
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel approaches to immobilize Candida rugosa lipase on nanocomposite membranes prepared by covalent attachment of magnetic nanoparticles on poly acrylonitrile membrane
Marzieh Aghababaie a,
Masoud Beheshti*b,
Abdol-Khalegh Bordbarc and
Amir Razmjoua
a,
Masoud Beheshti*b,
Abdol-Khalegh Bordbarc and
Amir Razmjoua
aBiotechnology Department, Faculty of Advanced Sciences and Technologies, University of Isfahan, Isfahan, Iran
bChemical Engineering Department, University of Isfahan, Isfahan, Iran. E-mail: masbeh@yahoo.com; Tel: +98 3137934529
cDepartment of Chemistry, University of Isfahan, Isfahan 81746-73441, Iran
First published on 25th January 2018
Abstract
Novel methods have been developed for lipase immobilization on poly acrylonitrile (PAN) membranes to increase the activity and stability of the immobilized lipase. In this study, poly acrylonitrile (PAN) membranes were aminated and then activated by glutaraldehyde or epichlorohydrine to be used for enzyme immobilization. In the other approach, magnetic nanoparticles (MNPs) which were functionalized with trichlorotriazine (TCT) or glutaraldehyde (GA) were attached to the membrane surface to prepare the nanocomposite membranes named TCT-MNP@PAN & GA-MNP@PAN membranes. Candida rugosa lipase (CRL) was covalently immobilized on this activated nanocomposite membrane. Nanoparticles and nanocomposite membranes were characterized with various techniques such as SEM, TEM, XRD, FTIR, FTIR-ATR, AFM, contact angle goniometry and surface free energy measurement. The evidence of immobilization was also done by FTIR-ATR, enzyme activity, and loading efficiency. It was found that the activity of immobilized lipase on GA and TCT functionalized NCPAN membrane were about 50% and 31% higher than that immobilized on GA-activated PAN membrane. The kinetic parameters of enzymatic membranes showed the better conformation of the lipase enzyme immobilized on the TCT-MNP@PAN membrane. The presented enzymatic nanocomposite membranes are easy to prepare with low cost and are good candidates for use in membrane bioreactors.
Introduction
Efficient enzyme immobilization on easy handling supports which can be used in bioreactors and biosensors is an interesting issue, nowadays. Lipases (EC 3.1.1.3) are biocatalysts for many reactions such as esterification, transesterification, oil hydrolysis, polymer synthesis, and biodiesel production.1 Candida rugosa lipase (CRL) is one of the most favorable lipases which is widely used in many industries due to its low cost and high activity.2–4 Although, for the application of enzymes in industry, immobilization of enzymes improves the stability of enzyme, providing easy biocatalyst recycling and easier product purification, elongating their life span.1,5,6 One of the favorable supports for this goal is membranes which in some cases can facilitate the use of enzymes in membrane bioreactors, enzymatic reactors and biosensors and also can act as a separation unit.7 Different membrane materials have been applied for lipase immobilization.5,8–14 In most reported cases, lipases have been physically immobilized on the membrane with the disadvantage of weak physical interactions. This reduces the reusability of the immobilized enzyme as a general feature of physical adsorption phenomena. However, direct covalent attachment of enzyme on the surface of the membrane, is more attractive while it would increase the stability and reusability of lipase. Although, immobilization of lipase on hydrophobic surfaces might improve lipase activity by rearranging their conformational structure (termed as ‘lid’) to open the active site.12 However, hydrophobic material has a negative effect on enzyme activity by denaturing the enzyme or by hampering the enzyme interaction with water-soluble substrates.15 Therefore, hydrophilic surfaces are more common to provide proper contact between enzyme and substrates. In this way, membrane with immobilized enzyme should have a moderate hydrophilic properties.12 Poly acrylonitrile (PAN) membrane is a hydrophilic membrane which is superior to many conventional membranes because of its reactive groups on the surface of membrane.16 The aim of this study is to immobilize lipase on the PAN membrane in hydrated microenvironment along with increasing the enzyme activity. Recently, enzyme immobilization on nanocomposite membranes has been performed due to the combination of the benefits of membrane filtration system and nanoparticle including larger surface area, high stability and flexibility.17 In most of the studies, nanoparticles have been presented in the membrane by blending and coating technique.18–20 Nanocomposite membranes with different nanoparticles such as TiO2,21 Al2O3,22 Fe3O4,18–20 SiO2,23 Ag24 and mesoporous carbon nanoparticles25 have been applied for different purposes. Silica coated magnetic nanoparticles (Fe3O4@SiO2NPs) have gained interests as enzyme carrier, due to their high surface area, high stability, and easy modification.26–28 The Fe3O4@SiO2 NPs can be aminated using amino propyltriethoxysilane (APTS) and then be modified using glutaraldehyde29 or trichlorotriazine (TCT).27 These activated magnetic nanoparticles can be use for lipase immobilization with enhanced stability and activity.27In our previous study, we immobilized lipase on Fe3O4@SiO2 dip-coated membrane.30 Dip-coating method along with low hydrothermal process was applied in this study which was time-consuming and difficult to be done for large membranes.30 Here, a novel activated and nanocomposite membranes were designed and prepared for covalent lipase immobilization. Two different approaches have been designed to immobilize lipase on PAN membrane. First, PAN membrane were amino functionalized and then were activated by glutaraldehyde (GA) or epichlorohydrine (EPI). CRL was immobilized on these heterofunctional supports which have several functionalities on their surfaces.31
On the other approach, nanocomposite membranes were prepared by covalent attachment of magnetic nanoparticles (MNPs), which was functionalized with GA or trichlorotriazine (TCT), on the membrane surface. To the knowledge of the authors of this work, there are no study regarding the preparation of nanocomposite membrane by covalent attachment of nanoparticles on the surface of membrane and also there is no study in the case of lipase immobilization on polyacrylonitrile membrane.
In this way, hydrophilic properties of the membranes and lipase microenvironment are different which can effect the activity of CRL. The membranes were characterized by various techniques such as SEM, TEM, XRD, FTIR, FTIR-ATR, AFM, contact angle goniometry and surface free energy measurement. Subsequently, CRL was covalently immobilized on these membranes and evidence of immobilization was done by FTIR-ATR, enzyme activity and loading efficiency.
Experimental
Materials
Poly acrylonitrile (PAN) ultrafiltration (UF) membrane was purchased from Sterlitech. Lipase from Candida rugosa (lyophilized powder, Type VII, nominal activity ≫ 700 U mg−1), Bovine Serum Albumin (BSA), para-nitrophenyl palmitate (p-NPP), Coomassie Brilliant Blue G-250, and amino propyltriethoxysilane (APTS) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). The iron(III) chloride hexahydrate (FeCl3·6H2O, 99.0%), iron(II) chloride tetrahydrate (FeCl2·4H2O, 99.7%), Triton X-100, isopropyl alcohol (99.7%) and Arabic gum were provided from Daejung Chemicals. Tetraethyl orthosilicate (TEOS), ammonia (NH3·H2O, 25 wt%), glutaraldehyde (50%), 1, 3, 5, trichlorotriazine (TCT) (cyanuric chloride) (>99%), epichlorohydrin (>99%), and tetrahydrofuran (THF) (99%) were obtained from Merck. Methanol (99.7%) and absolute ethanol were obtained from Carlo ERBA (99.9%). All other chemicals were of analytical grade and used without purifications.Magnetic nanoparticle preparation
Magnetic nanoparticles (MNPs) were synthesized by co-precipitation method and silica coated by sol–gel method as described previously.27,30,32 Briefly, the aqueous solution of FeCl3 and FeCl2 were mixed at a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in deionized water and stirred vigorously under a nitrogen atmosphere at 60 °C. Magnetic nanoparticles (MNPs) was produced by the addition of 6 mL of 25% ammonia. The MNPs was washed several times with deionized water and ethanol. The obtained MNPs were redispersed in 200 mL ethanol and after vigorous stirring under nitrogen atmosphere 30 mL of deionized water, 15 mL of NH4OH and TEOS (2 mL) were added to the solution and stirred and sonicated for 5 hours. Afterward, silica coated MNPs (Fe3O4@SiO2NPs) were washed with ethanol and water and dried at room temperature.
:1 in deionized water and stirred vigorously under a nitrogen atmosphere at 60 °C. Magnetic nanoparticles (MNPs) was produced by the addition of 6 mL of 25% ammonia. The MNPs was washed several times with deionized water and ethanol. The obtained MNPs were redispersed in 200 mL ethanol and after vigorous stirring under nitrogen atmosphere 30 mL of deionized water, 15 mL of NH4OH and TEOS (2 mL) were added to the solution and stirred and sonicated for 5 hours. Afterward, silica coated MNPs (Fe3O4@SiO2NPs) were washed with ethanol and water and dried at room temperature.
Silica-coated MNPs were dispersed in absolute ethanol and then APTS (20% (v/v)) was added and sonicated at 50 °C for 2 hours. The obtained aminated MNPs (Fe3O4@SiO2@APTS NPs) were washed with ethanol and distilled water, and then, dried at 40 °C oven to be used for further modifications.
The aminated MNPs were dispersed in THF and reacted with TCT (20 mg mL−1). The obtained TCT functionalized MNPs (Fe3O4@SiO2@APTS@TCT NPs) were washed with THF, ethanol, and water and then dried 40 °C oven.
In the other approach, the aminated MNPs were activated with 2% (v/v) glutaraldehyde in phosphate buffer solution (PBS) (50 mM; pH = 7.5). The obtained GA functionalized MNPs (Fe3O4@SiO2@APTS@GA NPs) were washed with PBS and DI water and then dried in a 40 °C oven.
Modification of the surface of PAN membrane
The outer surface of PAN membrane was hydrolyzed and aminated in an aqueous solution containing 1 N NaOH and 1 N 1,3-propanediamine at 60 °C for 20 min as shown in the below equation:33| PAN-CN + H2N(CH2)3NH2 + H2O → PAN–CONH–(CH2)3–NH2 + NH3 | (1) |
The resulting membranes were thoroughly rinsed with pH 7 buffer and DI water. In this regards, four different activated membranes were prepared for further lipase immobilization. Modified membranes were activated by glutaraldehyde (GA) solution (1% v/v) in PBS (50 mM, pH = 7.5) at 25 °C for 2 h or epichlorohydrine (EPI) solution (5 mL of EPI solution and 10 mL of 1.5 M sodium hydroxide) at 25 °C for 12 h. Furthermore, nanocomposite membranes (NCM) were prepared by immersing the aminated PAN membranes in a solution of TCT or GA functionalized MNPs (1 mg mL−1; in DI water) to obtain TCT-MNP@PAN and GA-MNP@PAN membranes, respectively.
Nanoparticles and nanocomposite membrane characterization
The size and structure of magnetic nanoparticles were determined using a field emission scanning electron microscope (FE-SEM) (JEOL 7001F FEG), at 15 kV acceleration voltages. To determine the size and morphology of Fe3O4@SiO2 NPs transmission electron microscopy (TEM, Philips CM30, operating at 200 kV) were used. Functional groupings of the nanoparticles were determined by Fourier transform infrared (FTIR) spectroscopy using an FTIR spectrometer (JASCO FT/IR-6300, Japan). In order to investigate the functional grouping of the PAN membranes, modified membranes, nanocomposite membranes and enzymatic nanocomposite membranes, FTIR-ATR (FTIR with ATR (attenuated total reflectance) accessory) was used. Bruker D8-advance X-ray diffractometer with Cu Kα radiation was used to investigated the X-ray powder diffraction (XRD) spectra of Fe3O4@SiO2 NPs.The morphology of the nanocomposite membrane and existence of Fe3O4@SiO2 NPs on the membrane surface was observed using SEM (environmental scanning electron microscope, Philips, XL30) and EDX (Energy dispersion of X-ray, Philips, XL30).
The roughness and surface morphology of the membranes were determined by atomic force microscopy (Scanning probe microscopy (SPM/DME) Dualscope C-26). The samples were cut into pieces of 3 cm × 3 cm and different parts of each sample were scanned in tapping mode at 5 μm × 5 μm and 2 μm × 2 μm. The average roughness was determined in the area of 2 μm × 2 μm.
In order to investigate the hydrophilicity of the membrane surfaces, the contact angles formed by water droplets on the membrane surface were measured using goniometer method (average of at least 5 measurements). The surface free energy of membranes was obtained using acid–base (Van-Oss) with contact angles of three liquids of water, glycerol, and formamide with known parameters.11,34
Enzyme immobilization
The prepared membranes were cut in the dimension of 1 cm × 1 cm and then immersed in lipase solution (0.2–1.8 mg in 1 mL of PBS; 50 mM, pH 7.5) for 2 hours at shaker incubator at 25 °C and then they were washed with PBS. Bradford method was used to estimate the amounts of lipase in the enzyme solution before and after immobilization.35 Immobilization efficiency was determined by the following equation:| Immobilization efficiency% = (C1V1 − C2V2) × 100/(C1V1) | (2) |
Activity assay
The hydrolysis of p-NPP as a substrate used to estimate the activities of free and immobilized lipases as it is reported previously.30 Briefly, a substrate solution which was a mixture of 9 volumes of solution B (0.4 mL of Triton X-100 and 0.1 g Arabic gum in 90 mL PBS; 50 mM, pH 7.5) and 1 volume of solution A (30 mg p-NPP in 10 mL isopropanol) was prepared. The reaction would take place at 37 °C by preincubating the substrate solution. For each activity assay, 1.8 mL of substrate solution and a specific amount of lipase or piece of membrane reacted for 2 minutes and released p-nitrophenol was measured at 405 nm in a UV/VIS spectrophotometer (Eppendorf) against the blank sample, which contains no enzyme. One unit of lipase activity (1 U) is defined as the amount of enzyme or biocatalyst, which liberates 1 μmol of p-nitrophenol per minute under the assay condition. Lipase activity and specific activity were determined as follows:| Activity (U cm−2) = (activity of immobilized enzyme/surface area of membrane) | (3) |
| Specific activity (U mg−1 enzyme) = (lipase activity/loading capacity) | (4) |
Relative activity was defined as the percentage of the maximum activity obtained in that series.
Kinetic parameters
Michaelis–Menten kinetics was used to describe the dependence of enzyme activity on substrate concentration. The kinetic parameters (Km and vmax) of free and immobilized lipase were determined in the concentration range of 0.1–0.5 mM pNPP using Lineweaver–Burk equation as follows:| 1/v = (1/vmax) + (Km/vmax[S]) | (5) |
Storage stability of immobilized lipase
Storage stability of immobilized enzyme on TCT-MNP@PAN membranes were evaluated at room temperature for two weeks. In this way, membranes with immobilized enzyme were dried and maintained at capped glass bottle and some of them were maintained at buffer solution.Results and discussion
Characterization of nanoparticles
According to the SEM and TEM of MNPs and Fe3O4@SiO2 NPs (Fig. 1(a) and (b)), respectively, the average size of Fe3O4@SiO2 NPs is about 30 nm and has not been considerably changed due to silica coating. Fig. 1(c) shows the XRD spectrum of Fe3O4@SiO2 NPs. In this spectrum, a broad peak around 2θ of 20–30° shows the amorphous phase of SiO2, and the 2θ peaks at 35.05, 41.65, 50.05, 67.37, and 74.25 are the characteristic diffraction of face centered cubic (fcc) of Fe3O4. The chemical composition of synthetized and modified MNPs were characterized by FTIR (Fig. 1(d)). In the spectra of Fe3O4 nanoparticles, the peak at 567 cm−1 corresponds to the Fe–O bond. According to the spectra of Fe3O4@SiO2 NPs, the peaks at 1091 and 797 cm−1 are referred to the asymmetric and symmetric stretching vibration of Si–O–Si bonds in an oxygen–silica tetrahedron, respectively.36,37 The peak at 3401 cm−1 in the spectra of Fe3O4@SiO2 MNPs, corresponds to the –OH stretching vibration bond. After the salinization of silica coated MNPs with APTS, the peak at around 2923.5 cm−1 are representing the C–H stretching vibration of APTS. However, the broad band at 3450 cm−1, which indicate the N–H stretching vibration, is overlapped with –OH bond. In the spectra of functionalized MNPs with TCT (Fe3O4@SiO2@APTS@TCT NPs), the fine peaks between 1000 and 1600 cm−1 were presented the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N and triazide ring and attachment of TCT to MNP.38 In the spectra of functionalized MNPS with GA (Fe3O4@SiO2@APTS@GA NPs), the peak at 2900 stands for the C–H stretching vibration of GA. Furthermore, in this spectra, the peak at 1710 is referred to the C
N and triazide ring and attachment of TCT to MNP.38 In the spectra of functionalized MNPS with GA (Fe3O4@SiO2@APTS@GA NPs), the peak at 2900 stands for the C–H stretching vibration of GA. Furthermore, in this spectra, the peak at 1710 is referred to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching bond of aldehyde group. After lipase immobilization, a peak at around 3430 and 1630 cm−1 were referred to amid group.
O stretching bond of aldehyde group. After lipase immobilization, a peak at around 3430 and 1630 cm−1 were referred to amid group.
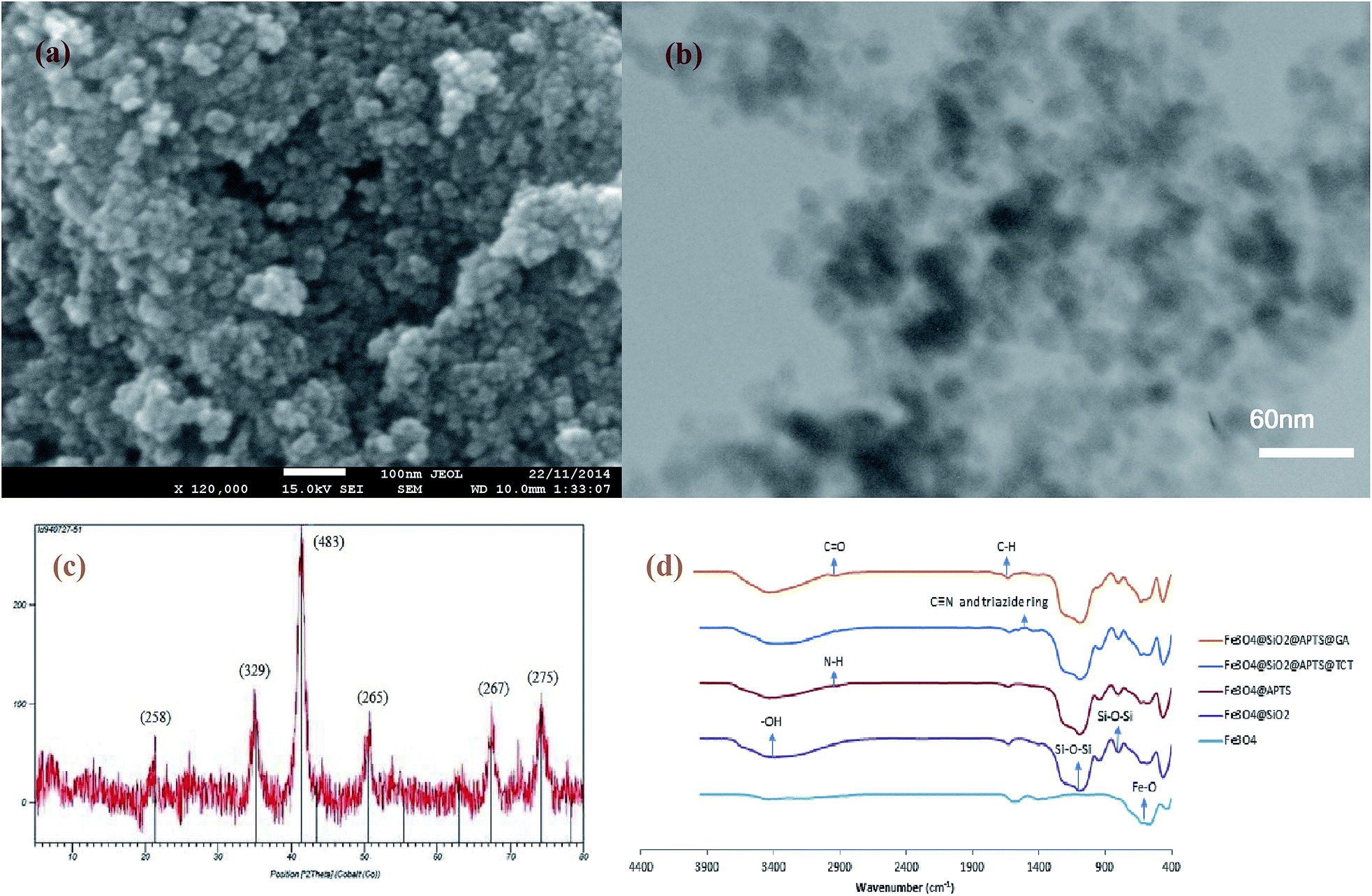 | ||
| Fig. 1 (a) SEM analysis of MNP (b) TEM of Fe3O4@SiO2 NPs (c) XRD pattern of Fe3O4@SiO2 NPs (d) FTIR spectra of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@APTS, Fe3O4@SiO2@APTS@TCT, and Fe3O4@SiO2@APTS@GA NP. | ||
Membrane characterization
 | ||
| Fig. 2 SEM images of the surface of (a) PAN membrane and (b) TCT-MNP@PAN membrane (c) EDX spectrum of NCM. | ||
(FTIR-ATR) of membranes
ATR of control PAN and aminated PAN membranes are shown in Fig. 3(a). A sharp band at 2242 represents a stretching vibration of CN band which can be seen in all the spectrums. A broad peak at about 3000–3300 in the spectra of aminated PAN is referred to N–H stretching vibration of the primary amine group. The peak at 2900 in the spectra of aminated PAN refers to the C–H stretching vibration of propyl group of diaminopropane. ATR of membranes which are activated by GA and EPI and enzymatic membranes are presented in Fig. 3(b). The peak at 2900 in the spectra of GA@PAN membrane represents the C–H stretching vibration of glutaraldehyde. The presence of epoxy group on EPI@PAN membranes can be identified by the change in peaks at around 1250, 815–950 and 700.39 However, some other peaks are observed in the spectra EPI@PAN which must be due to the crosslinking of epoxides, intramolecular cyclization, or epoxy ring opening which lead to the formation of different types of products.39
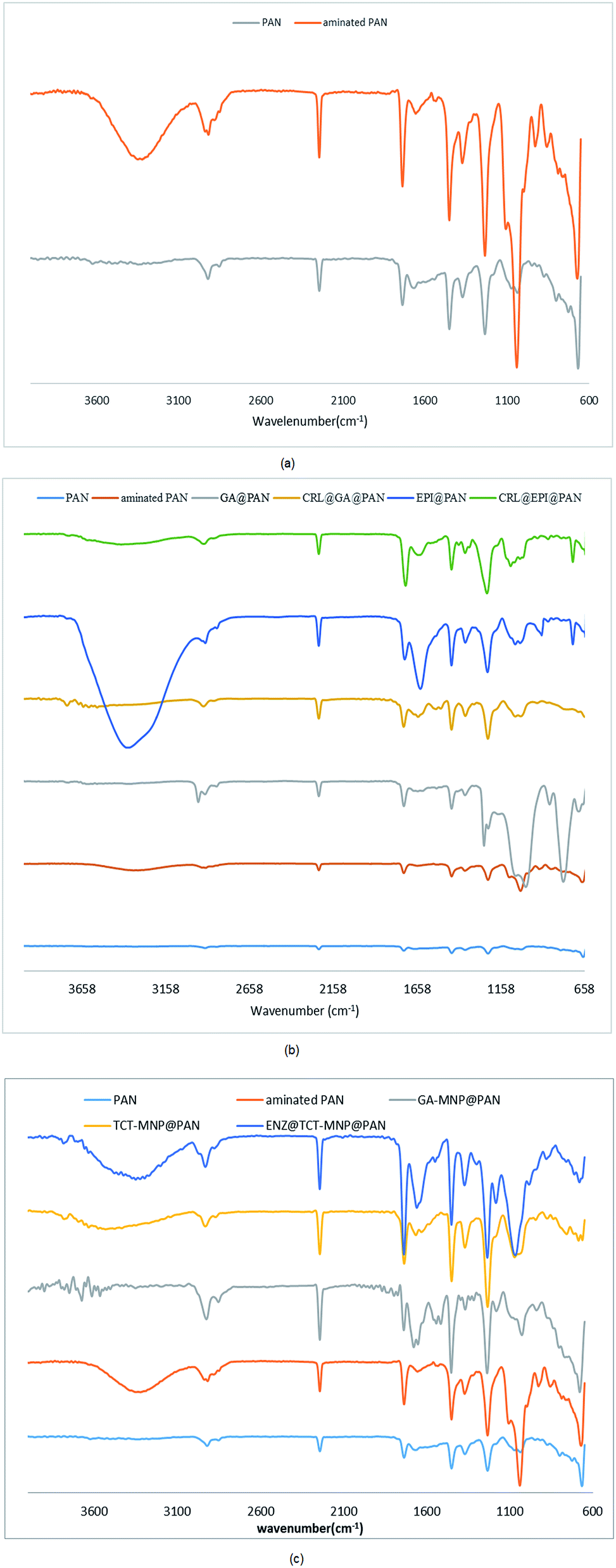 | ||
| Fig. 3 ATR spectra of membranes (a) PAN membrane and aminated PAN membrane (b) activated membranes and their enzymatic membranes (c) nanocomposite membranes and their enzymatic membranes. | ||
After immobilization of CRL, a peak at around 3430 and 1630 cm−1 were assigned to amid group. GA-MNP@PAN membrane TCT-MNP@PAN membrane and ENZ@TCT-MNP@PAN membrane are shown in Fig. 3(c). The peak at around 2900 in the spectra of GA-MNP@PAN membrane represents the C–H stretching vibration of GA and APTS on the MNP. In addition, the peak at 1710 in this spectra is referred to the CO stretching bond of aldehyde group. The peaks at 1677 and 1650 represent the bond between aldehyde group to the amine group of the membrane which forms a CN bond. In the spectra of TCT-MNP@PAN, the peak at about 700 corresponds to the C–Cl stretching bond of TCT on the MNPs. However, after enzyme immobilization, a broad peak at 3395 cm−1 from stretching –OH and –NH2 confirmed the presence of the enzyme.
Membrane roughness
Effect of membrane amination and coating of the membrane with MNPs were investigated by using AFM (Fig. 4). In this study, the average roughness (Ra) was estimated for a 1 μm × 1 μm, 2 μm × 2 μm and 5 μm × 5 μm surface area of control PAN, aminated PAN membrane, and TCT-MNP@PAN membrane through AFM results. The average roughness of the membranes were presented in Fig. 5(a). The Ra of PAN membrane and aminated PAN membrane were the same in all of the measuring scales. These results show that the chemical reaction only changed the functional groups on the surface of the membrane and did not change the morphology and structure of the membrane. However, the roughness of PAN membrane increased from 22, 65 and 102 nm to 45, 70 and 152 nm for TCT-MNP@PAN membrane in the scale of 1 μm × 1 μm, 2 μm × 2 μm and 5 μm × 5 μm, respectively. In our previous study regarding the preparing the nanocomposite membrane by the dip-coating of Fe3O4@SiO2 NPs in the polyethersulfone UF membrane, average roughness decreased after the dip-coating.30 It can be concluded that in the dip-coating method, nanoparticles would be trapped in the pores of membrane which causes the reduction in roughness. However, in the presented method, nanoparticles would be placed on the surface of the membrane and causes the increase in roughness.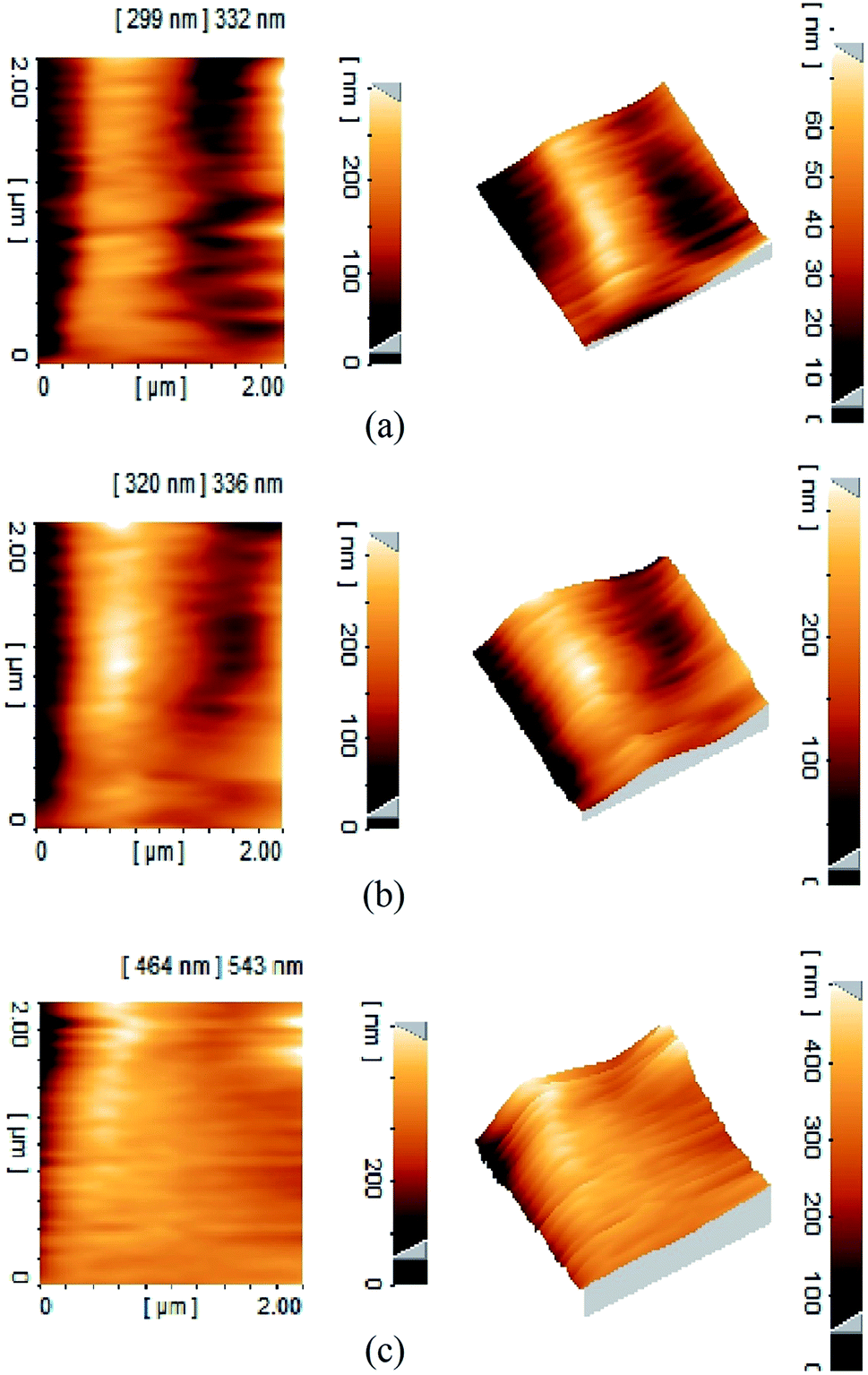 | ||
| Fig. 4 Three-dimensional AFM topography of membranes and their corresponding roughness images (a) PAN membrane (b) TCT-MNP@PAN membrane. | ||
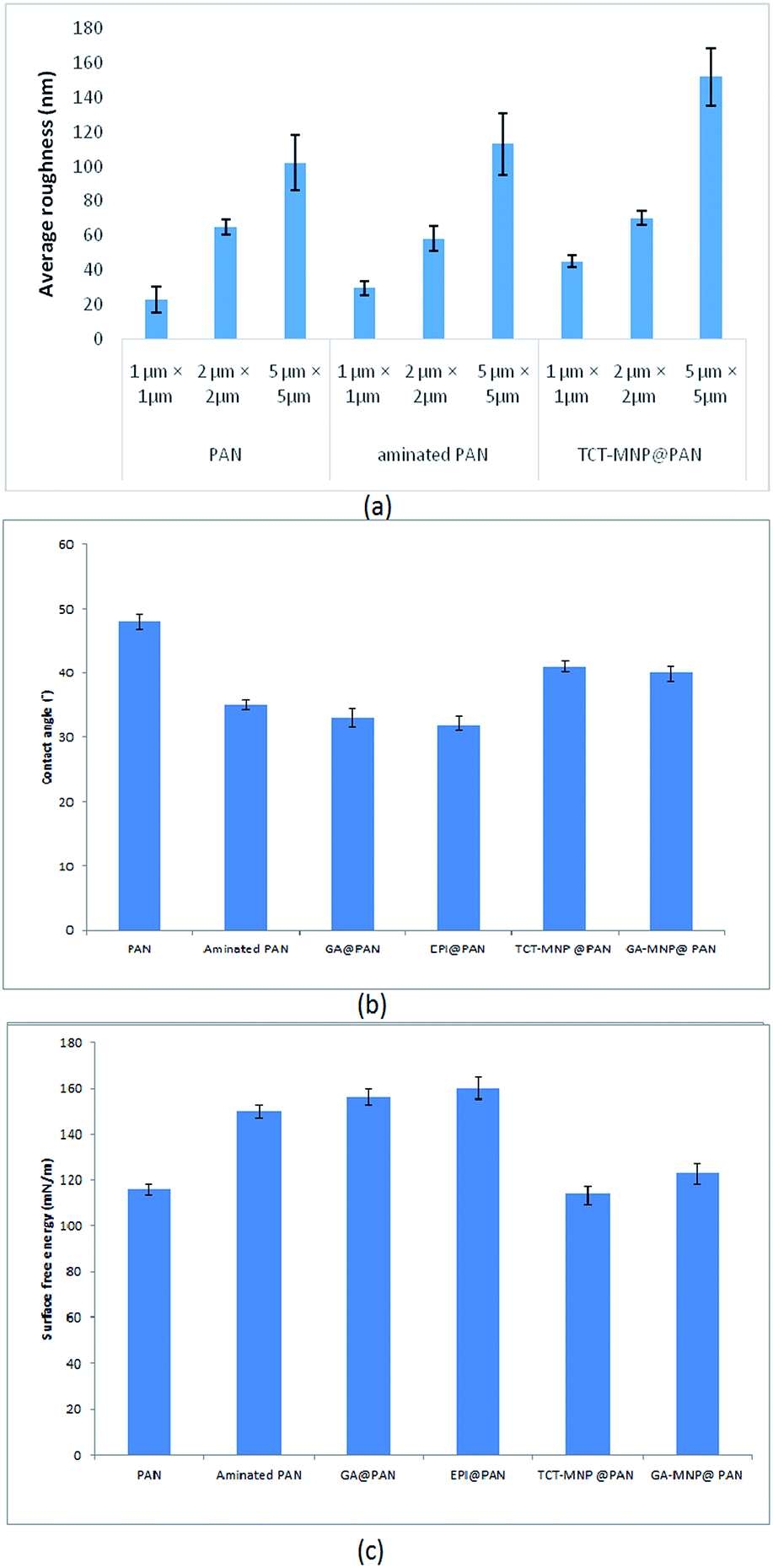 | ||
| Fig. 5 (a) Average roughness of PAN membrane, aminated PAN membrane, and TCT-MNP@PAN membrane (b) water contact angle and (c) surface free energy of membranes. | ||
Water contact angle and surface energy
Water contact angle and surface free energy of PAN membrane, aminated PAN membrane, activated membranes, and nanocomposite PAN membranes are presented in Fig. 5(b) and (c). PAN membrane had a water contact angle of about 48°, measured at 5th second after falling drop, respectively. A hydrophilic surface with low water contact angle lets the drop to spread and provides a high surface energy.40,41 The high surface energy of PAN membrane and aminated PAN membrane showed moderate hydrophilic properties of them. Functionalizing the membrane with amine groups decreased the water contact angle and increased the surface energy which showed the hydrophilic properties of the membrane after amination. Activating the membrane with GA or EPI, decreased the contact angle which showed the hydrophilic properties of these membrane which may be due the active functional groups of these molecules. After attaching the nanoparticles to the membrane, surface energy increased and water contact angle decreased. Based on the Wenzel model, roughening the hydrophilic surface (contact angle < 90°), transforms it into a superhydrophilic surface.42 Here, amination made the PAN membrane more hydrophilic and after increasing the roughness by attaching the nanoparticles to the surface, the hydrophilicity of membrane reduced. Nanocomposite membranes which have a higher average roughness are more hydrophilic than the PAN membrane.Lipase immobilization
CRL has been immobilized on PAN membrane with four different modifications. The relative activities and immobilization efficiency of immobilized lipases with same initial lipase concentration of 0.6 mg mL−1 are presented in Fig. 6(a). The activities and immobilization efficiency of immobilized lipase on GA and EPI activated PAN membrane was almost same. GA is widely used for enzyme immobilization which provides a heterofunctional supports.31,43 GA and EPI links to diaminopropane which has a three chain hydrocarbons and acts as a spacer arm on the membrane. EPI provides epoxy groups on the surface of membrane which has linked to amine groups of diaminopropane. These epoxy-amine groups are active for lipase immobilization via adsorption or covalent linkage that could occur between nucleophilic groups of the enzyme (amino, thiol, and hydroxyl) and the epoxy groups on the membrane surface.44 Lipase was immobilized on the membrane via covalent bonding to aldehyde group and epoxy groups of GA and EPI, respectively.44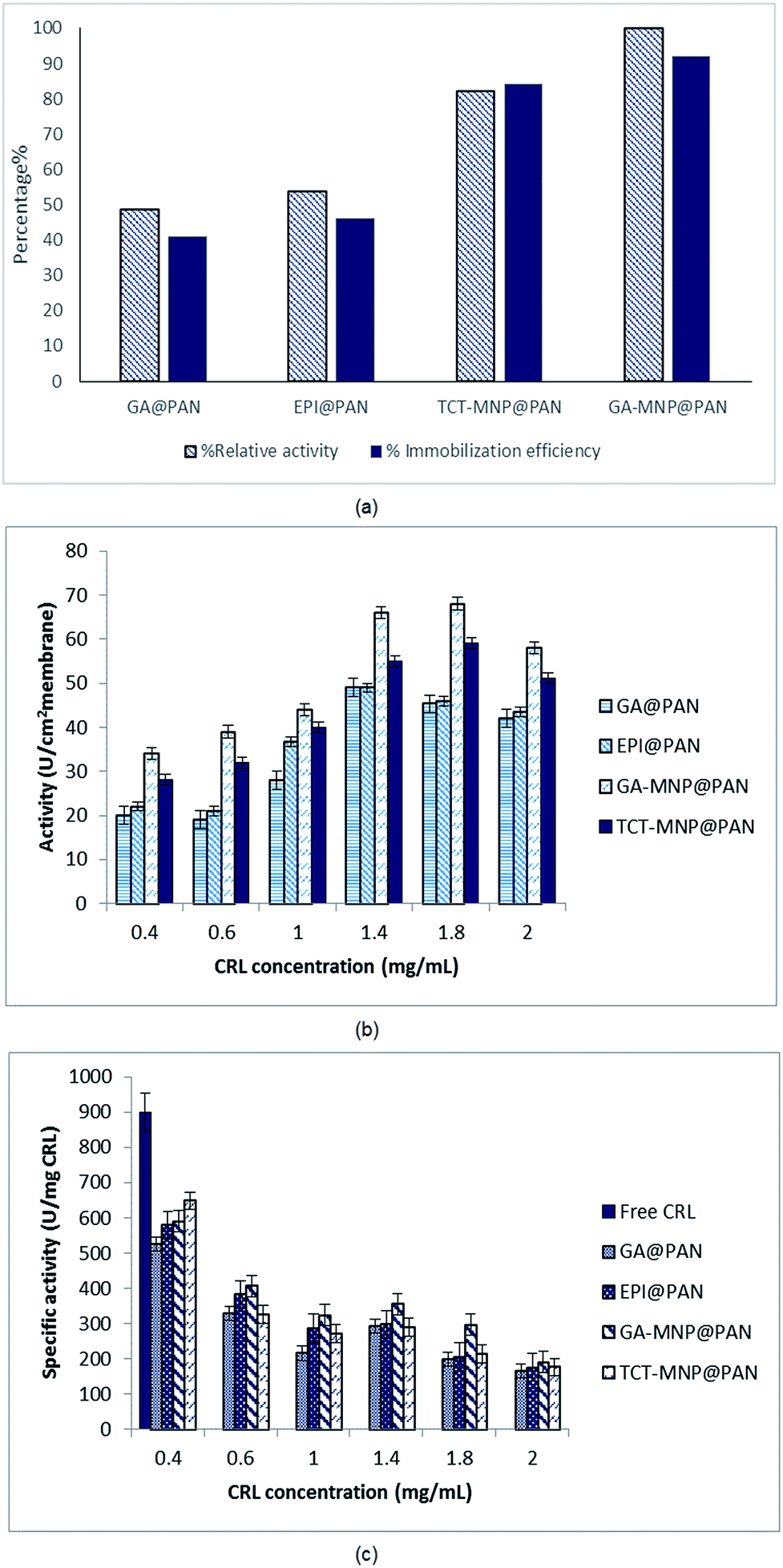 | ||
| Fig. 6 (a) Relative activity and loading efficiency of immobilized CRL on different membranes (b) activity of immobilized CRL on membranes at different lipase concentrations (c) specific activity of immobilized CRL on membranes at different lipase concentrations. | ||
However, these heterofunctional supports have provided two types of immobilization: (i) physical adsorption of lipases (e.g., by ionic exchange, by adsorption on immobilized metal chelates or hydrophobic surfaces); and (ii) covalent immobilization via aldehyde and epoxy groups.31,44
In the case of using MNPs, TCT and GA functionalized MNPs were linked to the spacer arms and provided heterofunctional supports for lipase immobilization. According to Fig. 5, NCPAN membranes had higher average roughness and more hydrophobic properties than GA and EPI activated PAN membranes. These properties have provided more options for lipase immobilization on membrane surface. Thus, it can be seen in Fig. 6(a) that the activity of immobilized lipase on GA and TCT functionalized NCPAN membrane were about 50% and 31% higher than that of immobilized on GA-activated PAN membrane. By increasing the CRL concentration, specific activity of immobilized lipases reduced due to high loading of the enzyme. Due to the immobilization, a conformational change in the 3D structure of enzyme, random attachment of enzyme from different amine residues and mass transfer limitation may be accrued which reduce the activity of the immobilized enzyme in comparison to free enzyme.
In the case of enzyme immobilization on the membrane, mass transfer limitations reduce the availability of substrate to enzyme.11,14,45 According to Fig. 6(b) and (c) different enzyme concentrations (0.4–2 mg mL−1) were used for immobilization to optimize the enzyme concentration. The activities of ENZ@GA-MNP@PAN and ENZ@TCT-MNP@PAN were enhanced with the increase of enzyme concentration to 1.8 mg mL−1. However, the specific activity reduced with the increase in enzyme concentration (Fig. 6(b) and (c)). It can be concluded that some enzymes have been overloaded on the membrane and increased the loading capacity. However, according to washing the membranes, the activity has reduced. Here, the activity was in the range of activities of our previous study regarding the lipase immobilization on Fe3O4@SiO2 dip-coated membrane.30 It can be concluded that the active site of the enzyme immobilized on GA-MNP@PAN and after that TCT-MNP@PAN membrane is more available than other immobilized enzymes. In addition, the presence of MNPs on the surface of the membrane changed the morphology and chemistry of the surface which helped the enzyme to be attached in proper structure and higher amount than that of membranes which was activated with GA or EPI.
Kinetic parameters
The kinetic parameters of free lipase and immobilized lipase on the membrane were determined using the Lineweaver–Burk plot and are shown in Table 1. The vmax shows the highest rate of enzymatic reaction when the enzyme is saturated with the substrate and the Km value shows the affinity of the enzyme for the substrate. Substrate concentration on the environment of enzyme affect the Km46 The results indicate a reduction in Vmax for immobilized lipases in comparison to free lipase which has been observed for CRL immobilized on silica, zirconia and niobium oxide47 and nanocomposite membrane.30 This may be due to the mass transfer limitation in the case of enzyme immobilization on membranes and has been reported for various supports.48,49 However, Vmax for immobilized lipase were in the same range which shows that the accessibility of substrate for immobilized enzyme on membrane was limited by the structure of membrane The Km values for immobilized lipases are lower than the free one, which showed a higher affinity of the substrate for immobilized lipase. This reduction has been perceived for lipase immobilization on magnetic nanoparticles27 and membrane.30 However, it was observed that Km value increased from free to immobilized lipase in some studies regarding the immobilization of lipase on membrane surface.1,7,45,50 It has been reported that immobilization of lipase on surfaces would improve lipase activity by rearranging the conformational structure (termed as ‘lid’) which opens its active site.12 Here, nanocomposite membranes had a lower Km and higher Vmax in comparison to activated membranes. It was observed that the activity and loading efficiency of lipase on nanocomposite membrane was higher than the activated membranes. This may be due to the presence of nanoparticles which supply more available active groups for enzyme immobilization and providing better conformation for lipase due to immobilization on nanoparticles. However, despite the lower activity of ENZ@TCT-MNP@PAN than ENZ@GA-MNP@PAN, immobilized enzyme on TCT-MNP@PAN membrane had a lower Km and higher Vmax which may be due to the better conformational change in the enzyme that leads to a higher possible formation of substrate–enzyme complex and more available active sites.| Sample | Km (mM) | vmax (U mg−1) |
|---|---|---|
| Free CRL | 0.213 | 4854 |
| ENZ@GA @PAN | 0.154 | 907.44 |
| ENZ@EPI@PAN | 0.112 | 915.67 |
| ENZ@GA-MNP@PAN | 0.0998 | 950.57 |
| ENZ@TCT-MNP@PAN | 0.0502 | 979.35 |
Effect of hydrophilicity on lipase activity
It is worth noting that, a moderate hydrophilic membrane is more suitable to supply a contact between substrate and lipase, while hydrophobic surfaces can increase the activity of lipase.12 According to Fig. 5(b) and (c), nanocomposite membranes which are more hydrophobic than activated membranes, have showed higher activity. This is in accordance with the results of Chen et al.14 which stated that the lipase exhibits a higher activity when the membrane has a higher hydrophobicity. Although, the kinetic parameters have confirmed the better lipase conformation on nanocomposite membranes. In this way according to the hydrophilic properties of activated membranes and nanocomposite membrane, nanocomposite membranes are good candidates for lipase immobilization.Storage stability of immobilized lipase on TCT-MNP@PAN membrane
The storage stability of immobilized lipase on TCT-MNP@PAN was examined in the dried and wet condition (Fig. 7). It can be seen that the stability of immobilized enzyme at room temperature is satisfying. While drying the membrane is more comfortable than drying the nanoparticles, using immobilized enzyme on nanocomposite membranes is more desirable.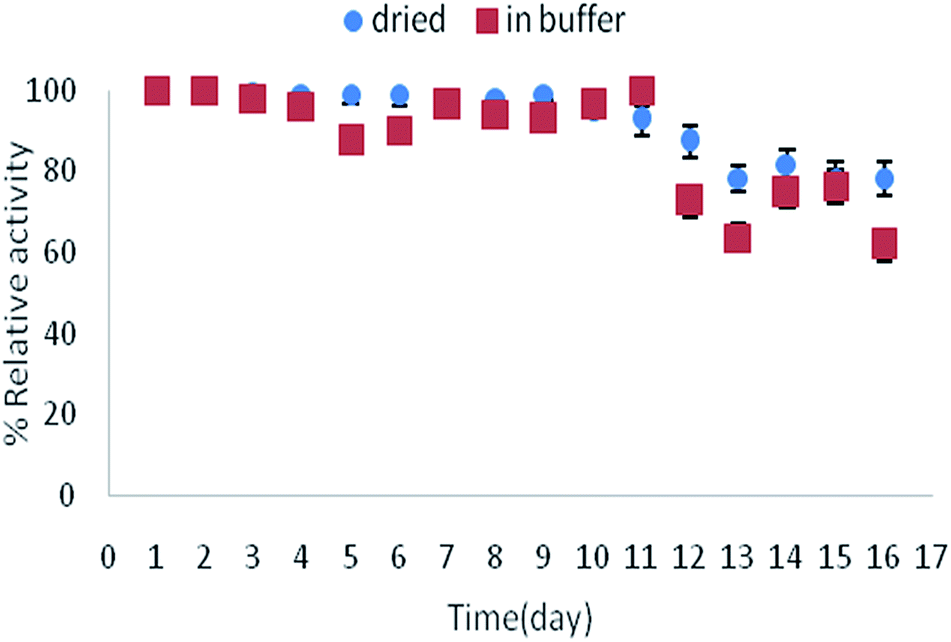 | ||
| Fig. 7 Storage stability of immobilized lipase on TCT-MNP@PAN at room temperature in buffer or dried state. | ||
Conclusions
In this work, easy, low cost and practical activated and nanocomposite membranes were prepared to covalently immobilize lipase enzyme. In this regards, PAN membranes were aminated and activated with GA or EPI. In the other method, TCT and GA functionalized magnetic nanoparticles were attached to the aminated PAN membrane and afterwards lipase was immobilized on these membranes. Various techniques such as SEM, TEM, XRD, FTIR, ATR, AFM, contact angle goniometry and surface free energy measurement were applied to characterize the nanoparticles and membranes. According to the relative activity, loading capacity and kinetic parameters of the enzymatic membrane, enzymatic nanocomposite membranes (TCT-MNP@PAN and GA-MNP@PAN) were improved in comparison to lipase immobilized on activated membrane (EPI@PAN and GA@PAN). These membranes have been applied for biodiesel production by the authors of this study which will be presented in the further work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the research council of the Isfahan University.Notes and references
- P. Ye, Z.-K. Xu, J. Wu, C. Innocent and P. Seta, Macromolecules, 2006, 39, 1041–1045 CrossRef CAS.
- L. T. Izrael Živković, L. S. Živković, B. M. Babić, M. J. Kokunešoski, B. M. Jokić and I. M. Karadžić, Biochem. Eng. J., 2015, 93, 73–83 CrossRef.
- M.-M. Zheng, L. Dong, Y. Lu, P.-M. Guo, Q.-C. Deng, W.-L. Li, Y.-Q. Feng and F.-H. Huang, J. Mol. Catal. B: Enzym., 2012, 74, 16–23 CrossRef CAS.
- M. Aghababaie, M. Beheshti, A. Razmjou and A. K. Bordbar, Biofuel, 2017, 1–7 Search PubMed.
- W. Yujun, X. Jian, L. Guangsheng and D. Youyuan, Bioresour. Technol., 2008, 99, 2299–2303 CrossRef PubMed.
- P. Ye, Z.-K. Xu, J. Wu, C. Innocent and P. Seta, J. Mol. Catal. B: Enzym., 2006, 40, 30–37 CrossRef CAS.
- S. Gupta, Yogesh, S. SJaviya, M. Bhambi, C. S. Pundir, K. Singh and A. Bhattachary, Int. J. Biol. Macromol., 2008, 42, 145–151 CrossRef CAS PubMed.
- S.-F. Li, Y.-H. Fan, R.-F. Hu and W.-T. Wu, J. Mol. Catal. B: Enzym., 2011, 72, 40–45 CrossRef CAS.
- P. Ye, J. Jiang and Z.-K. Xu, Colloids Surf., B, 2007, 60, 62–67 CrossRef CAS PubMed.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS.
- P. Ye, Z.-K. Xu, J. Wu, C. Innocent and P. Seta, Biomaterials, 2006, 27, 4169–4176 CrossRef CAS PubMed.
- N. S. Pujari, B. K. Vaidya, S. Bagalkote, S. Ponrathnam and S. Neneb, J. Membr. Sci., 2006, 285, 395–403 CrossRef CAS.
- N. Hilal, R. Nigmatullin and A. Alpatova, J. Membr. Sci., 2004, 238, 131–141 CrossRef CAS.
- G.-J. Chen, C.-H. Kuo, C.-I. Chen, C.-C. Yu, C.-J. Shieh and Y.-C. Liu, J. Biosci. Bioeng., 2012, 113, 166–172 CrossRef CAS PubMed.
- G. Vitola, D. Büning, J. Schumacher, R. Mazzei, L. Giorno and M. Ulbricht, Macromol. Biosci., 2017, 17, 1600381 CrossRef PubMed.
- X. Zhao, Y. Su, W. Chen, J. Peng and Z. Jiang, J. Membr. Sci., 2012, 415–416, 824–834 CrossRef CAS.
- J. Hou, G. Dong, Y. Ye and V. Chen, J. Membr. Sci., 2014, 469, 19–30 CrossRef CAS.
- P. Daraei, S. S. Madaeni, N. Ghaemi, E. Salehi, M. A. Khadivi, R. Moradian and B. Astinchap, J. Membr. Sci., 2012, 415–416, 250–259 CrossRef CAS.
- P. Jian, H. Yahui, W. Yang and L. Linlin, J. Membr. Sci., 2006, 284, 9–16 CrossRef CAS.
- Z.-Q. Huang, F. Zheng, Z. Zhang, H.-T. Xu and K.-M. Zhou, Desalination, 2012, 292, 64–72 CrossRef CAS.
- A. Razmjou, J. Mansouri and V. Chen, J. Membr. Sci., 2011, 378, 73–84 CrossRef CAS.
- L. Yan, Y. S. Li, C. B. Xiang and S. Xianda, J. Membr. Sci., 2006, 276, 162–167 CrossRef CAS.
- L.-Y. Yu, Z.-L. Xu, H.-M. Shen and H. Yang, J. Membr. Sci., 2009, 337, 257–265 CrossRef CAS.
- J. S. Taurozzi, H. Arul, V. Z. Bosak, A. F. Burban, T. C. Voice, M. L. Bruening and V. V. Tarabara, J. Membr. Sci., 2008, 325, 58–68 CrossRef CAS.
- Y. Orooji, M. Faghih, A. Razmjou, J. Hou, P. Moazzam, N. Emami, M. Aghababaie, F. Nourisfa, V. Chen and W. Jin, Carbon, 2017, 111, 689–704 CrossRef CAS.
- M. L. Verma, C. J. Barrow and M. Puri, Appl. Microbiol. Biotechnol., 2013, 97, 23–39 CrossRef CAS PubMed.
- E. Ranjbakhsh, A. K. Bordbar, M. Abbasi, A. R. Khosropour and E. Shams, Chem. Eng. J., 2012, 179, 272–276 CrossRef CAS.
- Y. Cui, Y. Li, Y. Yang, X. Liu, L. Lei, L. Zhou and F. Pan, J. Biotechnol., 2010, 150, 171–174 CrossRef CAS PubMed.
- S. Kanimozhi and K. Perinbam, Mater. Res. Bull., 2013, 48, 1830–1836 CrossRef CAS.
- M. Aghababaie, M. Beheshti, A. Razmjou and A.-K. Bordbar, Food Bioprod. Process., 2016, 100, 351–360 CrossRef CAS.
- O. Barbosa, R. Torres, C. Ortiz, Á. Berenguer-Murcia, R. C. Rodrigues and R. Fernandez-Lafuente, Biomacromolecules, 2013, 14, 2433–2462 CrossRef CAS PubMed.
- X.-J. Huang, P.-C. Chen, F. Huang, Y. Ou, M.-R. Chen and Z.-K. Xu, J. Mol. Catal. B: Enzym., 2011, 70, 95–100 CrossRef CAS.
- C.-C. Lin and M.-C. Yang, Biomaterials, 2003, 24, 549–557 CrossRef CAS PubMed.
- C. J. Van Oss, L. Ju, M. K. Chaudhury and R. J. Good, J. Colloid Interface Sci., 1989, 128, 313–319 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- G. E. P. Box and K. B. Wilson, J. Roy. Stat. Soc. B, 1951, 13, 1–45 Search PubMed.
- Y.-T. Zhu, X.-Y. Ren, Y.-M. Liu, Y. Wei, L.-S. Qing and X. Liao, Mater. Sci. Eng., C, 2014, 38, 278–285 CrossRef CAS PubMed.
- A. K. Bordbar, A. A. Rastegari, R. Amiri, E. Ranjbakhsh, M. Abbasi and A. R. Khosropour, Biotechnol. Res. Int., 2014, 2014, 6 Search PubMed.
- M. Mihailović, M. Stojanović, K. Banjanac, M. Carević, N. Prlainović, N. Milosavić and D. Bezbradica, Process Biochem., 2014, 49, 637–646 CrossRef.
- Z. Xu, J. Wang, L. Shen, D. Men and Y. Xu, J. Membr. Sci., 2002, 196, 221–229 CrossRef CAS.
- J. Wang, G. Meng, K. Tao, M. Feng, X. Zhao, Z. Li, H. Xu, D. Xia and J. R. Lu, PLoS One, 2012, 7, e43478 CAS.
- R. N. Wenzel, Ind. Eng. Chem., 1936, 28, 988–994 CrossRef CAS.
- O. Barbosa, C. Ortiz, A. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, RSC Adv., 2014, 4, 1583–1600 RSC.
- C. Mateo, R. Torres, G. Fernández-Lorente, C. Ortiz, M. Fuentes, A. Hidalgo, F. López-Gallego, O. Abian, J. M. Palomo, L. Betancor, B. C. C. Pessela, J. M. Guisan and R. Fernández-Lafuente, Biomacromolecules, 2003, 4, 772–777 CrossRef CAS PubMed.
- P. Ye, Z.-K. Xu, J. Wu, C. Innocent and P. Seta, J. Mol. Catal. B: Enzym., 2006, 40, 30–37 CrossRef CAS.
- R. C. Rodrigues, C. Ortiz, A. Berenguer-Murcia, R. Torres and R. Fernandez-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC.
- L. T. Izrael Živković, L. S. Živković, B. M. Babić, M. J. Kokunešoski, B. M. Jokić and I. M. Karadžić, Biochem. Eng. J., 2015, 93, 73–83 CrossRef.
- M. Kalantari, M. Kazemeini, F. Tabandeh and A. Arpanaei, J. Mater. Chem., 2012, 22, 8385–8393 RSC.
- C. J. van Oss, M. K. Chaudhury and R. J. Good, Adv. Colloid Interface Sci., 1987, 28, 35–64 CrossRef CAS PubMed.
- S.-H. Chiou and W.-T. Wu, Biomaterials, 2004, 25, 197–204 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |