′-dihydroxy-2,2′-bipyridine; DHPT = 4,7-dihydroxy-1,10-phenanthroline; Cp* = 1,2,3,4,5-pentamethylcyclopentadiene) showed remarkable catalytic efficiency in aqueous solutions (Scheme 1a).7 It has been claimed that the high activity of these catalysts was attributed to the strong electron donating property of generated O− ligand(s).8
 | ||
| Scheme 1 Representation of expected electron density increment on the Ir centers under basic condition. (a) [Cp*Ir(4DHBP)]2+; (b) [Cp*Ir(BiBzImH2)]2+. | ||
In order to enhance the above proposed electron donating effect of a ligand, here we designed an imidazole based bidentate 2,2′-bibenzimidazole ligand (BiBzImH2). The presence of six π-electrons in a 5-membered imidazole ring with NH functionality is expected to have a high electron donating property than the simple, unprotonated bipyridine ligand (Scheme 1b). Herein, synthesis, characterization and the catalytic reactivity of an iridium complex containing bibenzimidazole ligand [Cp*Ir(BiBzImH2)Cl]Cl (1) is reported for the hydrogenation of CO2 into formate.
Results and discussion
Synthesis and characterization of 1
Himeda et al. introduced various electron donating/withdrawing functional groups on N,N-bidentate ligated Ir complex and systematically studied their effects in the hydrogenation of CO2 to formate.9 An Ir complex containing simple bipyridine (bpy) ligand [Cp*Ir(Bpy)]2+ was shown to have an initial turnover frequency (TOF) of 0.3 h−1 at relatively milder temperature (50 °C).10 This TOF value was further increased to 8.6 h−1 when one of the pyridine rings of the bpy ligand was replaced with an electron rich imidazole moiety.10 A similar but more pronounced enhancement was observed while introducing OH functionality on the pyridine site(s).11 The TOF value was also improved when both of the pyridine sites were replaced with biimidazole ligand (TOF = 20 h−1);10 the generation of N− anion by the deprotonation of uncoordinated NH groups of imidazole rings and its strong electron donating character was found to be the origin of this enhancement. This was further confirmed by using N-methylated biimidazole ligands, where the activity was decreased drastically.11 These findings showed that the electron-rich ligands such as imidazole can significantly increase the reactivity of an iridium complex. More evidently, the Ir complex containing electron-donating methyl groups on the imidazole backbone [Cp*Ir(4MeBiImH2)]2+ (4MeBiImH2: 4,4′,5,5′-tetramethyl-biimidazole) demonstrated a highest TOF (36 h−1) among the tested imidazole ligands.10 Based on this understanding, we anticipated that the BiBzImH2 ligand which have a relatively rich π electrons than 4MeBiImH2 would significantly improve the efficiency of the Ir complex.BiBzImH2 was prepared through the coupling and elimination reaction between oxamide and o-phenylenediamine.12 Complex 1 was prepared by treating 2 equivalents of BiBzImH2 with a chloroform solution of [IrCp*Cl2]2 precursor. Yellow crystals of 1 was obtained in a chloroform/pentane system by the vapour diffusion method. X-ray crystallographic structure of 1 has the monoclinic space group P21/n with two chloroform molecules and a chloride counter anion in a unit cell (Fig. 1 and S1†) (the three-dimensional packing structure is shown in a Fig. S2†). The coordination environment of the Ir(III) center was composed of two nitrogen atoms of the BiBzImH2 ligand, Cp* moiety (bound in η5 fashion) and a chloride anion. One additional Cl− anion was located in the crystal lattice, balancing the charge state. All atoms in BiBzImH2 are placed approximately on one plane with an Ir–N1 and Ir–N2 respective distances of 2.127 (2) and 2.148 (2) Å. The bond distance of Ir–Cl was 2.3941 (7) Å, which was comparable to that of previously reported [Cp*Ir(BiImH2)Cl]Cl.13 The bond distances of N1–H and N2–H were 0.844 and 0.833 Å, respectively. The dihedral angle between BiBzImH2 ligand plane and the plane composed of the iridium atom and the two coordinated nitrogen atoms was 6.32 (19)°. The angle of N1–Ir–Cl and N2–Ir–Cl was 86.85° and 86.45° respectively.
 | ||
| Fig. 1 Crystal structure of 1 (a), with space filling model (b). Solvent molecules, and hydrogen atoms other than NH were omitted for clarity. | ||
UV-visible spectrum of 1 in chloroform solution showed a broad peak at 337 nm with extinction coefficient of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 M−1 (Fig. S3†); which corresponds to intraligand π–π* transitions.7b,14, An intensive peak at 281.7 m/z in the electrospray ionization (ESI) measurement was well matched with the expected [Cp*Ir(BiBzImH2)]2+ with formula C24H25IrN4 (Fig. S4†). This indicates that 1 maintain its composition in the solution medium. The 1H NMR spectral data showed that the symmetry of BiBzImH2 was broken while coordination to an iridium ion (Fig. S5 and S6b and c†). In addition, a downfield shift for the BiBzImH2 protons compared to that of bare BiBzImH2 ligand was observed, indicating that metalation shifts the electron density towards the Ir metal center. These results demonstrated that the structure of compound 1 was maintained in a solution also.
000 cm−1 M−1 (Fig. S3†); which corresponds to intraligand π–π* transitions.7b,14, An intensive peak at 281.7 m/z in the electrospray ionization (ESI) measurement was well matched with the expected [Cp*Ir(BiBzImH2)]2+ with formula C24H25IrN4 (Fig. S4†). This indicates that 1 maintain its composition in the solution medium. The 1H NMR spectral data showed that the symmetry of BiBzImH2 was broken while coordination to an iridium ion (Fig. S5 and S6b and c†). In addition, a downfield shift for the BiBzImH2 protons compared to that of bare BiBzImH2 ligand was observed, indicating that metalation shifts the electron density towards the Ir metal center. These results demonstrated that the structure of compound 1 was maintained in a solution also.
Structure of 1 in basic solutions
The hydrogenation of CO2 is usually proceeds under basic conditions; thus, the actual active form of the catalyst can be different from that of the initial compound. Since pKa1 and pKa2 of BiBzImH2 are 11.0 and >16 respectively, the catalyst is expected to have structure [Cp*Ir(BiBzimH)−Cl] (2) rather than 1 under the basic hydrogenation conditions (Scheme 2).15 To understand this, NMR of 1 with different equivalents of base (triethylamine, Et3N) was investigated (Fig. 2). In the presence of 1 equivalent of Et3N, the aromatic protons of 1 were slightly shielded (about 0.1 to 0.18 ppm) and the Cp* protons were slightly deshielded. In addition, it showed a new, broad peak at 3.6 ppm with integral value of 1 (enlarged figure is given in Fig. S6d†). The broad peak can be assigned to a proton held between two nitrogen atoms of BiBzImH− (Fig. S6d†). A similar change was observed with 4 equivalents of base; importantly, the peak at 3.6 ppm with integral value of 1 does not disappeared. These results indicated that the formation of dianionic (N2−) structure of 1 was difficult, and thus complex 1 may predominantly exists as an N− ligand (structure 2) under basic conditions. | ||
| Scheme 2 Deprotonation of 1 in the presence of a base. | ||
 | ||
| Fig. 2 1H NMR of (a) 1; (b) 1 in the presence of 1 eq. of Et3N; (c) 1 in the presence of 4 eq. of Et3N. | ||
Hydrogenation of CO2
As mentioned above, the hydrogenation of CO2 is usually performed in aqueous solution, with a base, to overcome the thermodynamic barrier.3 Because of the limited solubility of 1 in water, however, a mixture of water and methanol (1:1) was used in this study. Catalytic activity of 1 was compared with [Cp*Ir(Bpy)Cl]Cl and the results are shown in Table 1. As shown in entry 1, [Cp*Ir(Bpy)Cl]Cl produced a very low formate concentration {[HCO2−]} (2.5 mM) in 1 h with a turnover number (TON) of 5. Under similar reaction conditions, the catalyst 1 produced a substantial [HCO2−] of 50 mM with TON of 100. This value was 20 times higher than [Cp*Ir(Bpy)Cl]Cl catalyst, confirming that the high electron density of N− donor ligand results in a higher TON.
| Entry | Catalyst | Catalyst conc. (mM) | Time (h) | T (°C) | [HCO2−] (mM) | TONb |
|---|---|---|---|---|---|---|
| a The reaction was carried out in a 1 M KOH solution of MeOH/H2O (1:1) mixture under 4.0 MPa total pressure (CO2/H2 = 1).b TON = mole of formate/mole of Ir.c Initial TOF (h−1); calculated from the initial part of the reaction (after 15 min). |
||||||
| 1 | [Cp*Ir(Bpy)Cl]Cl | 0.5 | 1 | 50 | 2.5 | 5 |
| 2 | 1 | 0.5 | 1 | 50 | 50 | 100 |
| 3 | 1 | 0.2 | 0.25 | 80 | 75 | 375 (1500)c |
| 4 | 1 | 0.2 | 38 | 80 | 350 | 1750 |
In order to gain further insights into the system, time-dependent measurements were performed. As shown in Fig. 3, the rate of formate production was faster at an initial stage of the reaction (TON was 375 in 0.25 h) compared to the final stage of the reaction (TON was 1450 and 1750 in 20 and 38 h, respectively). To clearly visualize this behaviour, the TOF between the hours was plotted vs. time (Fig. S7†). It reiterated that the TOF was significantly higher for first 0.25 h (TOF was 1500 h−1) and was decreased rapidly while reaction proceeds (Table S1†).
 | ||
| Fig. 3 Time dependent formate generation by 1 (solid line) and [Cp*Ir(Bpy)Cl]Cl (dash line). | ||
To understand this, the reaction mixture was analyzed by Mass and NMR spectroscopy after 10 h of hydrogenation. 1H NMR spectrum showed that the intensity of Cp* peak was remarkably reduced compared to 1; the ratio between BiBzIm and Cp* was 5:1 (Fig. S8†). The ESI-mass measurement showed the presence of various fragments (Fig. S9†). These results suggested that a deformation of the catalyst has occurred during the hydrogenation. Currently we could not find and isolate the decomposed species of 1, and hence further efforts are needed to investigate the decomposed species and its pathway(s).
One of the major difference between 1 and [Cp*Ir(Bpy)Cl]Cl (in terms of structure) is the use of different N donor ligand. As shown in Table S2,† 1 has longer Ir–N (2.127 Å) and Ir–Cp* (1.789 Å) bond lengths compared to [Cp*Ir(Bpy)Cl]Cl (wherein the values were 2.076 and 1.785 Å, respectively). The electron density mapping of 1 (Fig. 1b) clearly suggests that the observed elongation of Ir–N and Ir–Cp* bond may originate from the steric effect between BiBzIm's benzene ring and the methyl group of Cp*. Since the bond length is inversely proportional to the bond strength, 1 may have weaker bonding interactions, which in turn might led to dissociation. These results shows that the initial excellent activity of catalyst was originated by the strong electron donating property of BiBzIm ligand, and a sharp diminish in activity when the catalytic cycle proceeds was due to its structural instability.
Conclusion
In this study, a bibenzimidazole ligated half-sandwich Ir complex [Cp*Ir(BiBzImH2)Cl]Cl was synthesized and characterized. The Ir complex demonstrated about 20 times higher activity than that of the typical bpy-based catalyst [Cp*Ir(Bpy)Cl]Cl. The generation of N− anion through the deprotonation of bibenzimidazole NH group is largely responsible for this high catalytic activity. Notwithstanding the forgoing, the activity of Ir complex was dramatically decreased while catalytic cycle proceeds this was found to be the low structural stability of Ir complex under hydrogenation conditions. Previously, it was reported that the decomposition of [Cp*Co(Bpy)]2+ catalyst was attributed to the steric effect caused by the small coordination sphere of cobalt.16 Iridium-based catalysts are usually known to have a relatively stable structure because of its large coordination sphere and rich coordination sites. Nevertheless, according to this study, in order to achieve a higher efficiency, iridium is required to maintain its stable structural arrangement during the hydrogenation. Thus, stability and electron donating ability are the important factors to be considered while designing the catalysts.Experimental section
General procedures
All the reagents were purchased from commercial sources and used without further purification unless otherwise described. BiBzImH2 and [IrCp*Cl2]2 were prepared according to literature procedures.13,17 Electrospray ionization mass spectrometry (ESI-MS) was measured on Agilent 6130 with single quadrupole LC/MS detector. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a Varian VNMRS500. UV-visible spectra were carried out using S-3100 of SCINCO Co. Ltd.Synthesis of [Cp*Ir(BiBzImH2)Cl]Cl (1)
A portion of [IrCp*Cl2]2 (0.100 g, 0.126 mmol) was mixed with a chloroform solution (20 mL) of BiBzImH2 (60.0 mg, 0.256 mmol) under a N2 atmosphere. After vigorous stirring for 12 h, the insoluble fractions were filtered out and the resulting dark orange filtrate was evaporated to dryness. Yellow crystals of 1 suitable for X-ray diffraction were obtained by vapour diffusion of pentane into the chloroform solution (5:2 v/v). Yield: 90.0%. 1H NMR (400 MHz, CDCl3, ppm): δ 14.74 (br s, 2H) 7.81 (dt, J = 7.98, 0.51 Hz, 2H), 7.71 (dt, J = 8.02, 0.74 Hz, 2H), 7.52 (td, J = 7.67, 1.18 Hz, 2H), 7.46 (td, J = 7.70, 1.19 Hz, 2H), 1.84 (s, 15H, Cp*); 13C NMR (100.6 MHz, CDCl3, ppm): 144.24, 139.13, 134.35, 126.44, 125.28, 116.65, 115.26, 87.81, 10.44; IR ν/cm−1: 3046(w), 2961(w), 2918(w), 2852(w), 1601(m), 1474(w), 1445(w), 1391(s), 1348(s), 1274(m), 1028(m), 742(s); UV λmax (acetonitrile) 217 (24675), 267 (4687), 337 (15046), 356 nm (11670); ESI-MS (MeOH) (+) (m/z, relative intensity%): 281.7 (100) [Cp*Ir(BiBzImH2)]2+, 562.2 (68.3) [Cp*Ir(BiBzIm-H2)H]+.
Note: During the manuscript preparation, Liang et al. reported the synthesis of 1 for the methylation of amines.18
X-ray crystallographic studies
Single crystal was mounted at room temperature on the tips of quartz fibres, coated with Partone-N oil, and cooled under a stream of cold nitrogen. Intensity data were collected on a Bruker Apex2 diffractometer running the SMART software package, with Mo Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods and refined on F2 by using the SHELXTL software package.19 Empirical absorption corrections were applied with SADABS,20 part of the SHELXTL program package, and the structures were checked for higher symmetry by the program PLATON.21 All non-hydrogen atoms and amine hydrogens were refined anisotropically. In general, other hydrogen atoms were assigned idealized position and given thermal parameters equivalent to 1.2 times the thermal parameter of the carbon atom to which they are attached. Data collection and experimental details for the complex are summarized in Table S3.†Hydrogenation activity
Hydrogenation was carried out in a 100 mL homemade stainless steel reactor with glass vessel insert. In a typical run, the complex was added to a water/methanol mixture of KOH solution (50 mL) and saturated the solution with CO2 for 20 min. The reactor was sequentially pressurized with CO2 and H2 (1:1) to 40 bar and heated to the desired temperature. The reaction was cooled to room temperature after appropriate time and slowly released the pressure. The concentration of formic acid was analysed by HPLC on an Aminex HPX-87H column with an RI detector using 5.00 mM H2SO4 eluent.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support by grants from Korea CCS R&D Center, funded by the Ministry of Education, Science and Technology of Korean government. CCDC 1419395† contains the supplementary crystallographic data for this paper.References
-
(a) P. G. Jessop, F. Joo and C. C. Tai, Coord. Chem. Rev., 2004, 248, 2425–2442 CrossRef CAS
; (b) P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 344–355 CrossRef CAS
-
(a) C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., 2010, 49, 9777–9780 CrossRef CAS PubMed
-
(a) P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef CAS
-
(a) Y. Inoue, Y. Sasaki and H. Hashimoto, J. Chem. Soc., Chem. Commun., 1975, 718–719 RSC
-
(a) P. G. Jessop, T. Ikariya and R. Noyori, Nature, 1994, 368, 231–233 CrossRef CAS
-
(a) W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed
-
(a) Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara and K. Kasuga, Organometallics, 2007, 26, 702–712 CrossRef CAS
-
(a) W.-H. Wang, J. F. Hull, J. T. Muckerman, E. Fujita and Y. Himeda, Energy Environ. Sci., 2012, 5, 7923–7926 RSC
-
(a) N. Onishi, S. Xu, Y. Manaka, Y. Suna, W.-H. Wang, J. T. Muckerman, E. Fujita and Y. Himeda, Inorg. Chem., 2015, 54, 5114–5123 CrossRef CAS PubMed
- Y. Manaka, W.-H. Wang, Y. Suna, H. Kambayashi, J. T. Muckerman, E. Fujita and Y. Himeda, Catal. Sci. Technol., 2014, 4, 34–37 CAS
- Y. Suna, Y. Himeda, E. Fujita, J. T. Muckerman and M. Z. Ertem, ChemSusChem, 2017, 10, 4535–4543 CrossRef CAS PubMed
- B. F. Fieselmann, D. N. Hendrickson and G. D. Stucky, Inorg. Chem., 1978, 17, 2078–2084 CrossRef
- R. Ziessel, J. Organomet. Chem., 1992, 441, 143–154 CrossRef CAS
- M. Kalidasan, R. Nagarajaprakash, S. Forbes, Y. Mozharivskyj and K. M. Rao, Z. Anorg. Allg. Chem., 2015, 641, 715–723 CrossRef CAS
- A. Kumar, H. K. Sinha and S. K. Dogra, Can. J. Chem., 1989, 67, 1200 CrossRef CAS
- Y. M. Badiei, W.-H. Wang, J. F. Hull, D. J. Szalda, J. T. Muckerman, Y. Himeda and E. Fujita, Inorg. Chem., 2013, 52, 12576–12586 CrossRef CAS PubMed
- C. White, A. Yates and P. M. Maltlis, Inorg. Synth., 1992, 29, 228–234 CrossRef CAS
- R. Liang, S. Li, R. Wang, L. Lu and F. Li, Org. Lett., 2017, 19, 5790–5793 CrossRef CAS PubMed
- SMART Version 5.618: Bruker Advanced X-ray Solution, Madison Inc., WI, 2002 Search PubMed
- G. M. Sheldrick, SHELXTL, Version 6.14: Bruker Advanced X-ray Solution, Madison Inc., WI, 2003 Search PubMed
- G. M. Sheldrick, SADABS, Area-Detector Absorption Correction, University of Göttingen, Göttingen, Germany, 2001 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1419395. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra12343d |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |