
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree successive and regiocontroled palladium cross-coupling reactions to easily synthesize novel series of 2,4,6-tris(het)aryl pyrido[1′,2′:1,5]pyrazolo[3,4-d]pyrimidines†
R. Belaroussiab,
A. Ejjoummanyab,
A. El Hakmaouib,
M. Akssirab,
G. Guillaumet *a and
S. Routier*a
*a and
S. Routier*a
aUniv Orleans, CNRS, Institut de Chimie Organique et Analytique, UMR 7311, BP 6759, 45067 Orléans Cedex 2, France. E-mail: sylvain.routier@univ-orleans.fr; gerald.guillaumet@univ-orleans.fr; Web: http://www.icoa.fr/fr
bLaboratoire de Chimie Physique et Chimie Bioorganique, FST Mohammedia, Université Hassan II de Casablanca, B. P. 146, 28800 Mohammedia, Morocco. E-mail: akssira@yahoo.fr; Web: http://www.fstm.ac.ma
First published on 2nd January 2018
Abstract
The first access to tris(het)arylated pyrido[1′,2′:1,5]pyrazolo[3,4-d]pyrimidine derivatives is reported. The series were generated from 4-chloroaminopyridinium, which afforded the key intermediate bearing three leaving groups, i.e. a C-2 methylsulfanyl, a lactame carbonyl group in C-4 and a chlorine atom in C-6. The regioselective reactions led to the tris(het)aryl derivatives with satisfying to high yields. The three successive cross-coupling reactions occurred first in C-6 by the displacement of chlorine, next in C-4 position by a sequential Pd-catalyzed phosphonium coupling and finally in C-2 under a Pd/Cu-catalyzed desulfitative cross-coupling reaction. The optimization and scope of each reaction are discussed and the original compounds characterized.
Introduction
Nitrogen-containing and fused heterocyclic compounds have attracted considerable attention in directed and targeted organic synthesis and many products have been used in materials and medicinal chemistry programs. With a view to increasing molecular diversity, our aim was to design rare tricyclic pyrido[1′,2′:1,5]pyrazolo[3,4-d]pyrimidines (PPPy), which incorporate a tricyclic system and four nitrogen atoms, two of which are vicinal. This fused tricyclic heterocycle contains a pyrazolopyridine core which has been used in diverse biologically and pharmaceutical active molecules including diuretic1 and antiherpetic drugs,2 and as p38 kinase inhibitors3 and melatonine receptor ligands.4 It also contains a pyrimidine natural moiety, which is known to possess a potential role in several biological processes and to have emerged as a significant motif in drug-discovery programs.5In order to build such a multi-heterocyclic compound, our group recently focused on developing new functionalization protocols6 to explore the chemical space, an indispensable key to design future useful molecules.
In order to build C-2 and C-4 disubstituted PPPy derivatives, we developed a straightforward strategy which included successive C-4 in situ C–O activation and a C-2 desulfurization through Liebeskind–Srogl cross-coupling reactions.7 To provide a versatile platform we then sought to design a new flexible route to access 2,4,6-trisubstituted PPPy's I from a unique intermediate i.e. the 6-chloro-2-(methylthio)pyrido[1′,2′:1,5]pyrazolo[3,4-d]pyrimidin-4(3H)-one 8.
We report herein the preparation of 8 and its regioselective functionalization by different types of palladium catalyzed reactions. After optimization of the reaction conditions, this strategy was achieved using three successive (het)arylations in the following order C-6, C-4 and then C-2 positions (Fig. 1). The scope and limits are discussed for each step.
 | ||
| Fig. 1 Access to 2,4,6-tris(het)arylated pyrido[1′,2′:1,5]pyrazolo[3,4-d]pyrimidines I from the unique platform 8. | ||
Results and discussion
Compound 8 was obtained via a 7-step sequence partially based on our previous work.7,8 Dimethyl 5-chloropyrazolo[1,5-a]pyridine-2,3-dicarboxylate 2 was obtained after a [3 + 2] cycloaddition between the 1-amino-4-chloropyridinium iodide 1 (ref. 9 and 10) with dimethyl acetylenedicarboxylate (DMAD). A regioselective saponification furnished the hemiester 3, which was subjected to an isocyanate generation under Curtius rearrangement. The unstable isocyanate function was quenched with tert-butanol to afford the Boc protected (het)aryl amine 4. After removal of the carbamate and benzoylisothiocyanate condensation, the thiourea 6 was subjected to annelation and finally a regioselective sulfur methylation led to 8 in satisfying yields (Scheme 1). This methodology proved to be highly efficient and amenable to multigram scale to access the unique and challenging tricyclic structure in sufficient amounts to pursue the optimization of the (het)arylation process. | ||
| Scheme 1 Synthetic route for 8: (a) DMAD (1.5 eq.), K2CO3 (1.4 eq.), DMF, r.t., 12 h, 86%; (b) NaOH (2N) (1.0 eq.), CH3OH, r.t., 6 h, 81%; (c) (i) Et3N (1.3 eq.), ClCO2Et (1.5 eq.), THF, −10 °C, 1.5 h, (ii) NaN3 (1.7 eq.), −10 °C, 1.5 h, (iii) t-butanol, 6 h, 67%; (d) TFA/CH2Cl2 (1/1), 4 h, r.t., quant.; (e) benzoylisothiocyanate (1.2 eq.), CHCl3, 12 h, r.t., 84%; (f) EtONa (1.1 eq.), EtOH, 8 h, reflux, 97%; (g) MeI (1.0 eq.), NaOH (1.0 eq.), EtOH, 6 h, r.t., 80%. | ||
1. Suzuki–Miyaura arylation in C-6
The functionalization of the C-4 position of compound 8 was first attempted via a C–O direct activation involving PyBrOP11 to generate the required in situ generated O-phosphonium leaving group (Scheme 2). However, this route proved to be unsuitable since only the disubstituted product 9 was isolated in 45% yield with no trace of the mono C-4-arylated molecule. This result seems to indicate that the 6-Cl atom is as reactive as the activated 4-O atom under palladium catalyzed conditions. | ||
| Scheme 2 Phosphonium-mediated Suzuki type cross coupling reaction from 8. | ||
In an attempt to selectively substitute one of the two positions (C-6 or C-4), we began by the regioselective functionalization of the C-6 position. Using several experimental coupling Suzuki–Miyaura conditions exploited in our previous research,12 this first test did not allow the formation of the C–C bond, as only the starting material 8 was recovered (Table 1, entry 1). The same behavior was observed using the conditions reported in Scheme 2, as if pyrimidinone exerted a negative impact on C-6 reactivity in basic and aqueous media.
|
||
|---|---|---|
| Entry | Conditions | Yield in 10 (%) |
| a Not detected. Starting material 8 was recovered at the end of the reaction.b Yield is indicated as isolated compound. | ||
| 1 | p-TolylB(OH)2 (1.2 eq.), DME/H2O (2/1), Na2CO3 (2 eq.), Pd(PPh3)4 (5% mol), 30 min, M.W, 120 °C | NDa |
| 2 | p-TolylB(OH)2 (1.1 eq.), 1,4-dioxane/H2O (4/1), Na2CO3 (3.0 eq.), PdCl2(dppf)·CH2Cl2 (10% mol), 1 h/M.W, 150 °C | NDa |
| 3 | p-TolylB(OH)2 (1.1 eq.), 1,4-dioxane, K2CO3 (2.0 eq.), Pd(OAc)2/xantphos (10%/20% mol), 1 h/M.W, 150 °C | 63b |

To circumvent this problem, we switched the palladium source and tested the catalytic system XantPhos/Pd(OAc)2.13 The reaction was carried out in dioxane under microwave irradiation at 150 °C using potassium carbonate as a base. To our delight these conditions provided the desired product 10 in 63% yield (Table 1, entry 3).
Next, the generalization of this approach was examined (Table 2). Results were found to be satisfactory as the majority of the boronic substrates employed provided the coupling products. Electron rich or deactivated aryl as well as naphthyl residues were introduced with good yields ranging from 56 to 73%. In all cases a full conversion of starting material and formation of a unique product were observed. The decrease in yield was mainly due to difficulties encountered during purification steps.





2. (Het)arylations under direct C–O activation in C-4
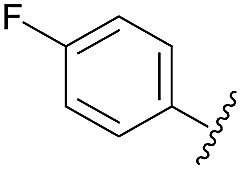
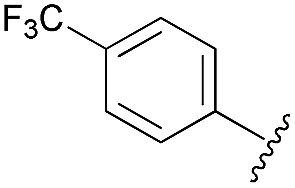
After having regioselectively introduced the C-6 aryl group, the direct C-4 arylation via PyBrOP amid activation was undertaken, applying the operating conditions previously established for the synthesis of 9.7 The reaction sequence involved first the activation of the C–O bond using Et3N to favor the formation of the oxanion which was in situ quenched by PyBrOP (1.2 eq.) activating agent. The second step, which required the addition of boronic acid and PdCl2(dppf)·CH2Cl2 as catalyst, was successfully achieved under thermal conditions. Initially, we carried out our investigations on 10, 11 and 13 (Table 3), which carry an electro-donor aryl group in C-6. The coupling reactions were achieved with representative boronic acids. Aryl and thiophenyl groups were easily introduced (entries 1, 4, 5, 7, 8, 9). The decreases in yield of purified product are mainly due to the purification step. Some difficulties occurred with the deactivated fluorophenyl and pyridinyl groups (entries 2, 6). The reactions appear incomplete and the corresponding starting materials were recovered. All modifications of the experimental conditions were inefficient. In the pyridinyl series, the introduction of a strong electrodonor OMe group restored the reactivity (entry 3).
|
||||
|---|---|---|---|---|
| Entry | Substrate | Boronic acids | Product | Yielda (%) (SM%)b |
| a Yields are indicated as isolated compounds.b SM starting material recovered.c Reaction time of the second step 48 h.d ND: Not detected.e Reaction time of the second step 72 h. | ||||
| 1 | 10 | 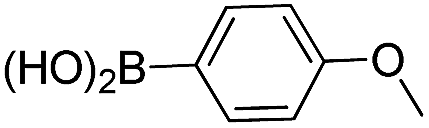 |
16 | 55 |
| 2 |  |
17 | 47c (28) | |
| 3 | 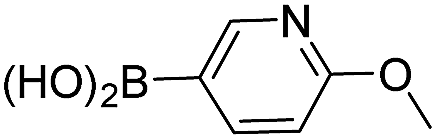 |
18 | 64c | |
| 4 | 11 |  |
19 | 78 |
| 5 |  |
20 | 70 | |
| 6 |  |
21 | 36 (46) | |
| 7 | 13 |  |
22 | 63 |
| 8 | 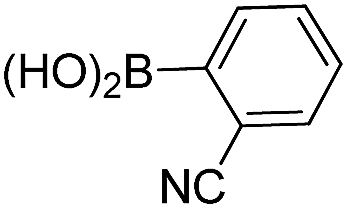 |
23 | 61 | |
| 9 |  |
24 | 52 | |
| 10 | 14 |  |
25 | 40 (21), 33e |
| 11 | 15 |  |
— | NDd |
While all reactions were inefficient starting from 15 (entry 11), we were able to introduce the 4-cyanophenyl group on 14 (entry 10) and derivative 25 was formed with an average yield of 40% whereas 21% of starting material still remained unchanged after 24 h. In order to improve the overall conversion, a longer reaction time (72 h), was used but degradation occurred.
Based on the results obtained, we can conclude that the electronic properties of the C-6 aryl moieties strongly affect the reactivity of the C-4 position. The best conversions were always obtained with the simultaneous presence of electron donor substituents (mesomeric or inductive effect) on the C-6 position as well as on the boronic acid used.
3. (Het)arylations under Liebeskind–Srogl in C-2
We next applied the third Liebeskind–Srogl14 method to the 4,6-bis(het)arylated derivatives in an attempt to furnish original 2,4,6-trisubstituted compounds. The reactivity of the three thioethers 18, 19, and 22 was explored in the presence of boronic acids, using an (het)arylboronic acid in the presence of Pd(PPh3)4 and copper(I) thiophene-2-carboxylate (CuTC) in THF at 100 °C under microwave irradiation.7 To our great satisfaction, the first test using 4-methoxyphenyl boronic acid as coupling partner gave 26 in a good 70% yield (Table 4). By changing both starting precursors (19 and 22) and the coupling partners, the tri-substituted tricycles 27 and 28 were readily isolated with 78% and 61% yields respectively (Table 4). The efficiency of this last reaction in C-2 seems insensitive to the nature of the 4,6-bis(het)arylated derivatives and boronic acids used.



Conclusions
In summary, we have described in this work several synthetic pathways for the preparation of a new pyrido[1′,2′:1,5]pyrazolo[3,4-d]pyrimidine scaffold, and have build several libraries of C-2, C-4, C-6 (het)arylated pyrido[1′,2′:1,5]pyrazolo[3,4-d]pyrimidines. First, the C-6 chlorine atom was substituted using a Suzuki–Miyaura cross coupling reaction. The resulting C-6-arylated products were engaged in a direct C–O activation from an amide function with PyBroP activation followed by a palladium catalyzed reaction to produce a series of C-4,6 bis-(het)arylated compounds. We proved that electron criteria governed the reactivity and efficiency. Finally, the 2-SMe residual function reacted under Liebeskind–Srogl conditions to afford the desired tri-substituted derivatives. The efficiency of the global sequence involving three successive and unprecedented cross coupling reactions will certainly prove useful to others in the future to develop poly functionalized heterocyclic PPPy series.Experimental
Reactions were monitored by thin-layer chromatography (TLC) analysis using silica gel (60 F254) plates. Compounds were visualized by UV irradiation. Flash column chromatography was performed on silica gel 60 (230–400.13 mesh, 0.040 0.063 mm). Melting points (Mp [°C]) were taken on samples in open capillary tubes and are uncorrected. The infrared spectra of compounds, recorded on a Thermo Scientific Nicolet iS10, are given in cm−1. 1H and 13C NMR spectra were recorded on a Bruker Avance II 250 (13C, 63 MHz), Bruker Avance 400 (13C, 101 MHz), or on a Bruker Avance III HD nanobay 400 (13C, 101 MHz). Chemical shifts are given in parts per million from tetramethylsilane (TMS) as internal standard. Coupling constants (J) are reported in hertz (Hz). High-resolution mass spectra (HRMS) were performed on a Maxis Bruker 4G.4. Characterization data
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc, 4:6) = 0.1; IR (ATR diamond, cm−1) ν 833, 950, 1119, 1471, 1607, 3054, 3167; 1H NMR (400 MHz, DMSO-d6) δ 8.18 (d, J = 7.0 Hz, 2H), 8.46 (s, 2H, NH2), 8.76 (d, J = 7.0 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 128.2 (2× CH), 139.4 (2× CH), 145.4 (Cq); HRMS (ESI) [M + H]+ calcd for C5H7ClN2: 129.0214, found: 129.0214.:EtOAc, 5:5) to give 2 as yellow crystals (523 mg, 86%). Mp 120–121 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.87; IR (ATR diamond, cm−1) ν 724, 811, 862, 1093, 1217, 1357, 1510, 1689, 3085; 1H NMR (400 MHz, CDCl3) δ 3.94 (s, 3H, CH3), 4.03 (s, 3H, CH3), 7.02 (dd, J = 7.3, 2.3 Hz, 1H), 8.16 (d, J = 2.3 Hz, 1H), 8.43 (d, J = 7.3 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 51.8 (CH3), 53.0 (CH3), 102.7 (Cq), 116.7 (CH), 118.7 (CH), 129.7 (CH), 135.2 (Cq), 141.5 (Cq), 148.1 (Cq), 162.0 (CO), 162.7 (CO); HRMS (ESI) [M + H]+ calcd for C11H10ClN2O4: 269.0324, found: 269.0321.:EtOAc, 2:8) = 0.1; IR (ATR diamond, cm−1) ν 664, 764, 856, 1105, 1209, 1379, 1480, 1621, 1750, 2651, 3083; 1H NMR (400 MHz, CDCl3) δ 4.15 (s, 3H, CH3), 7.17 (dd, J = 7.3, 2.3 Hz, 1H), 8.14 (d, J = 2.3 Hz, 1H), 8.64 (d, J = 7.3 Hz, 1H), 14.34 (s, 1H, OH); 13C NMR (101 MHz, DMSO-d6) δ 52.1 (CH3), 100.8 (Cq), 126.9 (CH), 127.8 (CH), 131.8 (CH), 135.1 (Cq), 140.8 (Cq), 150.4 (Cq), 132.3 (CO), 134.2 (CO); HRMS (ESI) [M + H]+ calcd for C10H8ClN2O4: 255.0167 [M + H]+, found: 255.0164.:EtOAc, 5:5) as a white solid (0.857 g, 67%). Mp 188–190 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.8; IR (ATR diamond, cm−1) ν 779, 850, 1123, 1221, 1268, 1369, 1434, 1534, 1685, 2980, 3338; 1H NMR (400 MHz, CDCl3) δ 1.55 (s, 9H, 3× CH3), 3.96 (s, 3H, CH3), 6.86 (dd, J = 7.3, 2.3 Hz, 1H), 7.85 (d, J = 2.3 Hz, 1H), 8.44 (d, J = 7.3 Hz, 1H), 8.97 (s, 1H, NH); 13C NMR (101 MHz, CDCl3) δ 28.6 (3× CH3), 51.9 (CH3), 82.1 (Cq), 90.0 (Cq), 114.9 (CH), 117.1 (CH), 130.5 (CH), 135.9 (Cq), 140.8 (Cq), 151.1 (Cq), 154.1 (CO), 165.1 (CO); HRMS (ESI) [M + H]+ calcd for C14H17ClN3O4: 326.0902 [M + H]+, found: 326.0901.:EtOAc, 4:6) = 0.67; IR (ATR diamond, cm−1) ν 776, 864, 1009, 1112, 1280, 1437, 1620, 1678, 3104, 3422; 1H NMR (400 MHz, CDCl3) δ 3.92 (s, 3H, CH3), 5.26 (s, 2H, NH2), 6.75 (dd, J = 7.3, 2.3 Hz, 1H), 7.78 (d, J = 2.3 Hz, 1H), 8.11 (d, J = 7.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 51.0 (CH3). 87.8 (Cq), 113.4 (CH), 116.2 (CH), 128.7 (CH), 134.3 (Cq), 141.8 (Cq), 160.3 (Cq), 164.7 (CO); HRMS (ESI) [M + H]+ calcd for C9H9ClN3O2: 226.0378 [M + H]+, found: 226.0375.:EtOAc, 4:6) as a yellow solid (1.44 g, 84%). Mp 201–202 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.90; IR (ATR diamond, cm−1) ν 698, 807, 1006, 1112, 1156, 1303, 1435, 1529, 1678, 3040; 1H NMR (400 MHz, CDCl3) δ 4.05 (s, 3H, CH3), 6.95 (dd, J = 7.3, 2.3 Hz, 1H), 7.59–7.47 (m, 2H), 7.71–7.62 (m, 1H), 7.98 (d, J = 7.3 Hz, 2H), 8.04 (d, J = 2.3 Hz, 1H), 8.49 (d, J = 7.3 Hz, 1H), 9.27 (s, 1H, NH), 13.85 (s, 1H, NH); 13C NMR (101 MHz, CDCl3) δ 52.2 (CH3), 93.45 (Cq), 115.7 (CH), 117.8 (CH), 128.1 (2× CH), 129.5 (2× CH), 130.6 (Cq), 131.9 (CH), 134.1 (Cq), 136.1 (CH), 141.2 (Cq), 152.3 (Cq), 163.9 (CO), 165.9 (CO), 176.6 (CS); HRMS (ESI) [M + H]+ calcd for C17H14ClN4O3S: 289.0470 [M + H]+, found: 389.0468.:EtOAc, 4:6) = 0.10; IR (ATR diamond, cm−1) ν 764, 824, 1106, 1208, 1379, 1480, 1621, 1750, 2652, 3083; 1H NMR (400 MHz, DMSO-d6) δ 7.36 (dd, J = 7.3, 2.3 Hz, 1H), 7.92 (d, J = 2.3 Hz, 1H), 8.95 (d, J = 7.3 Hz, 1H), 12.15 (s, 1H, NH), 13.36 (s, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ 92.8 (Cq), 115.9 (CH), 116.9 (CH), 132.1 (CH), 135.6 (Cq), 138.6 (Cq), 154.4 (Cq), 157.0 (CO), 177.0 (CS); HRMS (ESI) [M + H]+ calcd for C9H6ClN4OS: 252.9945 [M + H]+, found: 252.9942.:EtOAc, 4:6) = 0.57; IR (ATR diamond, cm−1) ν 648, 782, 861, 1121, 1252, 1432, 1522, 1635, 2820, 3033; 1H NMR (400 MHz, DMSO-d6) δ 2.58 (s, 3H, CH3), 7.38 (dd, J = 7.3, 2.3 Hz, 1H), 8.00 (d, J = 2.3 Hz, 1H), 8.96 (d, J = 7.3 Hz, 1H), 12.37 (s, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ 13.4 (CH3), 94.8 (Cq), 116.6 (CH), 117.0 (CH), 131.6 (CH), 134.0 (Cq), 138.0 (Cq), 158.7 (Cq), 161.9 (Cq), 162.4 (CO); HRMS (ESI) [M + H]+ calcd for C10H8ClN4OS: 267.0102 [M + H]+, found: 267.0100.
:EtOAc, 4:6) = 0.1; IR (ATR diamond, cm−1) ν 833, 950, 1119, 1471, 1607, 3054, 3167; 1H NMR (400 MHz, DMSO-d6) δ 8.18 (d, J = 7.0 Hz, 2H), 8.46 (s, 2H, NH2), 8.76 (d, J = 7.0 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 128.2 (2× CH), 139.4 (2× CH), 145.4 (Cq); HRMS (ESI) [M + H]+ calcd for C5H7ClN2: 129.0214, found: 129.0214.:EtOAc, 5:5) to give 2 as yellow crystals (523 mg, 86%). Mp 120–121 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.87; IR (ATR diamond, cm−1) ν 724, 811, 862, 1093, 1217, 1357, 1510, 1689, 3085; 1H NMR (400 MHz, CDCl3) δ 3.94 (s, 3H, CH3), 4.03 (s, 3H, CH3), 7.02 (dd, J = 7.3, 2.3 Hz, 1H), 8.16 (d, J = 2.3 Hz, 1H), 8.43 (d, J = 7.3 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 51.8 (CH3), 53.0 (CH3), 102.7 (Cq), 116.7 (CH), 118.7 (CH), 129.7 (CH), 135.2 (Cq), 141.5 (Cq), 148.1 (Cq), 162.0 (CO), 162.7 (CO); HRMS (ESI) [M + H]+ calcd for C11H10ClN2O4: 269.0324, found: 269.0321.:EtOAc, 2:8) = 0.1; IR (ATR diamond, cm−1) ν 664, 764, 856, 1105, 1209, 1379, 1480, 1621, 1750, 2651, 3083; 1H NMR (400 MHz, CDCl3) δ 4.15 (s, 3H, CH3), 7.17 (dd, J = 7.3, 2.3 Hz, 1H), 8.14 (d, J = 2.3 Hz, 1H), 8.64 (d, J = 7.3 Hz, 1H), 14.34 (s, 1H, OH); 13C NMR (101 MHz, DMSO-d6) δ 52.1 (CH3), 100.8 (Cq), 126.9 (CH), 127.8 (CH), 131.8 (CH), 135.1 (Cq), 140.8 (Cq), 150.4 (Cq), 132.3 (CO), 134.2 (CO); HRMS (ESI) [M + H]+ calcd for C10H8ClN2O4: 255.0167 [M + H]+, found: 255.0164.:EtOAc, 5:5) as a white solid (0.857 g, 67%). Mp 188–190 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.8; IR (ATR diamond, cm−1) ν 779, 850, 1123, 1221, 1268, 1369, 1434, 1534, 1685, 2980, 3338; 1H NMR (400 MHz, CDCl3) δ 1.55 (s, 9H, 3× CH3), 3.96 (s, 3H, CH3), 6.86 (dd, J = 7.3, 2.3 Hz, 1H), 7.85 (d, J = 2.3 Hz, 1H), 8.44 (d, J = 7.3 Hz, 1H), 8.97 (s, 1H, NH); 13C NMR (101 MHz, CDCl3) δ 28.6 (3× CH3), 51.9 (CH3), 82.1 (Cq), 90.0 (Cq), 114.9 (CH), 117.1 (CH), 130.5 (CH), 135.9 (Cq), 140.8 (Cq), 151.1 (Cq), 154.1 (CO), 165.1 (CO); HRMS (ESI) [M + H]+ calcd for C14H17ClN3O4: 326.0902 [M + H]+, found: 326.0901.:EtOAc, 4:6) = 0.67; IR (ATR diamond, cm−1) ν 776, 864, 1009, 1112, 1280, 1437, 1620, 1678, 3104, 3422; 1H NMR (400 MHz, CDCl3) δ 3.92 (s, 3H, CH3), 5.26 (s, 2H, NH2), 6.75 (dd, J = 7.3, 2.3 Hz, 1H), 7.78 (d, J = 2.3 Hz, 1H), 8.11 (d, J = 7.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 51.0 (CH3). 87.8 (Cq), 113.4 (CH), 116.2 (CH), 128.7 (CH), 134.3 (Cq), 141.8 (Cq), 160.3 (Cq), 164.7 (CO); HRMS (ESI) [M + H]+ calcd for C9H9ClN3O2: 226.0378 [M + H]+, found: 226.0375.:EtOAc, 4:6) as a yellow solid (1.44 g, 84%). Mp 201–202 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.90; IR (ATR diamond, cm−1) ν 698, 807, 1006, 1112, 1156, 1303, 1435, 1529, 1678, 3040; 1H NMR (400 MHz, CDCl3) δ 4.05 (s, 3H, CH3), 6.95 (dd, J = 7.3, 2.3 Hz, 1H), 7.59–7.47 (m, 2H), 7.71–7.62 (m, 1H), 7.98 (d, J = 7.3 Hz, 2H), 8.04 (d, J = 2.3 Hz, 1H), 8.49 (d, J = 7.3 Hz, 1H), 9.27 (s, 1H, NH), 13.85 (s, 1H, NH); 13C NMR (101 MHz, CDCl3) δ 52.2 (CH3), 93.45 (Cq), 115.7 (CH), 117.8 (CH), 128.1 (2× CH), 129.5 (2× CH), 130.6 (Cq), 131.9 (CH), 134.1 (Cq), 136.1 (CH), 141.2 (Cq), 152.3 (Cq), 163.9 (CO), 165.9 (CO), 176.6 (CS); HRMS (ESI) [M + H]+ calcd for C17H14ClN4O3S: 289.0470 [M + H]+, found: 389.0468.:EtOAc, 4:6) = 0.10; IR (ATR diamond, cm−1) ν 764, 824, 1106, 1208, 1379, 1480, 1621, 1750, 2652, 3083; 1H NMR (400 MHz, DMSO-d6) δ 7.36 (dd, J = 7.3, 2.3 Hz, 1H), 7.92 (d, J = 2.3 Hz, 1H), 8.95 (d, J = 7.3 Hz, 1H), 12.15 (s, 1H, NH), 13.36 (s, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ 92.8 (Cq), 115.9 (CH), 116.9 (CH), 132.1 (CH), 135.6 (Cq), 138.6 (Cq), 154.4 (Cq), 157.0 (CO), 177.0 (CS); HRMS (ESI) [M + H]+ calcd for C9H6ClN4OS: 252.9945 [M + H]+, found: 252.9942.:EtOAc, 4:6) = 0.57; IR (ATR diamond, cm−1) ν 648, 782, 861, 1121, 1252, 1432, 1522, 1635, 2820, 3033; 1H NMR (400 MHz, DMSO-d6) δ 2.58 (s, 3H, CH3), 7.38 (dd, J = 7.3, 2.3 Hz, 1H), 8.00 (d, J = 2.3 Hz, 1H), 8.96 (d, J = 7.3 Hz, 1H), 12.37 (s, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ 13.4 (CH3), 94.8 (Cq), 116.6 (CH), 117.0 (CH), 131.6 (CH), 134.0 (Cq), 138.0 (Cq), 158.7 (Cq), 161.9 (Cq), 162.4 (CO); HRMS (ESI) [M + H]+ calcd for C10H8ClN4OS: 267.0102 [M + H]+, found: 267.0100.5. General procedure A for Suzuki–Miyaura cross coupling reaction
A solution of compound 8 (100 mg, 0.37 mmol, 1.0 eq.) in 1,4-dioxane (4 mL) was degassed by argon bubbling. Potassium carbonate (103 mg, 0.74 mmol, 2.0 eq.), boronic acid (1.1 eq.), xantphos (43 mg, 0.074 mmol, 0.2 eq.) and Pd(OAc)2 (8.0 mg, 0.037 mmol, 0.1 eq.) were then added and the reaction mixture was heated to 150 °C for 1 h under microwave irradiation. The crude material was concentrated under reduced pressure, and the residue was diluted in CH2Cl2 (10 mL). The organic layers were washed with water (10 mL), dried over MgSO4, filtered and concentrated under reduced pressure. The crude residue was then purified by flash chromatography on silica to provide the desired compounds 10–15.6. General procedure B for the palladium catalyzed direct cross-coupling reactions involving a PyBroP-mediated C–O activation
A suitable compound 10–11, 13–15 and PyBroP (1.2 eq.) were dissolved in 1,4-dioxane (4 mL) and Et3N (3.0 eq.) was then added. Under stirring, the mixture was degassed by argon bubbling for 10 min and then heated at 80 °C for 18 h. After cooling, the required (het)arylboronic acid (2.0 eq.), Na2CO3 (5.0 eq.) in H2O (1 mL) and PdCl2(dppf)·CH2Cl2 (10 mol%) were added. After heating at 110 °C for 24 hours generally, volatiles were removed under reduced pressure and the crude material diluted in CH2Cl2 (20 mL). The combined organic extracts were dried over anhydrous Na2SO4, filtered, concentrated under reduced pressure, and purified by chromatography on silica gel to give the desired bis-arylated compounds 16–25.7. General procedure C for Liebeskind–Srogl cross-coupling reaction
A microwave vial with THF (4 mL) under inert atmosphere was charged with the corresponding 4,6-bis(het)aryl-2-(methylthio)-6-arylpyrido[1′,2′:1,5]pyrazolo[3,4-d]pyrimidines 18, 19 or 22, the corresponding aryl boronic acid (1.5 eq.), CuTC (3.0 eq.), and Pd(PPh3)4 (10 mol%). The mixture was heated under microwave irradiation at 100 °C for 1 h 30 min. After cooling to r.t., the solvent was removed under reduced pressure, the residue was diluted in aq. satd NaHCO3 solution (10 mL). The aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried with MgSO4, filtered, and concentrated under reduced pressure. The crude reaction mixture was purified by column chromatography on silica gel to provide the desired products 26–28.:EtOAc, 6:4) as a white solid (33 mg, 45%). Mp 222–223 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.80; IR (ATR diamond, cm−1) ν 796, 1005, 1163, 1244, 1452, 1578, 1641, 2919; 1H NMR (400 MHz, CDCl3) δ 2.42 (s, 3H, CH3), 2.50 (s, 3H, CH3), 2.74 (s, 3H, CH3), 7.31 (d, J = 7.8 Hz, 2H), 7.41 (d, J = 7.8 Hz, 2H), 7.48 (d, J = 7.8 Hz, 2H), 7.56–7.49 (m, 1H), 7.86 (d, J = 7.8 Hz, 2H), 8.16 (s, 1H), 8.83 (d, J = 7.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 14.5 (S–CH3), 21.2 (CH3), 21.6 (CH3), 101.9 (Cq), 115.9 (CH), 116.8 (CH), 126.7 (2× CH), 128.8 (2× CH), 129.5 (2× CH), 129.5 (CH), 129.6 (Cq), 130.1 (2× CH), 134.6 (Cq), 134.7 (Cq), 136.0 (Cq), 139.3 (Cq), 139.9 (Cq), 141.3 (Cq), 163.5 (Cq), 172.2 (Cq); HRMS (ESI) [M + H]+ calcd for C24H21N4S: 397.1481 [M + H]+, found: 397.1481.:EtOAc, 5:5) as a white solid (76 mg, 63%). Mp 302–303 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.62; IR (ATR diamond, cm−1) ν 666, 783, 873, 1001, 1122, 1210, 1363, 1447, 1580, 1643, 2829; 1H NMR (400 MHz, DMSO-d6) δ 2.39 (s, 3H, CH3), 2.59 (s, 3H, CH3), 7.36 (d, J = 7.8 Hz, 2H), 7.63 (dd, J = 7.3, 2.3 Hz, 1H), 7.87–7.70 (m, 2H), 8.14 (d, J = 2.3 Hz, 1H), 8.95 (d, J = 7.3 Hz, 1H), 12.34 (s, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ 13.4 (CH3). 21.2 (S–CH3), 94.7 (Cq), 113.5 (CH), 114.9 (CH), 127.2 (2× CH), 130.3 (2× CH), 130.4 (CH), 134.3 (Cq), 138.1 (Cq), 139.3 (Cq), 140.1 (Cq), 158.8 (Cq), 161.7 (Cq), 161.85 (CO); HRMS (ESI) [M + H]+ calcd for C17H15N4OS: 323.0961 [M + H]+, found: 323.0961.:EtOAc, 5:5) as a white solid (92 mg, 73%). Mp 295–296 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.50; IR (ATR diamond, cm−1) ν 956, 1033, 1113, 1251, 1453, 1518, 1661, 2835; 1H NMR (400 MHz, DMSO-d6) δ 2.59 (s, 3H, CH3). 3.84 (s, 3H, CH3), 7.18–6.99 (m, 2H), 7.61 (dd, J = 7.3, 2.3 Hz, 1H), 7.91–7.76 (m, 2H), 8.10 (d, J = 2.3 Hz, 1H), 8.92 (d, J = 7.3 Hz, 1H), 12.31 (s, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ 12.9 (S–CH3), 55.3 (O–CH3), 94.1 (Cq), 112.4 (CH), 114.3 (CH), 114.7 (2× CH), 128.2 (2× CH), 128.9 (Cq), 129.9 (CH), 137.7 (Cq), 139.4 (Cq), 158.3 (Cq), 160.1 (Cq), 161.2 (Cq), 161.3 (CO); HRMS (ESI) [M + H]+ calcd for C17H15N4O2S: 339.0910 [M + H]+, found: 339.0909.:EtOAc, 5:5) as a white solid (86 mg, 68%). Mp 266–267 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.57; IR (ATR diamond, cm−1) ν 775, 863, 1054, 1170, 1455, 1578, 1667, 2833; 1H NMR (400 MHz, DMSO-d6) δ 2.59 (s, 3H, CH3), 3.87 (s, 3H, CH3), 7.06 (d, J = 7.8 Hz, 1H), 7.37 (s, 1H), 7.41 (d, J = 7.8 Hz, 1H), 7.47 (t, J = 7.8 Hz, 1H), 7.64 (dd, J = 7.3, 2.3 Hz, 1H), 8.16 (d, J = 2.3 Hz, 1H), 8.97 (d, J = 7.3 Hz, 1H), 12.35 (s, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ 13.4 (S–CH3), 55.7 (O–CH3), 94.9 (Cq), 112.2 (CH), 114.2 (CH), 115.3 (CH), 115.4 (CH), 119.7 (CH), 130.4 (CH), 130.8 (CH), 138.0 (Cq), 138.8 (Cq), 140.1 (Cq), 158.8 (Cq), 160.3 (Cq), 161.8 (Cq), 161.9 (CO); HRMS (ESI) [M + H]+ calcd for C17H15N4O2S: 339.0910 [M + H]+, found: 339.0907.:EtOAc, 5:5) as a white solid (94 mg, 70%). Mp 311–312 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.65; IR (ATR diamond, cm−1) ν 746, 803, 960, 1181, 1475, 1582, 1644, 1739, 3044; 1H NMR (400 MHz, DMSO-d6) δ 2.60 (s, 3H, CH3), 7.71–7.51 (m, 2H), 7.80 (dd, J = 7.3, 2.3 Hz, 1H), 8.00 (dd, J = 9.9, 6.2 Hz, 2H), 8.10 (dd, J = 9.3, 4.5 Hz, 2H), 8.33 (d, J = 2.3 Hz, 1H), 8.49 (d, J = 2.0 Hz, 1H), 9.03 (d, J = 7.3 Hz, 1H), 12.38 (s, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ 13.4 (S–CH3), 94.9 (Cq), 114.3 (CH), 115.2 (CH), 124.9 (CH), 126.8 (CH), 127.2 (CH), 127.45 (CH), 128.0 (CH), 129.05 (CH), 129.4 (CH), 130.5 (CH), 133.4 (Cq), 133.6 (Cq), 134.5 (Cq), 138.1 (Cq), 139.9 (Cq), 158.8 (Cq), 161.8 (CO), 161.9 (CO); HRMS (ESI) [M + H]+ calcd for C20H15N4OS: 359.0961 [M + H]+, found: 359.0959.:EtOAc, 5:5) as a white solid (80 mg, 66%). Mp 319–320 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.62; IR (ATR diamond, cm−1) ν 658, 784, 870, 1107, 1233, 1445, 1647, 2819; 1H NMR (400 MHz, DMSO-d6) δ 2.59 (s, 3H, CH3), 7.37 (t, J = 8.6 Hz, 2H), 7.61 (dd, J = 7.3, 2.3 Hz, 1H), 7.99–7.88 (m, 2H), 8.13 (d, J = 2.3 Hz, 1H), 8.96 (d, J = 7.3 Hz, 1H), 12.34 (s, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ 13.4 (S–CH3), 94.8 (Cq), 114.0 (CH), 115.0 (CH), 116.6 (d, 2JC–F = 21.6 Hz, 2× CH), 129.7 (d, 3JC–F = 8.5 Hz, 2× CH), 130.4 (CH), 133.8 (d, 4JC–F = 3.1 Hz, Cq), 138.0 (Cq), 139.1 (Cq), 158.8 (Cq), 161.8 (Cq), 161.8 (CO), 163.2 (d, 1JC–F = 247 Hz, CF); HRMS (ESI) [M + H]+ calcd for C16H12FN4OS: 327.0710 [M + H]+, found: 327.0710.:EtOAc, 5:5) as a white solid (79 mg, 56%). Mp 342–343 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.66; IR (ATR diamond, cm−1) ν 811, 1015, 1109, 1330, 1574, 1650, 2833, 3044; 1H NMR (400 MHz, DMSO-d6) δ 2.58 (s, 3H, CH3), 7.67 (dd, J = 7.3, 2.3 Hz, 1H), 7.87 (d, J = 8.0 Hz, 2H), 8.08 (d, J = 8.0 Hz, 2H), 8.21 (d, J = 2.3 Hz, 1H), 9.00 (d, J = 7.3 Hz, 1H), 12.38 (s, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ 12.9 (S–CH3), 94.7 (Cq), 114.4 (CH), 114.5 (CH), 124.1 (q, 1JC–F3 = 272 Hz, CF3), 126.0 (q, 3JC–F3 = 3.3 Hz, 2× CH), 127.8 (2× CH), 129.4 (q, 2JC–F3 = 32 Hz, Cq), 130.2 (CH), 137.3 (Cq), 137.8 (Cq), 140.8 (Cq), 158.3 (Cq), 161.4 (Cq), 161.5 (CO); HRMS (ESI) [M + H]+ calcd for C17H12F3N4OS: 377.0678 [M + H]+, found: 377.0676.:EtOAc, 6:4) as a yellow solid (35 mg, 55%). Mp 266–267 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.85; IR (ATR diamond, cm−1) ν 775, 846, 1023, 1118, 1444, 1577, 1653, 2976; 1H NMR (400 MHz, CDCl3) δ 2.43 (s, 3H, CH3), 2.74 (s, 3H, CH3), 3.93 (s, 3H, CH3), 7.12 (d, J = 8.5 Hz, 2H), 7.32 (d, J = 7.8 Hz, 2H), 7.50 (d, J = 7.8 Hz, 2H), 7.56 (dd, J = 7.3 Hz, 1H), 7.95 (d, J = 8.5 Hz, 2H), 8.20 (s, 1H), 8.85 (d, J = 7.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 14.6 (S–CH3), 21.3 (CH3), 55.7 (CH3), 101.9 (Cq), 114.4 (2× CH), 115.9 (CH), 117.1 (CH), 126.9 (2× CH), 129.6 (Cq), 129.8 (CH), 130.3 (2× CH), 130.7 (2× CH), 134.7 (Cq), 136.3 (Cq), 139.6 (Cq), 140.3 (Cq), 162.2 (Cq), 162.9 (Cq), 163.5 (Cq), 171.8 (Cq); HRMS (ESI) [M + H]+ calcd for C24H21N4OS: 413.1431 [M + H]+, found: 413.1430.:EtOAc, 6:4) as a yellow solid (29 mg, 47%). Mp 225–226 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.89; IR (ATR diamond, cm−1) ν 799, 870, 1121, 1450, 1559, 1636, 1732, 2921; 1H NMR (400 MHz, CDCl3) δ 2.45 (s, 3H, CH3), 2.76 (s, 3H, CH3), 7.39–7.28 (m, 4H), 7.50 (d, J = 7.8 Hz, 2H), 7.58 (dd, J = 7.3, 2.3 Hz, 1H), 8.02–7.96 (m, 2H), 8.09 (d, J = 2.3 Hz, 1H), 8.87 (d, J = 7.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 14.4 (CH3), 21.2 (S–CH3), 101.8 (Cq), 115.5 (CH), 116.0 (d, 2JC–F = 21.8 Hz, 2× CH), 117.0 (CH), 126.7 (2× CH), 129.7 (CH), 130.2 (2× CH), 130.9 (d, 3JC–F = 8.7 Hz, 2× CH), 133.7 (d, 4JC–F = 3.2 Hz, Cq), 134.4 (Cq), 135.8 (Cq), 139.5 (Cq), 140.2 (Cq), 162.2 (Cq), 163.4 (Cq), 164.4 (d, 1JC–F = 248 Hz, CF), 172.2 (Cq); HRMS (ESI) [M + H]+ calcd for C23H18FN4S: 401.1231 [M + H]+, found: 401.1231.:EtOAc, 4:6) as a yellow solid (30 mg, 64%). Mp 230–231 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.82; IR (ATR diamond, cm−1) ν 798, 870, 1010, 1165, 1285, 1449, 1494, 1636, 2925; 1H NMR (400 MHz, CDCl3) δ 2.43 (s, 3H, CH3), 2.73 (s, 3H, CH3), 4.07 (s, 3H, CH3), 6.98 (d, J = 8.8 Hz, 1H), 7.32 (d, J = 7.8 Hz, 2H), 7.59–7.49 (m, 3H), 8.12 (s, 1H), 8.19 (d, J = 8.8 Hz, 1H), 8.81 (d, J = 2.3 Hz, 1H), 8.85 (d, J = 7.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 14.6 (CH3), 21.3 (S–CH3), 54.1 (O–CH3), 101.9 (Cq), 111.4 (CH), 115.6 (CH), 117.2 (CH), 126.9 (Cq), 126.9 (2× CH), 129.8 (CH), 130.2 (2× CH), 134.5 (Cq), 135.9 (Cq), 139.2 (CH), 139.6 (Cq), 140.5 (Cq), 147.9 (CH), 160.7 (Cq), 163.5 (Cq), 165.9 (Cq), 172.3 (Cq); HRMS (ESI) [M + H]+ calcd for C23H20N5OS: 414.1383 [M + H]+, found: 414.1381.:EtOAc, 4:6) as a yellow solid (47 mg, 78%). Mp 209–210 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.39; IR (ATR diamond, cm−1) ν 724, 1020, 1117, 1247, 1452, 1604, 1638, 2951; 1H NMR (400 MHz, CDCl3) δ 2.50 (s, 3H, CH3), 2.73 (s, 3H, CH3), 3.88 (s, 3H, CH3), 7.06–6.99 (m, 2H), 7.41 (d, J = 7.8 Hz, 2H), 7.57–7.48 (m, 3H), 7.90–7.84 (m, 2H), 8.13 (d, J = 2.2 Hz, 1H), 8.82 (d, J = 7.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 14.4 (CH3), 21.6 (S–CH3), 55.4 (O–CH3), 101.8 (Cq), 114.85 (2× CH), 115.3 (CH), 116.6 (CH), 128.1 (2× CH), 128.8 (2× CH), 129.5 (2× CH), 129.5 (CH), 129.7 (Cq), 134.7 (Cq), 136.0 (Cq), 139.6 (Cq), 141.3 (Cq), 160.5 (Cq), 163.4 (Cq), 163.5 (Cq), 172.1 (Cq); HRMS (ESI) [M + H]+ calcd for C24H21N4OS: 413.1431 [M + H]+, found: 413.1428.:EtOAc, 4:6) as a yellow solid (62 mg, 70%). Mp > 360 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.59; IR (ATR diamond, cm−1) ν 813, 1037, 1164, 1244, 1452, 1572, 1637, 2227, 3044; 1H NMR (400 MHz, DMSO-d6) δ 2.74 (s, 3H, CH3), 3.89 (s, 3H, CH3), 7.10–7.00 (m, 2H), 7.52–7.45 (m, 2H), 7.58 (dd, J = 7.3, 2.3 Hz, 1H), 7.97–7.89 (m, 3H), 8.13–8.05 (m, 2H), 8.86 (d, J = 7.3 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 14.6 (S–CH3), 55.6 (O–CH3), 101.8 (Cq), 114.65 (Cq), 115.0 (CH), 115.2 (2× CH), 117.3 (CH), 118.4 (Cq), 128.2 (2× CH), 129.4 (Cq), 129.7 (2× CH), 130.0 (CH), 132.8 (2× CH), 135.7 (Cq), 140.5 (Cq), 142.0 (Cq), 161.0 (Cq), 161.2 (Cq), 163.5 (Cq), 172.6 (Cq); HRMS (ESI) [M + H]+ calcd for C24H18N5OS: 424.1227 [M + H]+, found: 424.1224.:EtOAc, 4:6) as a yellow solid (21 mg, 36%). Mp 222–223 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.46; IR (ATR diamond, cm−1) ν 793, 1118, 1163, 1250, 1454, 1522, 1643, 2161, 2926; 1H NMR (400 MHz, CDCl3) δ 2.75 (s, 3H, CH3), 3.87 (s, 3H, CH3), 7.07–6.98 (m, 2H), 7.64–7.50 (m, 4H), 7.99 (d, J = 2.3 Hz, 1H), 8.29 (dd, J = 7.3, 2.3 Hz, 1H), 8.86 (d, J = 7.3 Hz, 2H), 9.22 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 14.6 (S–CH3), 55.6 (O–CH3), 102.1 (Cq), 115.0 (CH), 115.1 (2× CH), 117.3 (CH), 124.0 (CH), 128.3 (2× CH), 129.5 (Cq), 129.9 (CH), 135.9 (Cq), 133.7 (Cq), 136.6 (CH), 140.5 (Cq), 149.6 (CH), 151.9 (CH), 160.4 (Cq), 160.9 (Cq), 163.4 (Cq), 172.5 (Cq); HRMS (ESI) [M + H]+ calcd for C22H18N5OS: 400.1227 [M + H]+, found: 400.1226.:EtOAc, 5:5) as a yellow solid (39 mg, 63%). Mp 271–272 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.62; IR (ATR diamond, cm−1) ν 763, 795, 841, 1031, 1168, 1245, 1444, 1555, 1635, 2933; 1H NMR (400 MHz, CDCl3) δ 2.74 (s, 3H, CH3). 3.93 (s, 3H, CH3), 7.14 (d, J = 8.4 Hz, 2H), 7.60–7.53 (m, 2H), 7.72–7.65 (m, 2H), 8.02–7.86 (m, 5H), 8.07 (s, 1H), 8.33 (s, 1H), 8.89 (d, J = 7.0 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 14.5 (CH3). 55.5 (CH3), 101.6 (Cq), 114.2 (2× CH), 116.4 (CH), 117.0 (CH), 124.2 (CH), 126.4 (CH), 127.0 (CH), 127.1 (CH), 127.7 (CH), 128.4 (CH), 129.3 (CH), 129.6 (CH), 129.9 (Cq), 130.6 (2× CH), 133.2 (Cq), 133.4 (Cq), 134.7 (Cq), 136.0 (Cq), 139.7 (Cq), 162.0 (Cq), 163.0 (Cq), 163.5 (Cq), 172.2 (Cq); HRMS (ESI) [M + H]+ calcd for C27H21N4OS: 449.1430 [M + H]+, found: 449.1431.:EtOAc, 6:4) as a yellow solid (38 mg, 61%). Mp 258–259 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.80; IR (ATR diamond, cm−1) ν 726, 804, 908, 1124, 1165, 1276, 1445, 1558, 1637, 2228, 2925, 3061; 1H NMR (400 MHz, CDCl3) δ 2.75 (s, 3H, CH3), 7.61–7.53 (m, 3H), 7.79–7.68 (m, 3H), 8.03–7.82 (m, 7H), 8.90 (d, J = 7.0 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 14.5 (CH3). 102.4 (Cq), 112.2 (Cq), 116.0 (CH), 117.4 (Cq), 117.8 (CH), 124.1 (CH), 126.6 (CH), 127.0 (CH), 127.2 (CH), 127.7 (CH), 128.4 (CH), 129.3 (CH), 129.9 (CH), 130.0 (CH), 130.6 (CH), 132.9 (CH), 133.3 (Cq), 133.7 (Cq), 134.1 (CH), 134.5 (Cq), 135.7 (Cq), 140.4 (Cq), 140.6 (Cq), 159.4 (Cq), 163.1 (Cq), 172.6 (Cq); HRMS (ESI) [M + H]+ calcd for C27H18N5S: 444.1277 [M + H]+, found: 444.1278.:EtOAc, 6:4) as a yellow solid (31 mg, 52%). Mp 240–241 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.90; IR (ATR diamond, cm−1) ν 793, 959, 1168, 1229, 1349, 1444, 1557, 1638, 2921, 3074; 1H NMR (400 MHz, CDCl3) δ 2.71 (s, 3H, CH3), 7.30 (t, J = 4.2 Hz, 1H), 7.60–7.53 (m, 2H), 7.69 (m, 2H), 7.74 (m, 1H), 7.96–7.86 (m, 2H), 8.04–7.96 (m, 2H), 8.11 (s, 1H), 8.66 (s, 1H), 8.88 (d, J = 7.1 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 14.4 (CH3), 100.9 (Cq), 116.4 (CH), 117.2 (CH), 124.2 (CH), 126.5 (CH), 127.0 (CH), 127.1 (CH), 127.7 (CH), 127.8 (CH), 128.4 (CH), 129.3 (CH), 129.5 (CH), 129.7 (CH), 130.8 (CH), 133.3 (Cq), 133.4 (Cq), 134.7 (Cq), 135.7 (Cq), 140.0 (Cq), 141.3 (Cq), 156.4 (Cq), 163.6 (Cq), 171.9 (Cq); HRMS (ESI) [M + H]+ calcd for C24H17N4S2: 425.0889 [M + H]+, found: 425.0890.:EtOAc, 4:6) as a yellow solid (25 mg, 40%). Mp 343–344 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.43; IR (ATR diamond, cm−1) ν 817, 1048, 1166, 1448, 1574, 1732, 2229, 2918, 3079; 1H NMR (400 MHz, DMSO-d6) δ 2.65 (s, 3H, CH3), 7.40 (t, J = 8.6 Hz, 2H), 7.92–7.78 (m, 2H), 7.98–7.89 (m, 1H), 8.02 (d, J = 2.3 Hz, 1H), 8.20 (t, J = 6.8 Hz, 4H), 9.25 (d, J = 7.3 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 14.2 (CH3). 102.2 (Cq), 113.9 (Cq), 116.1 (CH), 116.8 (d, 2JC–F = 21.8 Hz, 2× CH), 118.5 (CH), 129.8 (d, 3JC–F = 8.5 Hz, 2× CH), 130.1 (2× CH), 131.05 (CH), 131.05 (Cq), 133.5 (2× CH), 133.65 (d, JC–F = 3.1 Hz, Cq), 135.3 (Cq), 139.1 (Cq), 141.6 (Cq), 161.5 (Cq), 163.0 (Cq), 163.3 (d, 1JC–F = 248.0 Hz, CF), 171.1 (Cq); HRMS (ESI) [M + H]+ calcd for C23H15FN5S: 412.1027 [M + H]+, found: 412.1024.:EtOAc, 4:6) as white solid (40 mg, 70%). Mp 239–240 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.50; IR (ATR diamond, cm−1) ν 800, 870, 1019, 1120, 1287, 1451, 1493, 1602, 1712, 2991; 1H NMR (400 MHz, CDCl3) δ 2.43 (s, 3H, CH3), 3.90 (s, 3H, CH3), 4.09 (s, 3H, CH3), 7.03 (d, J = 8.5 Hz, 3H), 7.32 (d, J = 7.8 Hz, 2H), 7.60–7.50 (m, 3H), 8.22 (s, 1H), 8.33 (dd, J = 8.5, 2.3 Hz, 1H), 8.72 (d, J = 8.5 Hz, 2H), 8.99–8.87 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 21.2 (CH3), 54.0 (CH3), 55.3 (CH3), 111.2 (CH), 113.7 (2× CH), 115.7 (CH), 117.2 (CH), 126.8 (2× CH), 127.8 (Cq), 129.6 (CH), 130.1 (2× CH), 130.6 (2× CH), 130.9 (Cq), 134.5 (Cq), 135.5 (Cq), 139.1 (CH), 139.2 (Cq), 139.3 (Cq), 139.7 (Cq), 143.2 (Cq), 147.9 (CH), 160.7 (Cq), 161.9 (Cq), 163.3 (Cq), 165.7 (Cq); HRMS (ESI) [M + H]+ calcd for C29H24N5O2: 474.1925 [M + H]+, found: 474.1921.:EtOAc, 4:6) as yellow solid (43 mg, 78%). Mp 207–208 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.40; IR (ATR diamond, cm−1) ν 835, 1033, 1278, 1455, 1605, 1731, 2849, 2918; 1H NMR (400 MHz, CDCl3) δ 2.54 (s, 3H, CH3), 2.71 (s, 3H, CH3), 3.89 (s, 3H, CH3), 7.04 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 7.7 Hz, 2H), 7.35 (d, J = 8.2 Hz, 1H), 7.58 (d, J = 7.8 Hz, 3H), 7.99 (d, J = 7.7 Hz, 2H), 8.29 (s, 1H), 9.09–8.89 (m, 2H), 9.86 (s, 1H); 13C DEPT NMR (101 MHz, DMSO-d6) δ 21.6 (CH3), 24.3 (CH3), 55.5 (CH3), 114.9 (2× CH), 115.1 (CH), 115.8 (CH), 117.3 (CH), 128.2 (2× CH), 129.0 (2× CH), 129.6 (2× CH), 129.7 (CH), 139.0 (CH), 149.8 (CH); HRMS (ESI) [M + H]+ calcd for C29H24N5O: 458.1975 [M + H]+, found: 458.1977.:EtOAc, 4:6) as white solid (34 mg, 61%). Mp 329–330 °C; Rf (petroleum ether:EtOAc, 4:6) = 0.75; IR (ATR diamond, cm−1) ν 802, 1026, 1119, 1254, 1510, 1605, 1685, 2224, 2918; 1H NMR (250 MHz, DMSO-d6) δ 3.90 (s, 3H, CH3), 7.29 (d, J = 8.5 Hz, 2H), 7.64–7.48 (m, 2H), 7.81 (d, J = 8.7 Hz, 1H), 8.22–7.88 (m, 8H), 8.35 (d, J = 7.2 Hz, 2H), 8.70 (d, J = 8.2 Hz, 2H), 9.28 (d, J = 7.1 Hz, 1H); HRMS (ESI) [M + H]+ calcd for C33H22N5O: 504.1819 [M + H]+, found: 504.1820.Conflicts of interest
No conflict of interest for authors.Acknowledgements
The authors acknowledge the OMJ (Office Mediterranéen de la Jeunesse) for financial support to Rabia Belaroussi. The authors thank the Ligue Contre le Cancer and the Labex IRON (ANR-11-LABX-0018-01) for supplies.Notes and references
- A. Akahane, H. Katayama, T. Mitsunaga, T. Kato, T. Kinoshita, Y. Kita, T. Kusunoki, T. Terai, K. Yoshida and Y. Shiokawa, J. Med. Chem., 1999, 42, 779–783 CrossRef CAS PubMed.
- B. A. Johns, K. S. Gudmundsson, E. M. Turner, S. H. Allen, V. A. Samano, J. A. Ray, G. A. Freeman, F. L. Boyd Jr, C. J. Sexton, D. W. Selleseth, K. L. Creech and K. R. Moniri, Bioorg. Med. Chem., 2005, 13, 2397–2411 CrossRef CAS PubMed.
- (a) K. L. Stevens, D. K. Jung, M. J. Alberti, J. G. Badiang, G. E. Peckham, J. M. Veal, M. Cheung, P. A. Harris, S. D. Chamberlain and M. R. Peel, Org. Lett., 2005, 7, 4753–4756 CrossRef CAS PubMed; (b) M. Cheung, P. A. Harris, J. G. Badiang, G. E. Peckham, S. D. Chamberlain, M. J. Alberti, D. K. Jung, S. S. Harris, N. H. Bramson, A. H. Epperly, S. A. Stimpson and M. R. Peel, Bioorg. Med. Chem. Lett., 2008, 18, 5428–5430 CrossRef CAS PubMed.
- T. Koike, T. Takai, Y. Hoashi, M. Nakayama, Y. Kosugi, M. Nakashima, S.-i. Yoshikubo, K. Hirai and O. Uchikawa, J. Med. Chem., 2011, 54, 4207–4218 CrossRef CAS PubMed.
- (a) W. Zhang, A. L. McIver, M. A. Stashko, D. DeRyckere, B. R. Branchford, D. Hunter, D. Kireev, M. J. Miley, J. Norris-Drouin, W. M. Stewart, M. Lee, S. Sather, Y. Zhou, J. A. Di Paola, M. Machius, W. P. Janzen, H. S. Earp, D. K. Graham, S. V. Frye and X. Wang, J. Med. Chem., 2013, 56, 9693–9700 CrossRef CAS PubMed; (b) C. Ríos-Luci, R. Domínguez-Kelly, L. G. León, E. Díaz-Rodríguez, R. Freire, A. Pandiella, I. Cikotiene and J. M. Padrón, Bioorg. Med. Chem. Lett., 2011, 21, 6641–6645 CrossRef PubMed; (c) D. W. Porter, M. Bradley, Z. Brown, S. J. Charlton, B. Cox, P. Hunt, D. Janus, S. Lewis, P. Oakley, D. O'Connor, J. Reilly, N. Smith and N. J. Press, Bioorg. Med. Chem. Lett., 2014, 24, 3285–3290 CrossRef CAS PubMed; (d) V. Sharma, N. Chitranshi and A. K. Agarwal, Int. J. Med. Chem., 2014, 31, 28–58 Search PubMed.
- (a) A. Tikad, S. Routier, M. Akssira and G. Guillaumet, Org. Biomol. Chem., 2009, 7, 5113–5118 RSC; (b) A. Tikad, S. Routier, M. Akssira, J.-M. Léger, C. Jarry and G. Guillaumet, Synthesis, 2009, 2379–2384 CAS; (c) S. Grosse, C. Pillard, S. Massip, J. M. Léger, C. Jarry, S. Bourg, P. Bernard and G. Guillaumet, Chem.–Eur. J., 2012, 18, 14943–14947 CrossRef CAS PubMed; (d) Z. Tber, M.-A. Hiebel, A. El Hakmaoui, M. Akssira, G. Guillaumet and S. Berteina-Raboin, J. Org. Chem., 2015, 80, 6564–6573 CrossRef CAS PubMed; (e) V. Prieur, M. D. Pujol and G. Guillaumet, Eur. J. Chem., 2015, 6547–6556 CAS; (f) M. Naas, S. ElKazzouli, E. M. Essassi, M. Bousmina and G. Guillaumet, Org. Lett., 2015, 17, 4320–4323 CrossRef CAS PubMed; (g) S. Grosse, C. Pillard, S. Massip, M. Marchivie, C. Jarry, P. Bernard and G. Guillaumet, J. Org. Chem., 2015, 80, 8539–8551 CrossRef CAS PubMed; (h) M. Loubidi, C. Pillard, A. El Hakmaoui, P. Bernard, M. Akssira and G. Guillaumet, RSC Adv., 2016, 6, 7229–7238 RSC; (i) M. El Hafi, M. Naas, M. Loubidi, J. Jouha, Y. Ramli, J. T. Mague, E. M. Essassi and G. Guillaumet, C. R. Chim., 2017, 20, 927–933 CrossRef CAS.
- R. Belaroussi, A. El Hakmaoui, M. Akssira, G. Guillaumet and S. Routier, Eur. J. Org. Chem., 2016, 3550–3558 CrossRef CAS.
- R. Belaroussi, A. El Bouakher, M. Marchivie, S. Massip, C. Jarry, A. El Hakmaoui, G. Guillaumet, S. Routier and M. Akssira, Synthesis, 2013, 45, 2557–2566 CrossRef CAS.
- J. D. Oslob, R. S. Mcdowell, R. Johnson, H. Yang, M. Evanchik, C. A. Zaharia, H. Cai and L. W. Hu, WO2014008197, 2014, Chem. Abstr., 2014, 160, 190128.
- (a) F. U. Schmitz, V. W. F. Tai, R. Rai, C. D. Roberts, A. D. M. Abadi, S. Baskaran, I. Slobodov, J. Maung and M. L. Neitzel, WO2009023179, 2009, Chem. Abstr., 2009, 150, 260191; (b) Y. Feng, Y. T. Chen, H. Sessions, J. K. Mishra, S. Chowdhury, Y. Yin, P. Lograsso, J.-L. Luo, T. Bannister and T. Schroeter, WO2011050245, 2011, Chem. Abstr., 2011, 154, 540472; (c) M. Henrich, T. Weil, S. Mueller, V. Kauss, E. Erdmare and R. Zemribo, WO2009095253, 2009, Chem. Abstr., 2009, 151, 245654; (d) T.-S. Chung, X. Qiao and R. Liu, WO2006068626, 2006, Chem. Abstr., 2006, 145, 104617.
- (a) F. A. Kang, Z. Sui and W. V. Murray, J. Am. Chem. Soc., 2008, 130, 11300–11302 CrossRef CAS PubMed; (b) F. A. Kang, Z. Sui and W. V. Murray, Eur. J. Org. Chem., 2009, 461–479 CrossRef CAS; (c) F. A. Kang, J. C. Lanter, C. Cai, Z. Sui and W. V. Murray, Chem. Commun., 2010, 46, 1347–1349 RSC; (d) E. Frérot, J. Coste, A. Pantaloni, M.-N. Dufour and P. Jouin, Tetrahedron, 1991, 47, 259–270 CrossRef; (e) J. Coste, E. Frerot, P. Jouin and B. Castro, Tetrahedron Lett., 1991, 32, 1967–1970 CrossRef CAS; (f) J. Coste, E. Frerot and P. Jouin, J. Org. Chem., 1994, 59, 2437–2446 CrossRef CAS.
- R. Belaroussi, A. El Hakmaoui, N. Percina, A. Chartier, M. Marchivie, S. Massip, C. Jarry, M. Akssira, G. Guillaumet and S. Routier, Eur. J. Org. Chem., 2015, 4006–4017 CrossRef CAS.
- C. Copin, N. Henry, F. Buron and S. Routier, Eur. J. Org. Chem., 2012, 3079–3083 CrossRef CAS.
- (a) Q. Sun, F. Suzenet and G. Guillaumet, J. Org. Chem., 2010, 75, 3473–3476 CrossRef CAS PubMed; (b) H. Prokopcová and C. O. Kappe, Angew. Chem., Int. Ed., 2009, 48, 2276–2286 CrossRef PubMed; (c) F.-A. Alphonse, F. Suzenet, A. Keromnes, B. Lebret and G. Guillaumet, Synlett, 2002, 447–450 CrossRef CAS; (d) C. Kusturin, L. S. Liebeskind, H. Rahman, K. Sample, B. Schweitzer, J. Srogl and W. L. Neumann, Org. Lett., 2003, 5, 4349–4352 CrossRef CAS PubMed.
- J. Mendiola, J. A. Rincon, C. Mateos, J. F. Soriano, O. de Frutos, J. K. Niemeier and E. M. Davis, Org. Process Res. Dev., 2009, 13, 263–267 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12246b |
| This journal is © The Royal Society of Chemistry 2018 |