
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chetracins E and F, cytotoxic epipolythiodioxopiperazines from the marine-derived fungus Acrostalagmus luteoalbus HDN13-530†
Guihong Yua,
Yanjuan Wanga,
Rilei Yua,
Yanyan Fenga,
Lu Wanga,
Qian Chea,
Qianqun Gua,
Dehai Li ab,
Jing Liab and
Tianjiao Zhu*a
ab,
Jing Liab and
Tianjiao Zhu*a
aKey Laboratory of Marine Drugs, Chinese Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, Qingdao 266003, P. R. China. E-mail: zhutj@ouc.edu.cn
bLaboratory for Marine Drugs and Bioproducts of Qingdao National Laboratory for Marine Science and Technology, Qingdao, 266237, P. R. China
First published on 19th December 2017
Abstract
Two new epipolythiodioxopiperazines, named chetracins E and F (1 and 2), along with the known chetracin C (3), were isolated from the fungus Acrostalagmus luteoalbus HDN13-530. Their structures were elucidated based on the NMR, MS and CD data, as well as chemical conversion. All of the compounds exhibited cytotoxicity against the tested five cancer lines in low-micromolar or nanomolar IC50 values. The computational docking indicated that compounds 1–3 could bind to the C-terminal of heat shock protein 90 (Hsp90), which was in line with the experimental observation of decreases in levels and active forms of Hsp90 client proteins.
Introduction
The epidithiodioxopiperazine alkaloids (ETPs) characterized by a bridged polysulfide piperazine ring, represent a large family of secondary metabolites with various structure types and interesting biological activities.1,2 Among them, the representative chaetocin displayed a wide spectrum of antitumor activities in vivo in nanomolar IC50 level.1–6 Furthermore, recent research showed that chaetocin and the analogue chetracin B (discovered by our group), could function as heat shock protein 90 (Hsp90) inhibitors binding to the C-terminal.7 Hsp90 has proved to be an important target for cancer treatment, and is known as a crucial facilitator of oncogene addiction and cancer cell survival.7–9 Although most of the Hsp90 inhibitors undergoing clinic evaluations bind to the N-terminal (1-275 aa), those binding to the C-terminal (444-677 aa) are believed to have more potential without the tendency to induce expression of the undesired cytoprotective Hsp70 proteins.7Encouraged by the discovery of chetracin B as a novel C-terminal Hsp90 inhibitor from the fungus Oidiodendron truncatum GW3-13,1,7 the ETP alkaloids attracted our particular attention. Due to the difficulty and complexity in synthesizing this kind of alkaloid,10 we looked for more analogues from more producing fungi. As the characteristic of containing multi-sulfur atoms, the ETPs will show special isotope peaks in the LC-MS analysis. During the screening of our marine-derived microorganisms' library using LC-MS, the fungus, Acrostalagmus luteoalbus HDN13-530 isolated from soil of Liaodong Bay, was selected. The LC-MS profile of its fermentation extract showed significant M + 2 isotope peak which suggests the presentation of sulfur-containing metabolites (Fig. S1†). Further investigation showed that the EtOAc extract has potent cytotoxicity against P388 cells (66% inhibition of P388 cells at 100 μg mL−1). The LC-MS-UV guided fractionation of the fermentation extract led to the discovery of two new ETPs, named chetracins E (1) and F (2), together with the known chetracin C (3) (Fig. 1). In this report, we describe the isolation, structure elucidation, and activity evaluation of these compounds.
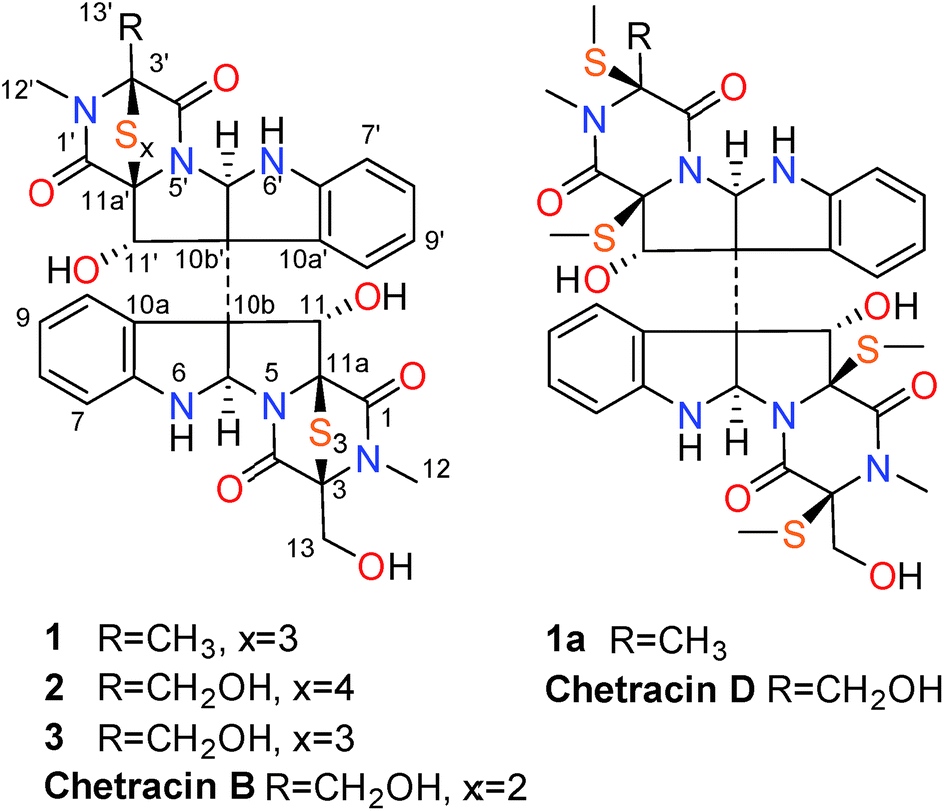 | ||
| Fig. 1 Structures of compounds 1–3, 1a, chetracins B and D. | ||
Results and discussion
The fungal strain A. luteoalbus HDN13-530 was fermented (30 L) under static conditions at 28 °C for 4 weeks. Guided by LC-MS-UV data, the EtOAc extract (40 g) of the fermentation was fractionated by silica gel vacuum liquid chromatography, C-18 ODS column chromatography, Sephadex LH-20 column chromatography, ODS MPLC, and finally HPLC to yield compounds 1 (20.0 mg), 2 (15.0 mg) and 3 (15.0 mg).Chetracin E (1) was isolated as a pale yellow, amorphous powder. Based on the HRESIMS adduct ion detected at m/z 777.0432 [M + H]+, its molecular formula was established as C30H28N6O7S6, requiring 20 degrees of unsaturation. The major 1D NMR resonances was categorized into three methyls with two nitrogenized ones (δC 27.5 and 28.2), one oxygenated methylene (δC 60.1), twelve methines (including eight aromatic ones), fourteen non-protonated carbons including four carbonyls (δC 163.1, 164.1, 167.5, and 167.8) (Table 1). The 1D NMR data of 1 were nearly superimposable to those of chetracin C (3) (Table 1).1 The only difference was that one oxygenated methylene in 3 was replaced by a methyl in 1, which was also confirmed by the HMBC correlations from H3-13′ to C-3′, C-4′ (Fig. 2).
| Position | 1 | 2 | 3b | |||
|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | |||
| a Signals were overlapped.b The assignment of 13C and 1H NMR data of C/H-5a and C/H-11 in the previous report1 was revised as those in this Table. | ||||||
| 1 | 167.8, qC | 167.9, qC | 167.9, qC | |||
| 3 | 75.8, qC | 75.9, qC | 75.9, qC | |||
| 4 | 163.1, qC | 163.1, qC | 163.3, qC | |||
| 5a | 80.8, CH | 4.69 s | 80.5, CH | 4.87 s | 80.9, CH | 4.72 s |
| 6a | 152.9, qC | 152.9, qC | 153.0, qC | |||
| 7 | 109.8, CH | 6.50–6.59a | 109.6, CH | 6.58–6.62a | 109.9, CH | 6.50–6.60a |
| 8 | 130.5, CH | 7.01–7.07a | 130.5, CH | 7.05 t (7.1) | 130.7, CH | 7.04 dd (7.1, 7.7) |
| 9 | 118.0, CH | 6.50–6.58a | 118.3, CH | 6.47–6.50a | 118.1, CH | 6.50–6.60a |
| 10 | 129.2, CH | 7.47 d (7.5) | 128.8, CH | 7.53 d (7.4) | 129.5, CH | 7.50 d (7.7) |
| 10a | 126.8, qC | 126.5, qC | 126.9, qC | |||
| 10b | 63.8, qC | 65.1, qC | 64.1, qC | |||
| 11 | 83.5, CH | 5.37 s | 84.2, CH | 5.37 d (5.4) | 83.7, CH | 5.38 d (5.5) |
| 11a | 85.2, qC | 84.8, qC | 85.4, qC | |||
| 12 | 27.5, CH3 | 3.09 s | 27.5, CH3 | 3.12 s | 27.6, CH3 | 3.11 s |
| 13 | 60.1, CH2 | 3.65 d (12.3) | 60.1, CH2 | 3.67 d (12.3) | 60.2, CH2 | 3.66 dd (4.9, 12.7) |
| 4.00 d (12.4) | 4.02 d (12.3) | 4.01 dd (6.1, 12.7) | ||||
| 11-OH | — | 6.15 d (5.0) | ||||
| 1′ | 167.5, qC | 167.3, qC | ||||
| 3′ | 72.0, qC | 79.8, qC | 6.52 d (4.9) | |||
| 4′ | 164.1, qC | 164.7, qC | 6.88 s | |||
| 5a′ | 80.8, CH | 4.68 s | 82.1, CH | 5.05 s | 5.68 t (5.5, 6.0) | |
| 6a′ | 152.9, qC | 151.0, qC | ||||
| 7′ | 109.9, CH | 6.50–6.59a | 109.6, CH | 6.58–6.62a | ||
| 8′ | 130.5, CH | 7.01–7.07a | 129.6, CH | 6.89–6.93a | ||
| 9′ | 117.9, CH | 6.50–6.58a | 118.2, CH | 6.47–6.50a | ||
| 10′ | 129.3 CH | 7.45 d (7.5) | 128.2, CH | 7.59 d (7.6) | ||
| 10a′ | 126.7, qC | 126.5, qC | ||||
| 10b′ | 64.0, qC | 64.8, qC | ||||
| 11′ | 83.6, CH | 5.35 s | 82.8, CH | 5.11 s | ||
| 11a′ | 85.8, qC | 81.2, qC | ||||
| 12′ | 28.2, CH3 | 3.04 s | 28.6, CH3 | 2.94 s | ||
| 13′ | 21.5, CH3 | 1.67 s | 60.9, CH2 | 3.84 d (11.2) | ||
| 3.97 d (11.1) | ||||||
| 11′-OH | — | 6.58–6.62a | ||||
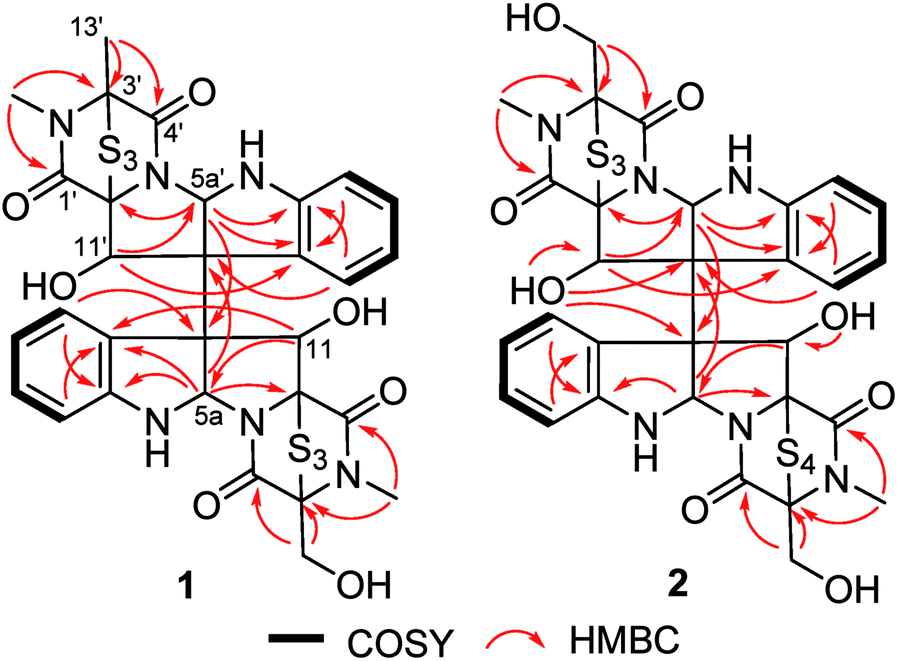 | ||
| Fig. 2 Key HMBC and 1H–1H COSY correlations of 1 and 2. | ||
The relative configuration of 1 was established based on the NOESY experiments (Fig. 3). The NOESY correlations from H-10 to H-11 and H-10′ to H-11′ indicated that H-11 and H-11′ faced to the same orientation to C-10a–C-10b bond and C-10a′–C-10b′ bond, respectively. Since the H-5a/H-5a′ and the C-10b–C-10b′ bond must be on the same side of disubstituted indole fragment because of structural rigidity, H-5a and the C-10b–C10b′ bond should be trans to H-11, H-5a′ and the C-10b–C10b′ bond also should be trans to H-11′.1 In order to determine the relative configuration of sulfur-bridged section, the tetrakis(methyl-sulfanyl) derivative (1a) (Fig. 1 and S2, Table S1†), was produced by treatment of 1 with NaBH4 and MeI. The NOESY correlations from 11a-SCH3 to H-11 and 3a-SCH3, as well as 11a′-SCH3 to H-11′ and 3a′-SCH3 indicated that H-11 and H-11′ were cis to sulfur-bridged (Fig. 3). The absolute configuration of 1 was determined to be the same as 3, evidenced by the similar CD spectra of 1 and 3, as well as the almost identical CD curves between 1a and chetracin D1 which was the tetrakis(methyl-sulfanyl) derivative of 3 (Fig. 4).
 | ||
| Fig. 3 Key NOESY correlations of 1 and 1a. | ||
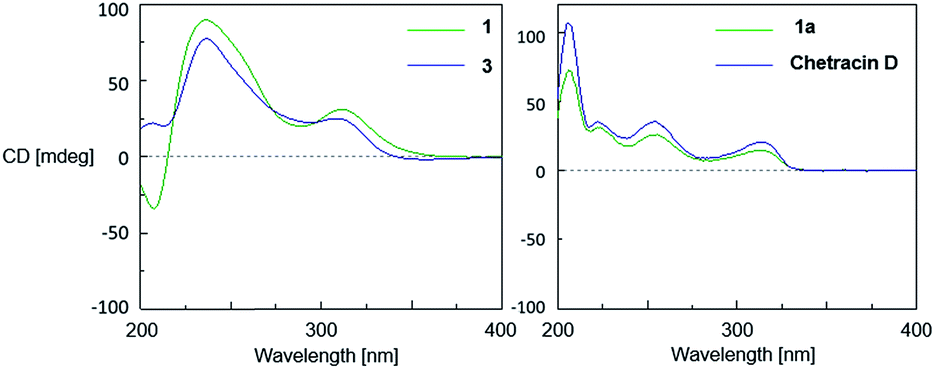 | ||
| Fig. 4 CD spectra of 1, 3, 1a and chetracin D (in MeOH). | ||
Chetracin F (2) was isolated as a pale yellow, amorphous powder. The molecular formula was assigned as C30H28N6O8S7 by HRESIMS adduct ion detected at m/z 825.0105 [M + H]+, indicating the presence of one additional sulfur atom in the molecule compared to 3. Unlike compound 3 which was composed by two symmetric monomers, the asymmetric NMR signals of 2 indicated the existence of a tetrasulfide bridge containing the additional sulfur atom. The tetrasulfide bridge was assigned in the second monomeric subunit (between C-1′ and C-13′) according to the chemical shifts (Fig. 1, Table 1), and the planar structure of compound 2 was also confirmed by the COSY and HMBC correlations (Fig. 2). The stereochemistry of compound 2 was deduced by chemical conversion. The tetrakis(methylsulfanyl) derivative of 2 showed identical spectroscopic data to those of chetracin D, which indicated the same absolute configuration of them. In addition, when kept at room temperature for two weeks, compound 2 could convert to 3 partially in DMSO induced by free radical reaction (Fig. S3†),1,11 suggesting that they share the same absolute configuration.
Biological evaluation using an MTT method showed that 1–3 exhibited extensive cytotoxicity against all the five tested cancer cell lines (Table 2). Among them, compound 1 showed the strongest cytotoxicity on H1975 cells with IC50 value 0.2 μM.
In light of discovery of the novel C-terminal Hsp90 inhibitors chaetocin and chetracin B, the interactions between compounds 1–3 and Hsp90 were investigated primarily in silico. The docking results displayed that 1–3 could bind to the 526–570 region (C-terminal) of Hsp90α by forming hydrogen bonds and hydrophobic interactions, with the average binding energy of −9.58 kcal, −6.21 kcal and −9.59 kcal, respectively. Distinguished from the phenotypic cytotoxicity, compound 2 showed a high binding energy, which possibly because of ignoring a potential cation–pi interaction between side chain K(570) of Hsp90 and the aromatic ring of 2 (Fig. 5). The cation–pi interaction is about −4 kcal that was not taken into account by the docking software. Anyway, the docking data suggest that all the compounds will be potential C-terminal Hsp90 inhibitors. To confirm the docking result, we estimated the levels of expression and phosphorylation of Hsp90 client oncoproteins induced by compounds 1–3 (with chetracin B used as reference drug). Similar to chetracin B, the treatment of 1–3 at the concentration of 0.5 μM reduced the expressions of EGFR, Akt, and the active forms of EGFR, Stat3, Akt and Erk in H1975 cells (Fig. 6). These results suggested that compounds 1–3 also could inhibit Hsp90 by binding to the C-terminal, which may subsequently induce the degradation of a serious of client oncoproteins. In addition, as chetracin B and compounds 1–3 show the effect of similar levels (Fig. 6), the number of sulfur atoms in the bridge and the hydroxyl group at C-13/C-13′ seem to had little influence on the interactions between this kind of ETPs and Hsp90.
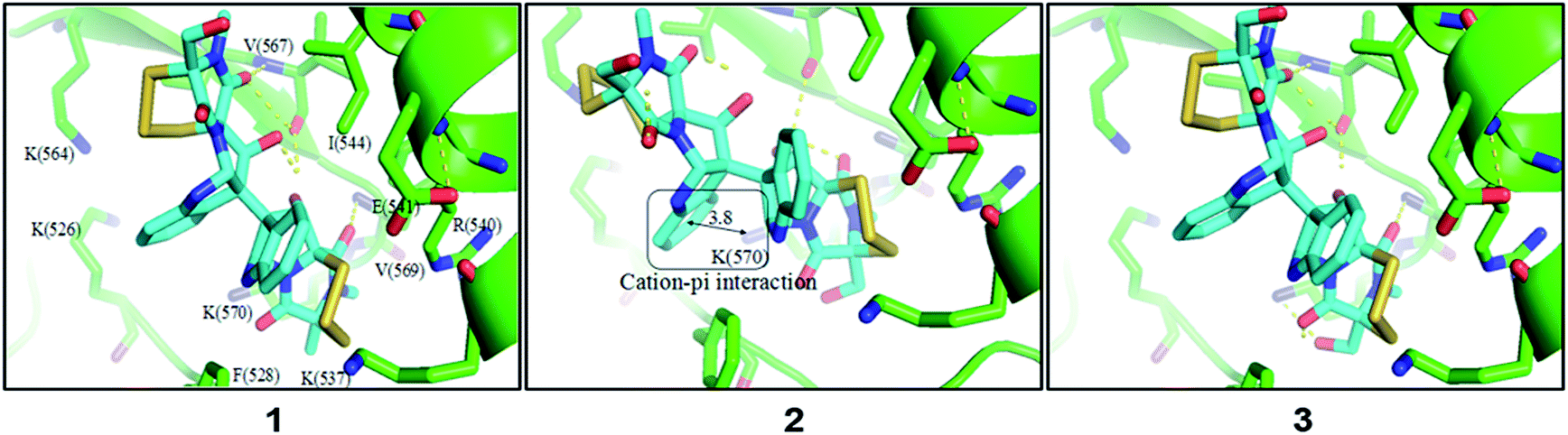 | ||
| Fig. 5 Amplified view of 1–3 binding to Hsp90α. | ||
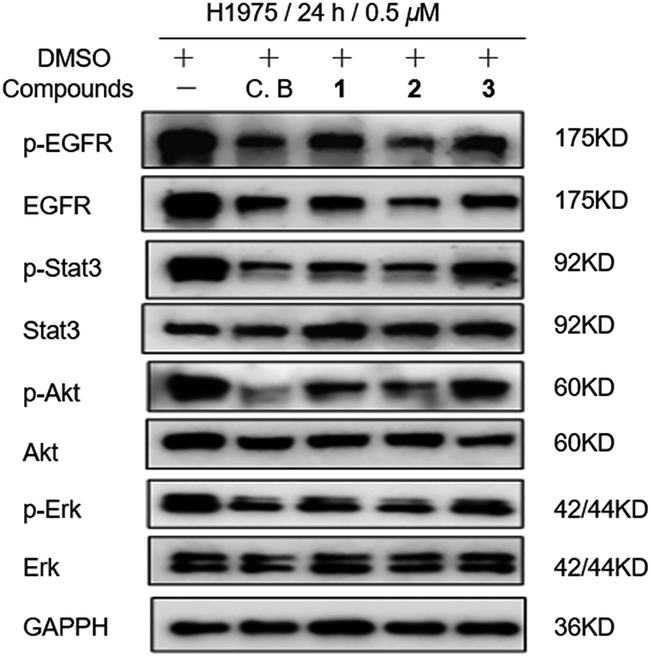 | ||
| Fig. 6 Compounds 1–3 (0.5 μM) treatment induced expression levels of Hsp90 client proteins in H1975 cells with chetracin B (C. B) used as reference drug. | ||
Experimental
General experimental procedures
Specific rotations were measured on a JASCO P-1020 digital polarimeter. UV spectra were recorded on Beckman DU 640 spectrophotometer. IR spectra were taken on a Bruker tensor-27 spectrophotometer in KBr discs. ESIMS data were obtained on a Thermo Scientific LTQ Orbitrap XL mass spectrometer or a Micromass Q-TOF ULTIMA GLOBAL GAA076 LC Mass spectrometer. CD spectra were measured on JASCO J-715 spectropolarimeter. NMR spectra were recorded on Agilent 500 MHz DD2 spectrometer using TMS as internal standard and chemical shifts were recorded as δ-values. Semipreparative HPLC was performed on an ODS column [HPLC (YMC-Pack ODS-A, 10 × 250 mm, 5 μm, 3 mL min−1)]. Medium-pressure preparation liquid chromatography (MPLC) was performed on a Bona-Agela CHEETAH™ HP100 (Beijing Agela Technologies Co., Ltd). Column chromatography (CC) were performed with silica gel (200–300 mesh, Qingdao Marine Chemical Inc., Qingdao, People's Republic of China), and Sephadex LH-20 (Amersham Biosciences), respectively. All the cell lines including A549 (human lung cancer cell line), HCT116 (human colorectal cancer cell line), K562 (human chronic myeloblastic leukemia), H1975 (human non-small cell lung cancer cell line) and HL-60 (human promyelocytic leukemia cell line) were purchased from Institute of Biochemistry and Cell Biology, CAS.Fungal material
The fungal strain A. luteoalbus HDN13-530 was isolated from soil of Liaodong Bay, China. It was identified by ITS sequence and the sequence data have been submitted to GenBank (accession number KP969081). The voucher specimen was deposited in our laboratory at −80 °C.Fermentation and extraction
The fungus A. luteoalbus HDN13-530 was cultured under static conditions at 28 °C in 1 L Erlenmeyer flasks containing 300 mL of liquid culture medium, composed of glycerin (20.0 mL L−1), peptone (2.0 g L−1), yeast extract, power (2.0 g L−1), and seawater (Huiquan Bay, Yellow Sea). After 4 weeks of cultivation, 30 L of whole broth was filtered through cheesecloth to separate the supernatant from the mycelia. The former was extracted three times with EtOAc, while the latter was extracted three times with acetone and concentrated under reduced pressure to afford an aqueous solution, which was extracted three times with EtOAc. Both EtOAc solutions were combined and concentrated under reduced pressure to give the organic extract (40 g).Isolation
The organic extract was subjected to vacuum liquid chromatography over a silica gel column using a gradient elution with petroleum ether–CH2Cl2–MeOH to give six fractions (fractions 1–6). Guided by LC-MS-UV data, fraction 3 (5.1 g) eluted with CH2Cl2–MeOH (97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) was applied on a C-18 ODS column using a stepped gradient elution of MeOH–H2O yielding five subfractions (fractions 3.1–3.5). Fraction 3.4 that eluted with MeOH–H2O (80:20) was chromatographed on SephadexLH-20 with CH2Cl2–MeOH (1:1) and further separated by MPLC (C-18 ODS) using MeOH–H2O (70:30) to furnish four subfractions (fractions 3.4.1–3.4.4). Fractions 3.4.1 and 3.4.3 were purified by semi-preparative HPLC (MeOH–H2O (60:40), MeCN–H2O (55:45), 3 mL min−1) to afford compounds 1 (20.0 mg), 2 (15.0 mg) and 3 (15.0 mg).
:3) was applied on a C-18 ODS column using a stepped gradient elution of MeOH–H2O yielding five subfractions (fractions 3.1–3.5). Fraction 3.4 that eluted with MeOH–H2O (80:20) was chromatographed on SephadexLH-20 with CH2Cl2–MeOH (1:1) and further separated by MPLC (C-18 ODS) using MeOH–H2O (70:30) to furnish four subfractions (fractions 3.4.1–3.4.4). Fractions 3.4.1 and 3.4.3 were purified by semi-preparative HPLC (MeOH–H2O (60:40), MeCN–H2O (55:45), 3 mL min−1) to afford compounds 1 (20.0 mg), 2 (15.0 mg) and 3 (15.0 mg).
Characterisation of compounds
ε): 221 (4.10), 292 (2.66) nm; 1H and 13C NMR data, see Table 1; HRESIMS [M + H]+ m/z 777.0432 (calcd for C30H29O7N6S6, 777.0416).ε): 221 (4.11), 298 (2.62) nm; CD (MeOH) λ [nm] (Δε): 357 (−5.2), 309 (60.0), 296 (50.9), 236 (187.4), 215 (34.0), 207 (60.0); IR (KBr) νmax 3397, 2930, 1671, 1367, 1205, 1064, 748 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS [M + H]+ m/z 825.0105 (calcd for C30H29O8N6S7, 825.0086).Formation of tetrakis(methylsulfanyl) derivative from compound 1
In the pyridine (0.1 mL) and MeOH (0.16 mL) solution of compound 1 (5 mg), MeI (0.1 mL) and NaBH4 (2 mg) were added, after stirring for 30 min at room temperature, the reaction mixture was then diluted with water and extracted with diethyl ether, and the residue evaporated under reduced pressure was purified by HPLC (CH3OH:H2O = 50–100%, 3 mL min−1) to afford compound 1a (2.0 mg). 1a: pale yellow powder; [α]20D + 76.5 (c 0.1, MeOH); UV (MeOH) λmax (logε): 221 (4.10), 292 (2.50) nm; CD (MeOH) λ [nm] (Δε): 315 (11.4), 286 (5.6), 255 (20.3), 238 (13.8), 222 (24.5), 218 (22.1), 206 (56.6); IR (KBr) νmax 3382, 2924, 2854, 1654, 1427, 1386, 1194, 1095, 748 cm−1; 1H and 13C NMR data, see Table S1;† HRESIMS [M + H]+ m/z 773.1925 (calcd for C34H41O7N6S4, 773.1914).
Formation of tetrakis(methylsulfanyl) derivative from compound 2
In the pyridine (0.1 mL) and MeOH (0.16 mL) solution of compound 2 (4 mg), MeI (0.1 mL) and NaBH4 (2 mg) were added, after stirring for 30 min at room temperature, the reaction mixture was then diluted with water and extracted with diethyl ether, and the residue evaporated under reduced pressure was purified by HPLC (CH3OH:H2O = 50–100%, 3 mL min−1) to afford the known compound chetracin D (2.0 mg).
Cytotoxicity assay
Cytotoxic activities of 1–3 were evaluated by an MTT method using A549, HCT116, K562, H1975 and HL-60 cell lines. Dox (doxorubicin hydrochloride) was used as reference drug. The detailed methodology for biological testing had already been described in a previous report.12Computational modeling
The 3D structures of Hsp90 (PDB: 2CGE)13 was taken from the Protein Data Bank (http://www.rcsb.org/pdb). The initial structures of compounds 1–3 were sketched in Sybyl 2.0 and their 3D structures were minimized using 3000 steps of conjugated minimization method in Sybyl 2.0. Ligand docking of compounds 1–3 to Hsp90 was performed using AutoDock 4.2.14 Gasteiger charges were used and non-polar hydrogens of the macromolecule and the ligand were merged. A grid box with dimensions of 60 × 60 × 60 Å and a grid spacing of 0.375 Å was set up and centered on the binding pocket of Hsp90. Docking was performed using a Lamarckian Genetic Algorithm (LGA), with the receptor treated as rigid. For each of the above compounds, 50 complexes were generated and the best-ranked score from the largest cluster (with the RMSD threshold set at 2.0 Å) was selected as the final pose.Western blotting assay for expression and activation levels of multiple oncoproteins
The detailed methodology has already been described in a previous report.7 Chetracin B (C. B) was used as reference drug.Conclusions
In summary, three ETPs including two new ones were isolated from the fungus A. luteoalbus HDN13-530 by UPLC-MS-UV guided fractionation. Compounds 1–3 exhibited extensive cytotoxicity in low-micromolar or nanomolar IC50 values and could function as Hsp90 C-terminal inhibitors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the National Natural Science Foundation of China (21372208, 81673450 and 41676127), the Shandong Provincial Natural Science Fund for Distinguished Young Scholars (JQ201422), AoShan Talents Program Supported by Qingdao National Laboratory for Marine Science and Technology (2015ASTP-ES09), NSFC-Shandong Joint Fund for Marine Science Research Centers (U1606403), Scientific and Technological Innovation Project Financially Supported by Qingdao National Laboratory for Marine Science and Technology (2015ASKJ02), Shandong province key research and development program (2016GSF201204), Fundamental Research Funds for the Central Universities (201564026).Notes and references
- L. Li, D. Li, Y. Luan, Q. Gu and T. Zhu, J. Nat. Prod., 2012, 75, 920–927 CrossRef CAS PubMed.
- L. Du, A. J. Robles, J. B. King, S. L. Mooberry and R. H. Cichewicz, J. Nat. Prod., 2014, 77, 1459–1466 CrossRef CAS PubMed.
- C. R. Isham, J. D. Tibodeau, A. R. Bossou, J. R. Merchan and K. C. Bible, Br. J. Cancer, 2012, 106, 314–323 CrossRef CAS PubMed.
- H. Chaib, A. Nebbioso, T. Prebet, R. Castellano, S. Garbit, A. Restouin, N. Vey, L. Altucci and Y. Collette, Leukemia, 2012, 26, 662–674 CrossRef CAS PubMed.
- C. R. Isham, J. D. Tibodeau, W. Jin, R. F. Xu, M. M. Timm and K. C. Bible, Blood, 2007, 109, 2579–2588 CrossRef CAS PubMed.
- Y. M. Lee, J. H. Lim, H. Yoon, Y. S. Chun and J. W. Park, Hepatology, 2011, 53, 171–180 CrossRef CAS PubMed.
- X. Song, Z. Zhao, X. Qi, S. Tang, Q. Wang, T. Zhu, Q. Gu, M. Liu and J. Li, OncoTargets Ther., 2015, 6, 5263–5273 Search PubMed.
- L. Whitesell and N. U. Lin, Biochim. Biophys. Acta, 2012, 1823, 756–766 CrossRef CAS PubMed.
- T. Taldone, H. J. Patel, A. Bolaender, M. R. Patel and G. Chiosis, Expert Opin. Ther. Pat., 2014, 24, 501–518 CrossRef CAS PubMed.
- J. Kim, J. A. Ashenhurst and M. Movassaghi, Science, 2009, 324, 238–241 CrossRef CAS PubMed.
- T. L. Pickering, K. J. Saunders and A. V. Tobolsky, J. Am. Chem. Soc., 1967, 89, 2364–2367 CrossRef CAS.
- L. Du, T. Zhu, H. Liu, Y. Fang, W. Zhu and Q. Gu, J. Nat. Prod., 2008, 71, 1837–1842 CrossRef CAS PubMed.
- M. M. U. Ali, S. M. Roe, C. K. Vaughan, P. Meyer, B. Panaretou, P. W. Piper, C. Prodromou and L. H. Pearl, Nature, 2006, 440, 1013–1017 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The MS, NMR and IR spectra of 1, 2 and 1a; M + 2 negative ion isotope peaks of many sulfur-containing metabolites in fermentation extract of A. luteoalbus HDN13-530; 13C NMR and 1H NMR spectroscopic data of 1a; key HMBC correlations of 1a; HPLC analysis of conversions of 2 to 3 and standard samples. See DOI: 10.1039/c7ra12063j |
| This journal is © The Royal Society of Chemistry 2018 |