
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into thermal reduction of the oxidized graphite from the electro-oxidation processing of nuclear graphite matrix†
Gengyu Zhang,
Mingfen Wen*,
Shuwei Wang,
Jing Chen and
Jianchen Wang *
*
Institute of Nuclear and New Energy Technology, Tsinghua University, Beijing 100084, China. E-mail: wangjianchen@mail.tsinghua.edu.cn; wenmingfen@mail.tsinghua.edu.cn; Tel: +86 13910738328 Tel: +86 13910638139
First published on 2nd January 2018
Abstract
Electro-oxidation disintegration is a potential technology pathway to detach graphite waste from high-level radioactive wastes in the spent fuel reprocessing of high temperature gas cooled reactors (HTGR). But this electrochemical treatment can introduce oxygen functional groups into the graphite lattice. Further disposal will require the development of safe and cost-effective methods to achieve the reduction and deoxygenation of the oxidized graphite wastes. In general, oxygen species can be removed from oxidized graphite directly in an easy-to-operate way by thermal annealing. In this paper, the thermal reduction process of oxidized graphite from the electro-oxidation processing of graphite matrix from simulative element of HTGR was researched. The thermodynamic state and kinetic process of the deoxygenation reaction of electrolytic graphite oxide (EGO) have also been investigated for better understanding of the reduction mechanism. According to thermal analysis results, the oxygen functional groups were removed mostly between 160 °C and 250 °C, and the main weight loss of EGO during thermal annealing was caused by evolved volatile gases (CO2, CO and H2O). The activation energy of deoxygenation reaction for EGO is calculated to be 115.6 kJ mol−1 (1.20 eV per atom). And the persistent residual oxygen species on reduced EGO (REGO) is determined to be from two parts, i.e. the stubborn built-in oxygen species of pristine nuclear graphite and the newly formed C–O moieties on the basal plane of graphite sheet during thermal annealing process. In addition, an attempt was also made to elucidate how the thermal treatment acted on the electronic structure of EGO. Meanwhile, the mechanism of dissociation of oxygen species (–OH, C–O–C, ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –COOH) on the thermal reduction of oxidized graphite has been suggested in this paper.
O, –COOH) on the thermal reduction of oxidized graphite has been suggested in this paper.
1. Introduction
In the nuclear industry, graphite has been widely utilized in HTGR where graphite and carbon materials are used for in-core structural components, such as fuel blocks, reflectors, core support structures and a fuel element matrix material.1–4 Hence, the decommissioning of HTGR will produce a large amount of irradiated graphite waste. In recent years, electro-oxidation method with salts as electrolyte has been developed for the separation of the graphite matrix from simulated spent fuel elements of HTGR in Institute of Nuclear and New Energy Technology (INET), Tsinghua University.5–7 The so-called “electrochemical disintegration” method has been achieved using graphite matrix as an anode and inert counter electrode in concentrated nitrates, sulfates, phosphates, sulphuric acid and nitric acid. Results showed that the graphite matrix could be detached from spent fuel elements effectively and the dissolution of the fuel kernel could be avoided in this process.6,7However, the electro-oxidation process may induce oxygen-containing groups into the graphite structure, and lead to the conversion of sp2 carbon atoms into sp3 ones. And the oxidized graphite is widely regarded as an energetic material which undergoes explosive decomposition if heated.8–10 Thus, the oxygen-rich derivative of graphite from the electrochemical process exhibits a weak chemical-stability. These reactive features represent important safety concerns in industry that makes further processing more difficult. To increase the stability of the graphite waste and make it safer in reprocessing, storing or disposal, the very first thing is to remove the oxygen species from the graphite backbone. It has been well demonstrated in previous researches that oxidized graphite can be deoxygenated by thermal annealing, chemical reduction or photo-reduction.11–15 However, both chemical reduction and photo-reduction suffers from a relatively low C/O element content, with a sizeable amount of residual oxygen species. In comparison, thermal treatment is considered as the most simple and cost-effective method to achieve the reduction & de-oxygenation of oxidized graphite with no introduction of any impurities. In thermal reduction process, oxygen species of graphite oxide decompose exothermically upon heating, and produce a gas mainly composed of H2O, CO, and CO2.12,16,17 Thermal “reduction” is actually a thermolysis or pyrolysis process. Technically, it should not be called a reduction process since there is no external reductant participation in reaction. Instead, it is more like a chemical disproportionation reaction, in which the original carbon atoms are decomposed into reduced forms in solid reduced graphite oxide and oxidized forms that are primarily consisted of carbonous gas-phase (i.e. CO, CO2).
In addition, thermal reduction of oxidized graphite is a common processing step in the fabrication of many graphene-related materials and devices. Currently, many researches are focused on the application of graphite oxide and on the materials obtained by its modification (e.g. thermally modified graphene oxide). And the thermal behavior of graphite oxide prepared by traditional methods (Staudenmaier, Hofmann and Hummers methods) has been extensively investigated.12,18–20 These researches can help us understand the thermal treatment process of oxidized graphite from the electro-oxidation processing of simulated nuclear graphite matrix.
However, the detailed mechanisms of the thermal reduction process of EGO stay unclear, and the thermal behavior of graphite oxide prepared by electrochemical methods has rarely been investigated. Therefore, it is highly desirable to understand the thermal reduction process and to be able to provide new guides for controlling the properties of resulting reduction product.
In this work, we strive to give a comprehensive thermal analysis of EGO in inert atmosphere. Meanwhile, we compared the thermal stability of the pristine nuclear graphite and EGO in two different atmospheres. The material's structural evolution during the thermal treatment in inert nitrogen atmosphere was investigated using X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), transmission electron microscopy (TEM), and selected area electron diffraction (SAED). These results can make us get a better understanding of the physical and chemical properties of electrochemical oxidized nuclear-grade graphite and offer us the guide for applying the thermal methods to the reduction of EGO from the reprocessing procedure of HTGR. Combining these analytical results and concluding from the previous research, we discussed the implications of thermal treatment for safe EGO processing. And at last, an explanation of reduction mechanism of the thermal process and a formation pathway of the evolved gases were proposed.
2. Experimental
2.1 Material
2.2 Characterization
TGA/MS experiment was carried out using another instrument (TGA Q500), different from the previous one. It was coupled to a Pfeiffer Vacuum Thermo-Star mass spectrometer equipped with a channeltron detector. This experiment was recorded under constant nitrogen flow of 100 mL min−1.
XRD patterns of the samples were collected using an advance diffractometer (D8, Bruker, Germany) equipped with a Cu-Kα radiation, at steps of 0.02°, and with a scan range between 10° and 80°.
SEM micrographs were recorded using a Merlin FESEM equipped with an energy-dispersive X-ray spectrometer at 50 kV. Energy dispersive X-ray spectroscopy (EDX) was applied to monitor the major element composition of the oxidized graphite and its thermal reduction products.
TEM measurements were conducted with a TECNAIG2 20 microscope at an acceleration voltage of 300 kV. Meanwhile, SAED pattern was performed using the TEM instrument, and the electron beam size was set to about 500 nm.
XPS analyses were done with a PHI Quantera SXM spectrometer equipped with a monochromatized Al Kα X-ray source. And XPS peak software was used for data processing and analysis, in which Gaussian–Lorentzian waveforms were used to fit C 1s and O 1s core-level spectra after deducting a Shirley background.
Nitrogen adsorption/desorption isothermal analysis was measured by using BEL mini-instrument, and the specific surface area was obtained by Brunauer Emmett Teller (BET) analyses of the adsorption isotherm. All surface area values were recorded only when linear BET correlation factor was at least R2 = 0.9999.
3. Results and discussions
3.1 Thermochemistry of oxidized graphite upon thermal process
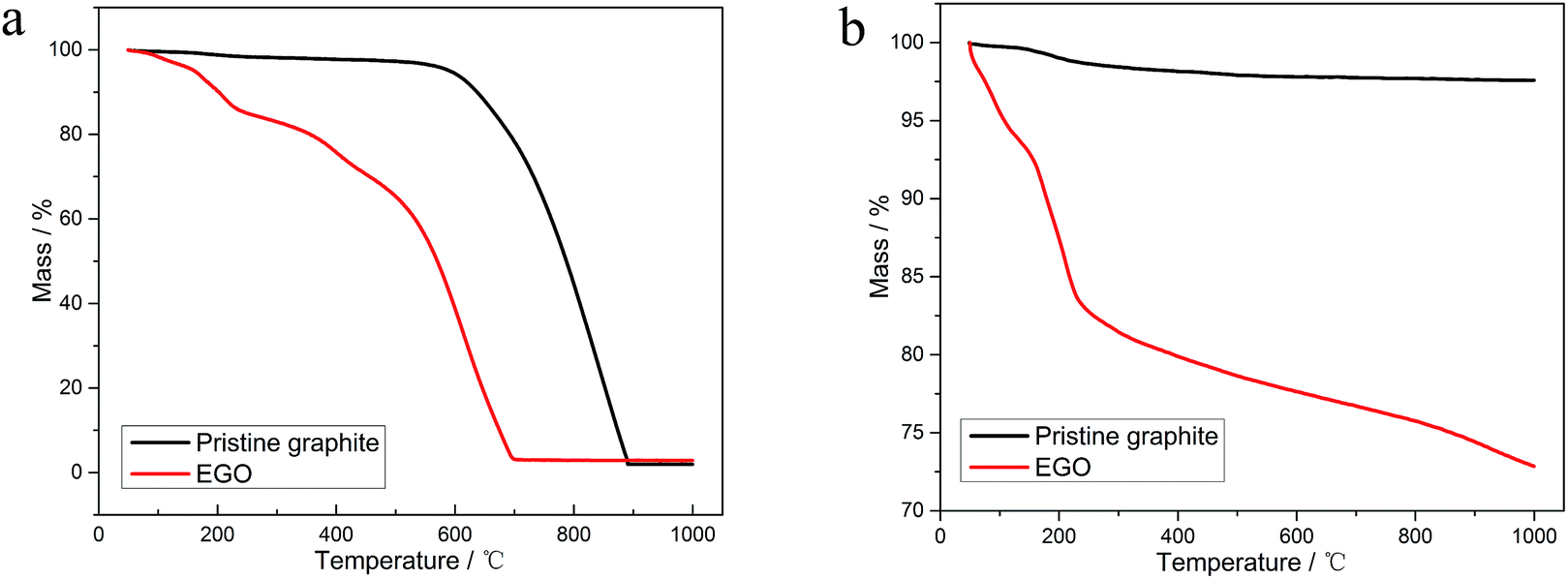 | ||
| Fig. 1 TGA thermograms with a heating rate of 10 °C min−1 of (a) EGO thermal reduction compared with the pristine graphite oxidation in air atmosphere; (b) EGO thermal reduction compared with the pristine graphite oxidation in N2 atmosphere. | ||
Fig. 2a shows the detailed TGA results of EGO in insert atmosphere. The mass in percentage with increasing temperature is displayed on the left axis, along with the derivative mass percentage related to the temperature on the right axis. The EGO began to lose weight upon thermal treatment even below 100 °C, which was associated with the escape of adsorbed water and gas molecules as stated earlier. With temperature rising, two main peaks and two small peaks in the derivative weight are observed near 91 °C, 211 °C, 383 °C and 541 °C, which indicated that there were two large mass transitions and two minor mass fluctuations as EGO was heated. The peak near 91 °C is widely considered due to the loss of interstitial H2O in graphite lattice. At around 216 °C, the weight loss rate of EGO reached a maximum. This may be due to the decomposition of oxygen functional groups in the EGO. The weight loss at around 383 °C originated from the pyrolysis of stubborn oxygen-containing species. The peak at 541 °C can be attributed to the sublimation of the carbon skeletons of EGO.11 The detailed causes of weight loss will be presented in the TGA-MS results in next section. In Fig. 2b, there is only one obvious peak in the derivative weight of PG at 204 °C during the thermal analysis. This weight transition can be attributed to the dissociation of absorbed oxygen atoms from the backbone of pristine graphite.
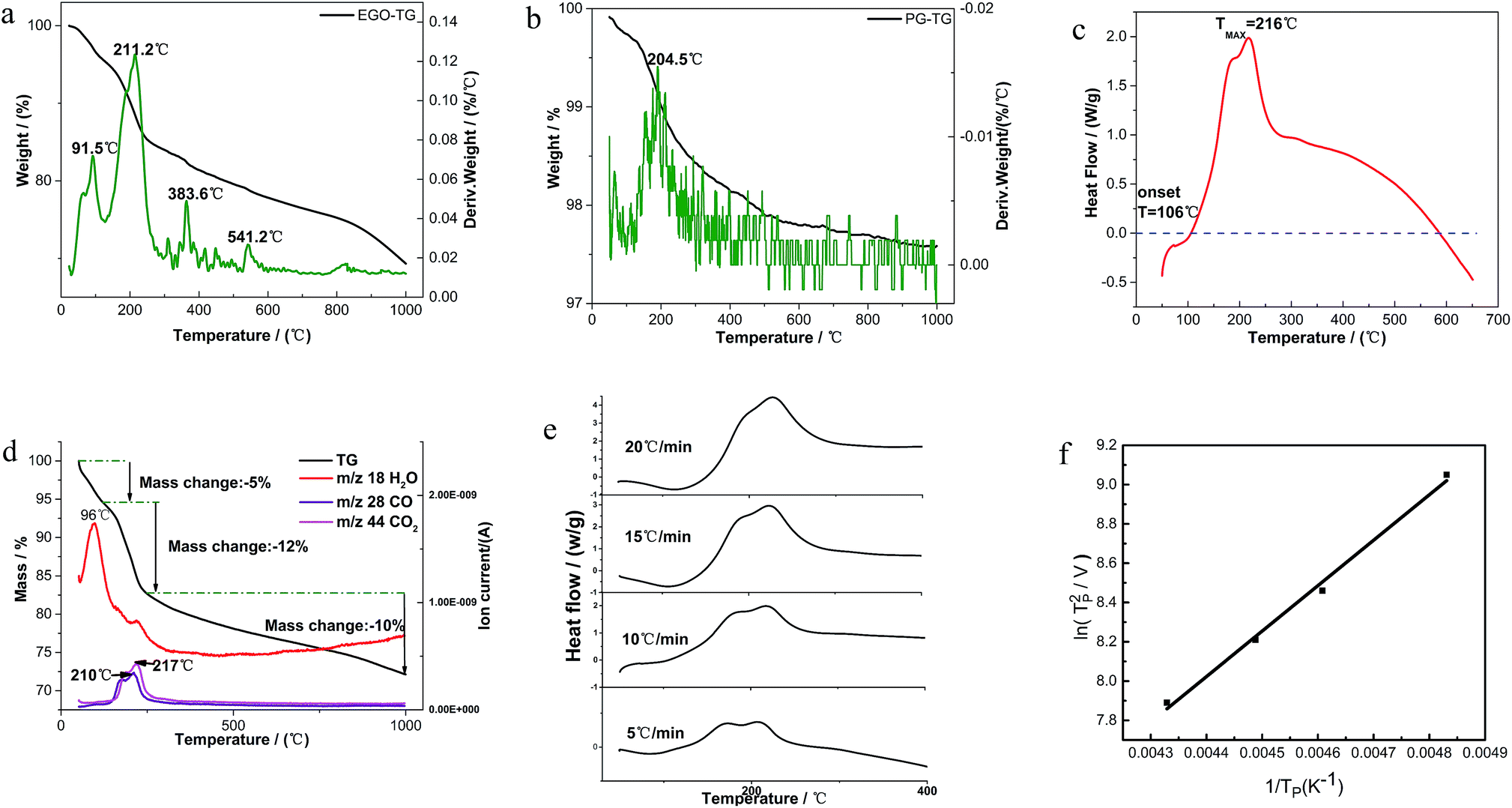 | ||
Fig. 2 (a) TGA thermograms with a heating rate of 10 °C min−1 of derivative weight of EGO; (b) TGA thermograms with a heating rate of 10 °C min−1 of derivative weight of pristine graphite; (c) DSC thermogram of EGO in 100 mL min−1 N2 flow; heating rate 10 °C min−1; (d) TGA-MS spectra of EGO with a heating rate of 10 °C min−1 between rt and 1000 °C; (e) dynamic mode DSC thermograms with systematic variation in heating rate; (f) 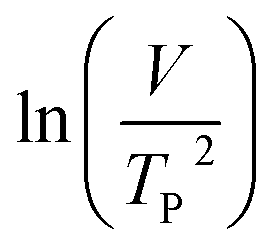 are as a function of 1/TP. are as a function of 1/TP. | ||
A typical DSC scan of EGO with the heating rate of 10 °C min−1 performed at varying temperatures is shown in Fig. 2c. There was a dramatic weight loss at an onset temperature of approximately 106 °C accompanied by a strongly-exothermic DSC peak between 106 °C and 600 °C. The features correspond to the transformation of thermolabile function groups and the formation of evolved gases.
where V is heating rate, TP is the temperature at which the heat flow reaches maximum, Ea is activation energy, A is frequency factor and R is universal gas constant. Plotting
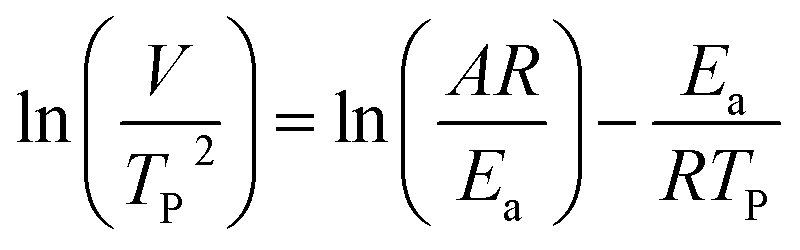
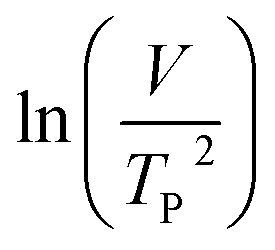 vs.
vs. 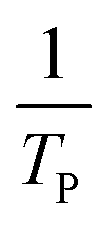 offers linear regression, from which activation energy Ea and frequency factor A are calculated. In this research, four different heating rates (5, 10, 15, and 20 °C min−1) were adopted. The results of
offers linear regression, from which activation energy Ea and frequency factor A are calculated. In this research, four different heating rates (5, 10, 15, and 20 °C min−1) were adopted. The results of 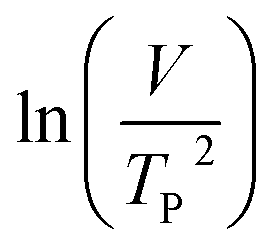 vs.
vs. 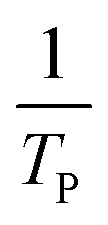 are plotted in Fig. 2f. The activation energy originated from these data was about 115.6 kJ mol−1 (1.20 eV per atom). The activation energy of electrochemical produced GO obtained in this work (115.6 kJ mol−1) was lower than the GO reported previously using traditional synthetic methods.9,10,16
are plotted in Fig. 2f. The activation energy originated from these data was about 115.6 kJ mol−1 (1.20 eV per atom). The activation energy of electrochemical produced GO obtained in this work (115.6 kJ mol−1) was lower than the GO reported previously using traditional synthetic methods.9,10,163.2 Morphology and structural evolution of oxidized graphite during thermal annealing process in insert atmosphere
The morphology of the thermal treated EGO was examined using a field-emission SEM equipment. For the parent EGO samples, high-resolution SEM images could be easily obtained without any pretreatments, suggesting that the products from the electro-treatment process had strong conductivity. This feature reveals that the pristine nuclear graphite was partially oxidized during the electrochemical process, which is also confirmed by XRD data (seen in Fig. 3e).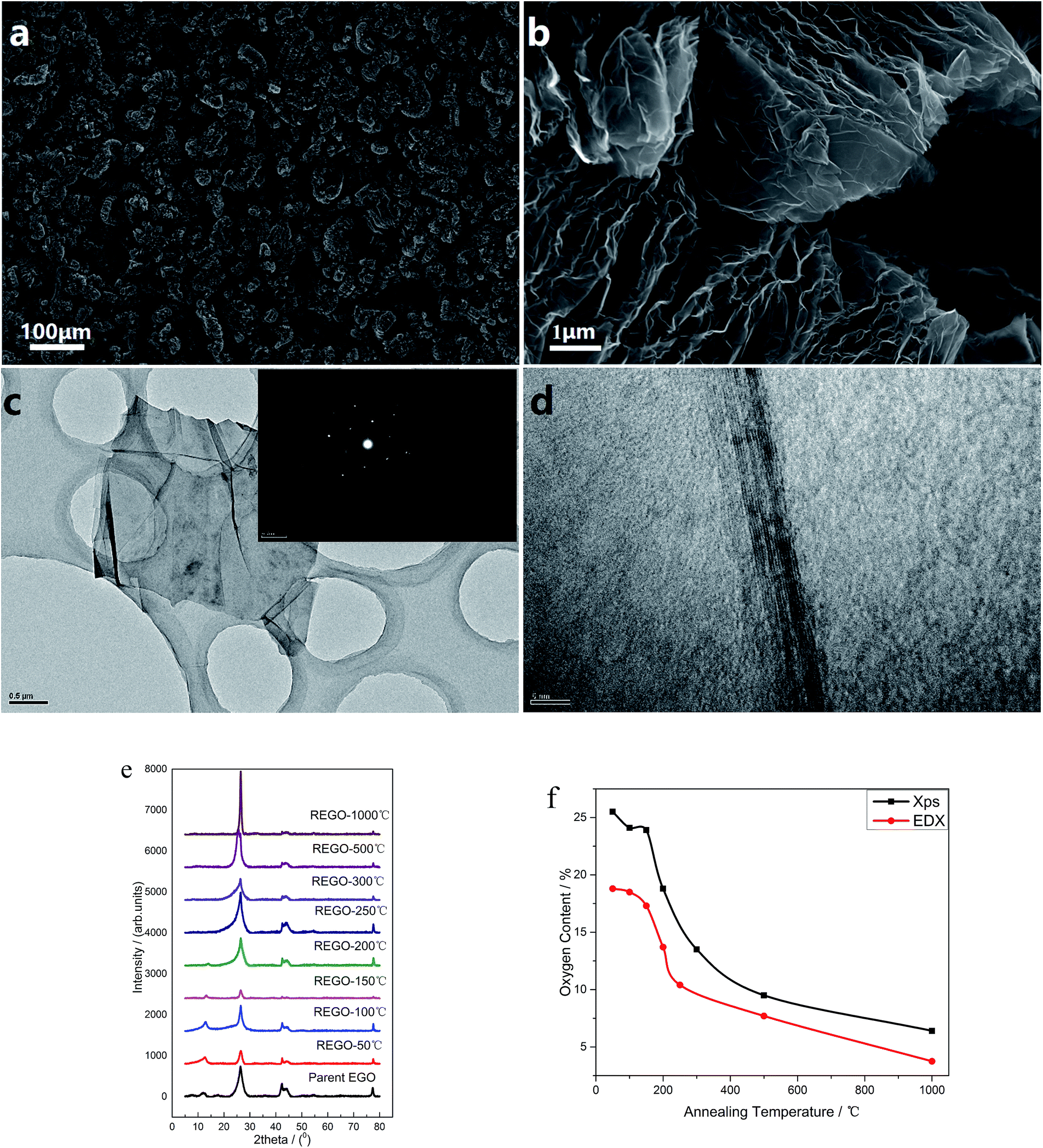 | ||
Fig. 3 (a) Morphology of REGO-1000 °C (at 100× magnification); (b). Morphology of REGO-1000 °C (at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× magnification); (c) low-magnification TEM image of REGO-1000 °C and corresponding SAED patterns; (d) high-resolution TEM image of REGO-1000 °C (e) XRD spectra of REGO annealed at different temperature, compared with that of pristine graphite; (f) temperature-dependence of oxygen content of reduced product measured by XPS and EDX. 000× magnification); (c) low-magnification TEM image of REGO-1000 °C and corresponding SAED patterns; (d) high-resolution TEM image of REGO-1000 °C (e) XRD spectra of REGO annealed at different temperature, compared with that of pristine graphite; (f) temperature-dependence of oxygen content of reduced product measured by XPS and EDX. | ||
Some representative SEM images of the thermally-treated EGO at different temperatures and the results of nitrogen adsorption/desorption isothermal analysis are shown in ESI data.† The morphology of REGO (Fig. S1†) tends to be more and more fluffy with the annealing temperature rising, besides the results of BET N2 surface area clearly confirmed that the porosity of REGO continues to increase along with the increasing of annealing temperature. The BET N2 surface area of the parent EGO is only 6.87 m2 g−1, while we obtain higher area values when the product had been thermal-treated. And the fastest surface area rising period happened between 150 °C and 200 °C, which is exactly the time of the occurrence of the decomposition of the main oxygen moieties. And ultimately the REGO presents a worm-like morphology in the low magnification (Fig. 3a) and an accordion-like structure in the high magnification (Fig. 3b) when the annealing temperature reaches 1000 °C. Meanwhile, the BET surface area of REGO-1000 °C increased to 45.7 m2 g−1. In addition, the elemental composition of thermally-reduced EGO at different temperatures was investigated by SEM-EDX. According the EDX data (Fig. 3f), O-content of thermally-reduced EGO were significantly diminished with annealing temperatures rising. Similar trends could be observed from the XPS data, although there were some numerical differences in the two observation results because of the different principles of observation method and instruments.
Fig. 3c depicts the transmission electron microscopy (TEM) and corresponding SAED characterization results of REGO annealed at 1000 °C. Low-magnification TEM (Fig. 3c) observation indicates that the reduced products were inhomogeneous micrometer-sized flakes. And some sheet-like graphitic stackings and discontinued fringes with corrugated structure can be observed from the high-resolution TEM. The discontinued fringes in the HRTEM image (Fig. 3d) were attributed to the defective structures, and it indicates the interlayered coherence was destroyed. As the EGO was identified to be a heterogeneous and disordered system by previous research,5 the distorted structure of REGO could not be determined from the thermal reduction process or the prior electrolytic process. The typical well-defined diffraction spots in SAED pattern depicts that the amorphous state of the EGO was restored, confirming that the well-crystallized structure of REGO-1000 °C as XRD data had presented (see below). But the crystallinity was not as perfect as pristine graphite, because of the defects from the thermal-reduction process of O-moieties that still remained on the graphitic structure.
The reduction and crystalline degree of thermally-treated oxidized graphite were further characterized by X-ray diffraction (XRD). As validated by the XRD spectra, the starting pure nuclear graphite before thermal treatment exhibited a characteristic peak (002) located at around 26.4° (seen in ESI, Fig. S2a†). The parent EGO exhibits a main peak at 26.51°, and another smaller peak (001) can be observed at 12.52° associated with the oxide phase of EGO caused by electro-oxidation treatment (Fig. S2b†). The XRD patterns of the samples of EGO heated at different temperatures are shown in Fig. 3e. After thermal treatment, the peak at 12.52° becomes smaller and smaller with the temperature rising and completely disappears when the temperature rises to 250 °C. Meanwhile, the peak around 26° becomes sharper and continuously shifts to the right with temperature rising. The REGO sample at 1000 °C exhibited a structure closer to the pristine nuclear graphite as showed by the shifting of (002) peak back to 26.4°. However, its FWHM still remained larger than that of pristine graphite, which suggested the presence of strains or defects. Overall, the results of XRD indicate that oxygen species on the oxidized graphite was mostly removed during the thermal annealing process and the amorphous phase of oxidized graphite was largely transformed into crystalline phase.
XPS was used to analyze the changes of the structure and the surface chemical composition of EGO during the thermal process. The C 1s spectra of pristine graphite (PG) (Fig. 4a) before electro-treatment shows two peaks, which were related to sp2-hybridized (284.5 eV) and hydroxyl configurations (285.6 eV) respectively. This significant C–O peak (around 285.6 eV) in pristine graphite was associated with atmospheric oxidation during the manufacturing processes, which had previously been reported.22–24 The XPS spectra of the PG sample after high temperature (1000 °C) processing in inert atmosphere (Fig. 4b) exhibit a similar graphical shape as the untreated one (Fig. 4a), and the intensity of C–O single bond had not lessened. Consequently, this phenomenon suggests that the inherent C–O of PG was super-stable and hard-to-remove during heat treatment process. At the same time, we found four different bond types accounting for the C 1s spectra of parent EGO. In accordance with the results of prior literature, the XPS C 1s spectrum of the parent EGO was deconvoluted into four peaks: the first peak occurring at 284.6 eV could be assigned to be sp2-hybridized carbons; the second peak at 285.4 eV was assigned to C–O or C–O–C (hydroxyl or epoxide) groups; and the peak at 287.1 eV for the CO (carbonyl) bond types, and 288.1 eV for HO–CO (carboxylic) bond types.5,25 Referring to the previous researches of graphite oxide, epoxide and hydroxyl were considered to be highly likely distributed on the basal planes of EGO's skeleton, while carbonyl and carboxyl were located at the edges of oxidized graphite.26–29 Noting that except sp2 fraction, CO groups was the maximum contribution of C 1s, which was more than the two other bond types C–O (hydroxyl or epoxide) and O–CO (carboxylic). Additionally, the total CO double bonded contribution (including CO and COOH groups) was more than C–O single bonded groups. ([CO + COOH]/[C–OH + C–O–C] = 1.7). This can be interpreted that the C–O single bonded groups tended to be oxidized to become CO double bonded species at harsh electro-oxidization conditions.5
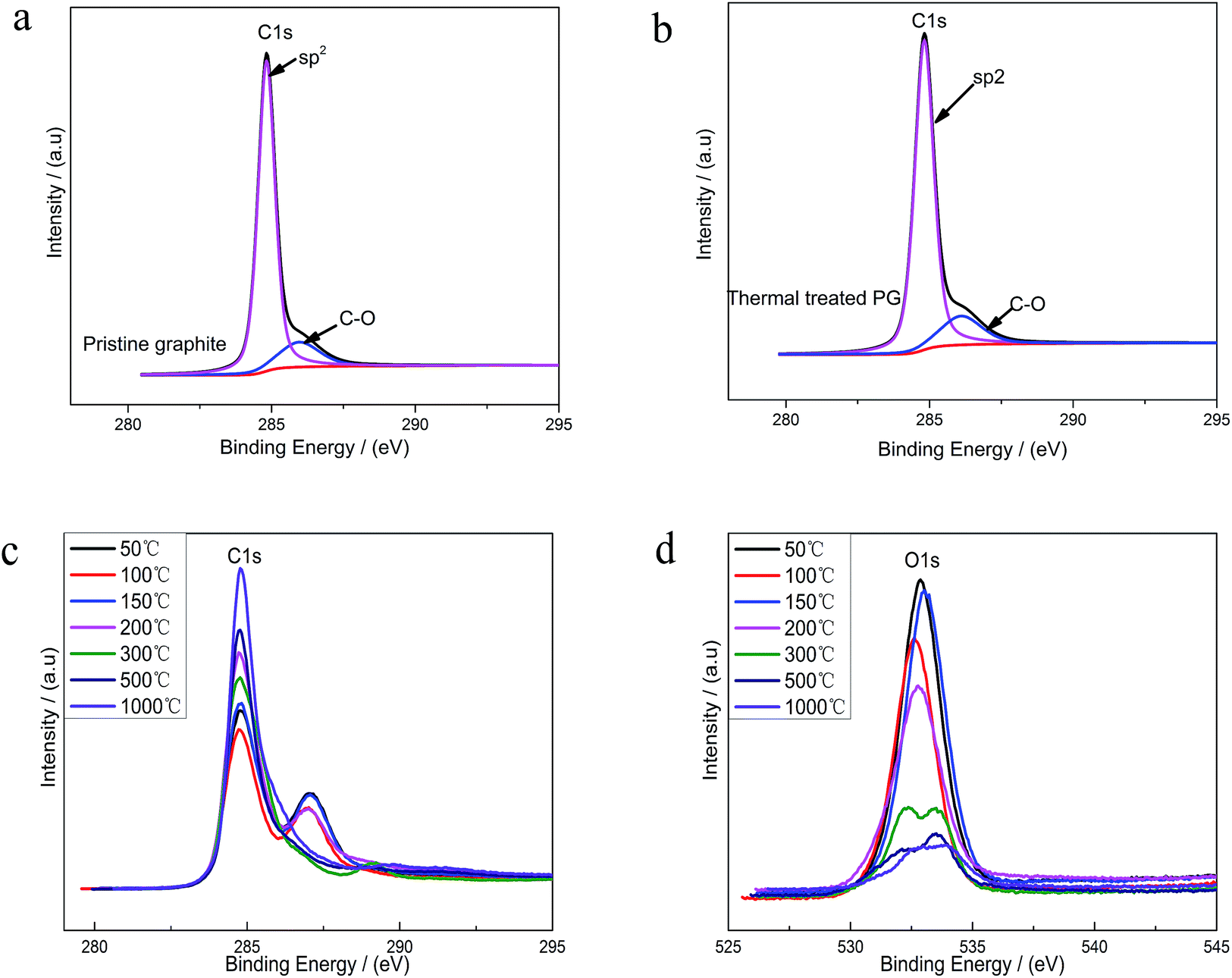 | ||
| Fig. 4 (a) C 1s XPS spectra for pristine graphite (PG); (b) O 1s XPS spectra for PG; (c) C 1s spectra measured on EGO after annealing at increasing temperatures; (d) O 1s spectra measured on EGO after annealing at increasing temperatures. | ||
Upon thermal treatment, the C 1s spectrum of EGO presented a transformation from a double peak to a single sharp peak with annealing temperature rising (Fig. 4c). The evolution of the C 1s spectrum reveals an increase of C-content with annealing temperature rising, and a corresponding decrease of the O-species. 96% C-content of REGO could be achieved with temperature reaching 1000 °C, with only about 4% O contribution remained.
The C 1s XPS spectra of REGO annealed at different temperatures are illustrated in Fig. 5, and the changes of relative contribution of the bond types with increasing temperature are presented in Fig. 7a. Between 50 °C and 150 °C, the relative intensities of the four bond types undulate slightly and almost maintains a certain level. From 150 °C to 200 °C, the edge-plane HO–CO suffers a sharp decline and becomes extinct at 200 °C accompanied with a slight rise of C–O bond types. And a π–π* shakeup satellite peak at around 290 eV appears at the same time, which indicates that the delocalized π conjugation of aromatic carbon structure on REGO was on some level restored.25 Then, the C–O shows a slight increase from 200 °C to 300 °C and subsequently diminishes between 300 °C and 500 °C. And the upward trend of C–O in the range of 200 °C to 300 °C has also been reported in previous researches.25,30,31 The rise of C–O at initial stage was interpreted to be contributed by newly-formed phenolic group. And these phenolic groups were distributed between graphite sheets and intercalated into the interlayer galleries between intact conjugated domains. Due to its special intercalating position, the completely thermal decomposition of such C–O is thermodynamically difficult.25,31 And the transformation of O 1s spectrum provides further robust evidence on the generation of phenolic groups (later in the paper). The subsequent relative decline of C–O between 300 °C and 500 °C could be attributed to the elimination of C–OH which distribute at the edge of graphite sheets. Meanwhile, the CO started a continued decline from 150 °C, and ultimately disappeared at the temperature of 1000 °C.
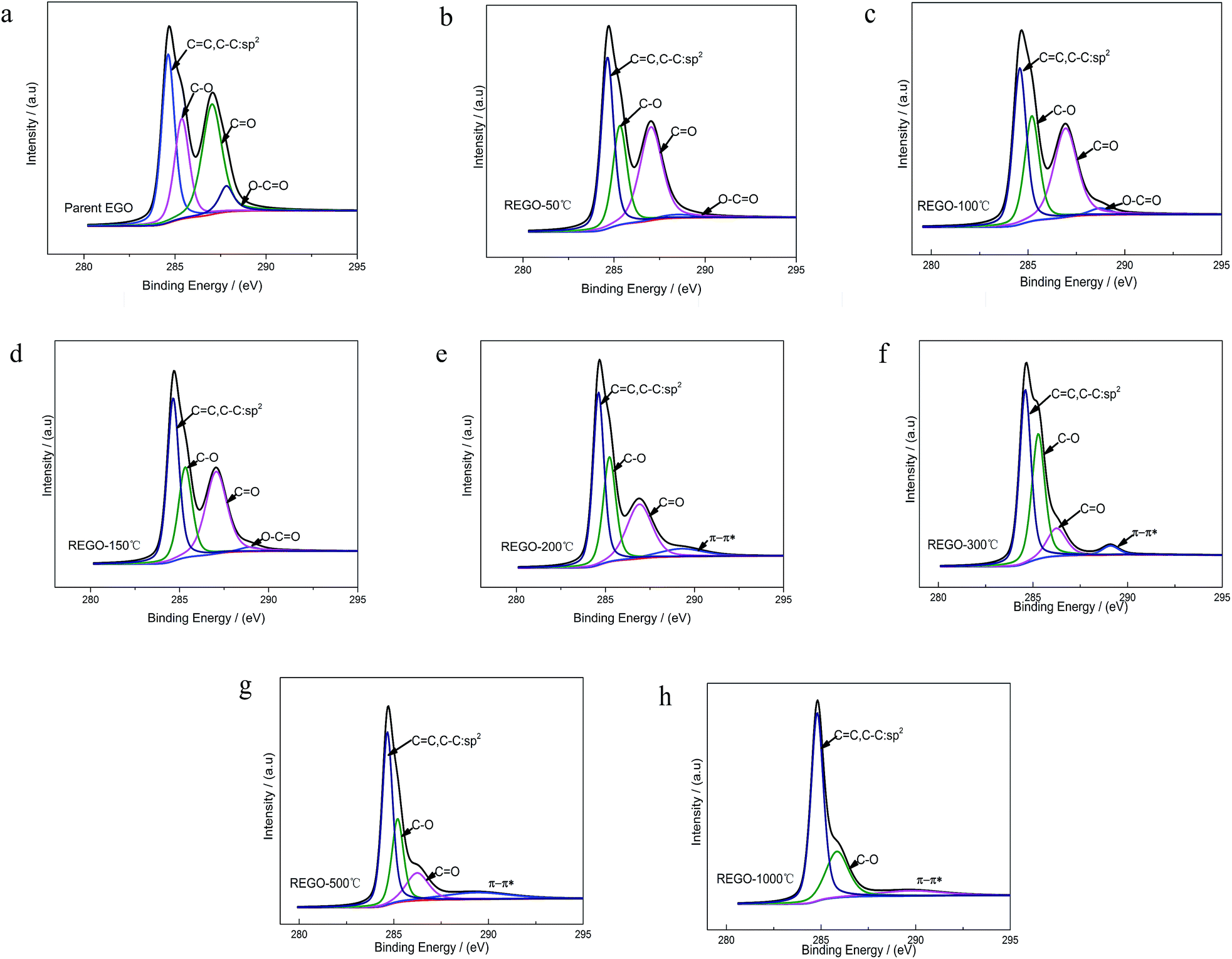 | ||
| Fig. 5 C 1s XPS spectra deconvoluted peaks with increasing annealing temperature. | ||
The internal conversion of CO to C–O at low reduction temperature was considered to be the reason of the initial increase of C–O single bond.25 The dispersion of the peak positions as a function of annealing temperature is shown in Fig. 7b. In general, all the peaks fluctuate within a fixed range. Notable among them is that the CO peak turned to shift to lower binding energy side representative of C–O single bond between 200 °C to 300 °C, which indirectly reconfirmed the transformation of CO to C–O.
As shown in Fig. 6, the O 1s spectra of parent EGO can be deconvoluted into three main peaks: the first peak at 531.4 eV is assigned to CO (O atom doubly bonded to aromatic carbon labelled as O1), the second peak (532.6 eV) is assigned to C–O (O atom singly bonded to aliphatic carbon labeled as O2), and the third peak (533.5 eV) can be due to the presence of phenolic group (O atom singly bonded to aromatic carbon labeled as O3).25,32–35 These assignments are consistent with the previous researches, indicating that our results are reliable. Thermal reduction of EGO creates a shift of O 1s spectra from low energy side to the high, and from a single peak to a double feature with the rise of temperature (Fig. 4d). From 50 °C to 150 °C, both the relative intensity and integrated intensity show no obvious changes (Fig. 7c). The integrated intensity of oxygen moieties started to decrease since the temperature reached 200 °C, and simultaneously the relative intensity of O3 increased rapidly with the decrease of O1 and O2 between 200 °C and 500 °C. In the following, the intensity of O-moieties (both the relative intensity and integrated intensity) changed marginally from 500 °C to 1000 °C.
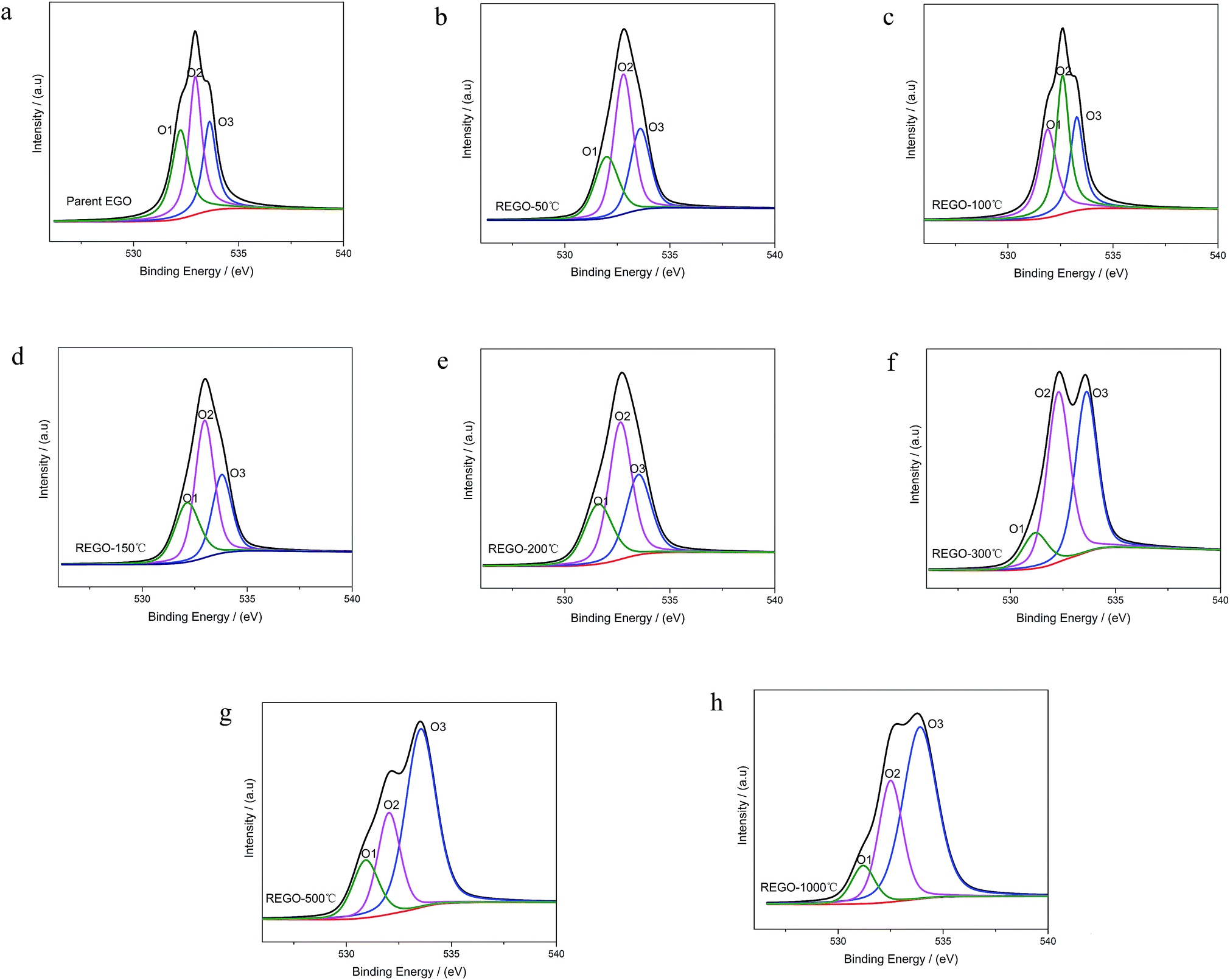 | ||
| Fig. 6 O 1s XPS spectra deconvoluted peaks with increasing annealing temperature. | ||
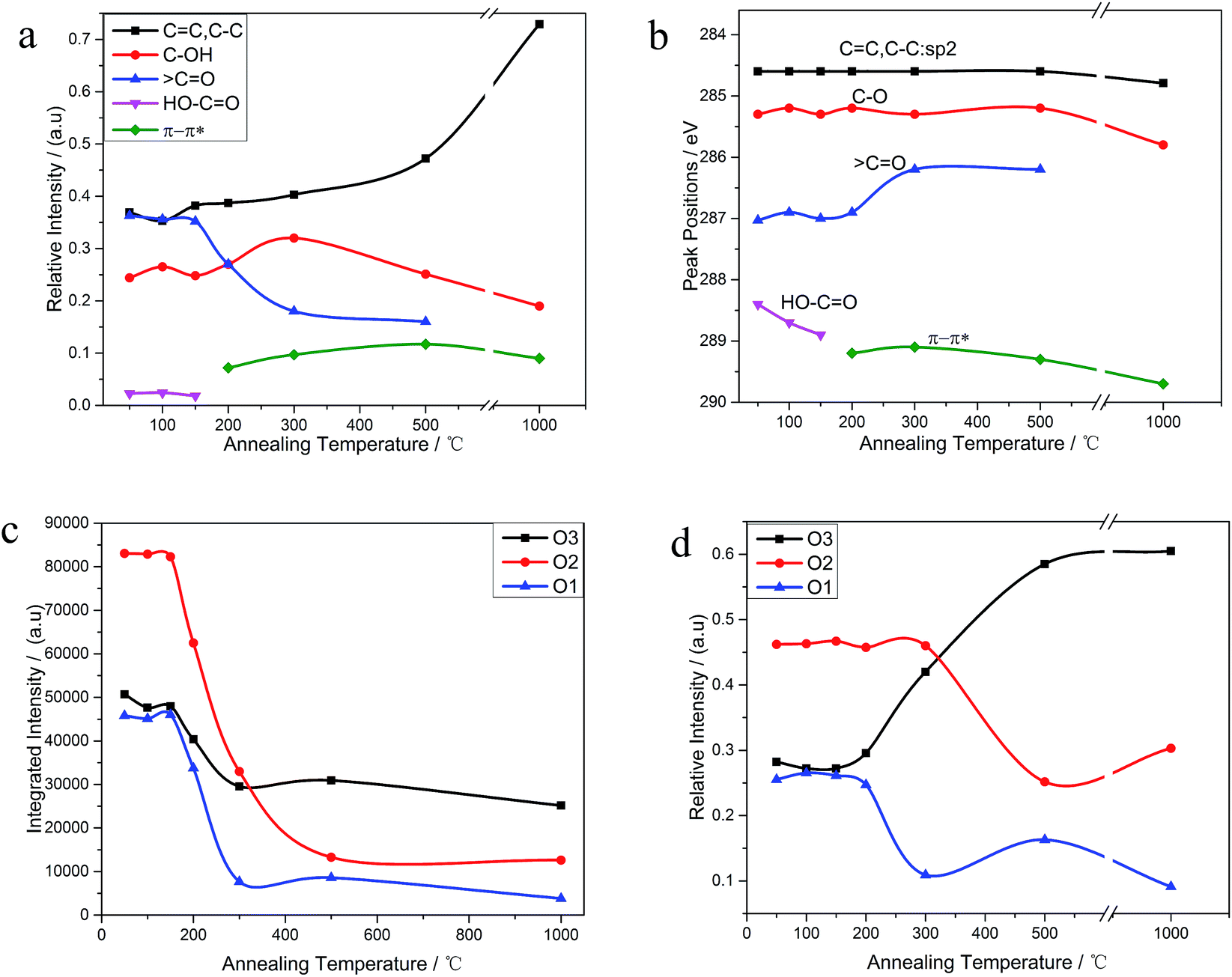 | ||
| Fig. 7 (a) The relative contribution of C 1s peak components estimated by dividing the area under each component; (b) temperature-dependence of the peak-position of C 1s peak components; (c) temperature-dependence of the integrated intensity of O 1s peak components; (d) the corresponding relative contribution estimated by dividing the area under each component by whole O 1s peak-area. | ||
In summary, the peak of the graphitic carbon sp2 component became largely dominant with the annealing temperature rising, along with the removal of main oxygen groups (including C–O–C, CO and HO–CO). However, the C 1s XPS spectrum of REGO-1000 °C still exhibits oxygen-containing groups. The nature of this persistent residual oxygen that last remains in REGO was confirmed to be C–O bond types by XPS spectra (Fig. 5h). During the overall process, the relative intensity of O3 (phenol group) sustained a general upward trend although the integrated intensity of all O-moieties decreased. Hence, it can be envisaged that the increase of relative intensity of C–O in C 1s peak was mainly contributed by newly-formed phenolic groups. Furthermore, the C–O staying in the REGO structure was confirmed to be extremely similar to that observed in the pristine graphite material according to their peak positions. Meanwhile, the intensities of remaining C–O were much smaller than those in parent EGO but a little more than those in pristine graphite. These features suggest that the thermally-stable O-species residues in the REGO might partly come from the built-in oxygen moieties of pristine graphite, and the rest thermally-stable C–O single bond was generated during the heat treatment process.
3.3 Discussion of results
On the basis of the above results, four stages can be proposed to describe the continuous thermal reduction process of EGO: (1) the dehydration stage, in which water intercalated or absorbed in the EGO sheets is volatilized between 30 °C and 120 °C, while the carbon framework of EGO remains thermally stable;21 (2) the stable stage, in which most of the O-species remain thermally stable in the temperature range of 120–160 °C, and there are no obvious physical and chemical changes during this period; (3) the formal reduction stage, in which the conjugated structure is restored due to the decomposition of the carboxyl and carbonyl groups in the temperature region 160–250 °C, and leading to the release of carbon oxide gases and the crystallization of oxidized graphitic structure accompanied with an exothermic process. Additionally, thermally super-stable C–O single bonded in the phenolic form is generated from the internal conversion of CO double bonded. In the meantime, complex carbon fragments, such as aliphatic, aromatic hydrocarbons and even some other carcinogenic organic compounds, are very likely released from the EGO during thermal degasification process as the prior literatures described;11,36,37 and (4) the rearrangement of carbon framework period, in which the remnant stubborn O-containing group involve in rearrangement or disproportionation reaction, and the sublimation of the carbon framework occurs simultaneously. At this period, the temperature generally remains above 260 °C. More complex inorganic molecules are generated, and permanent defects of carbon skeleton are created that include missing carbon atoms or stable O-containing species.38–41 A schematic illustration of the temperature evolution of EGO is shown in Fig. 8 in accordance with the results we described above.
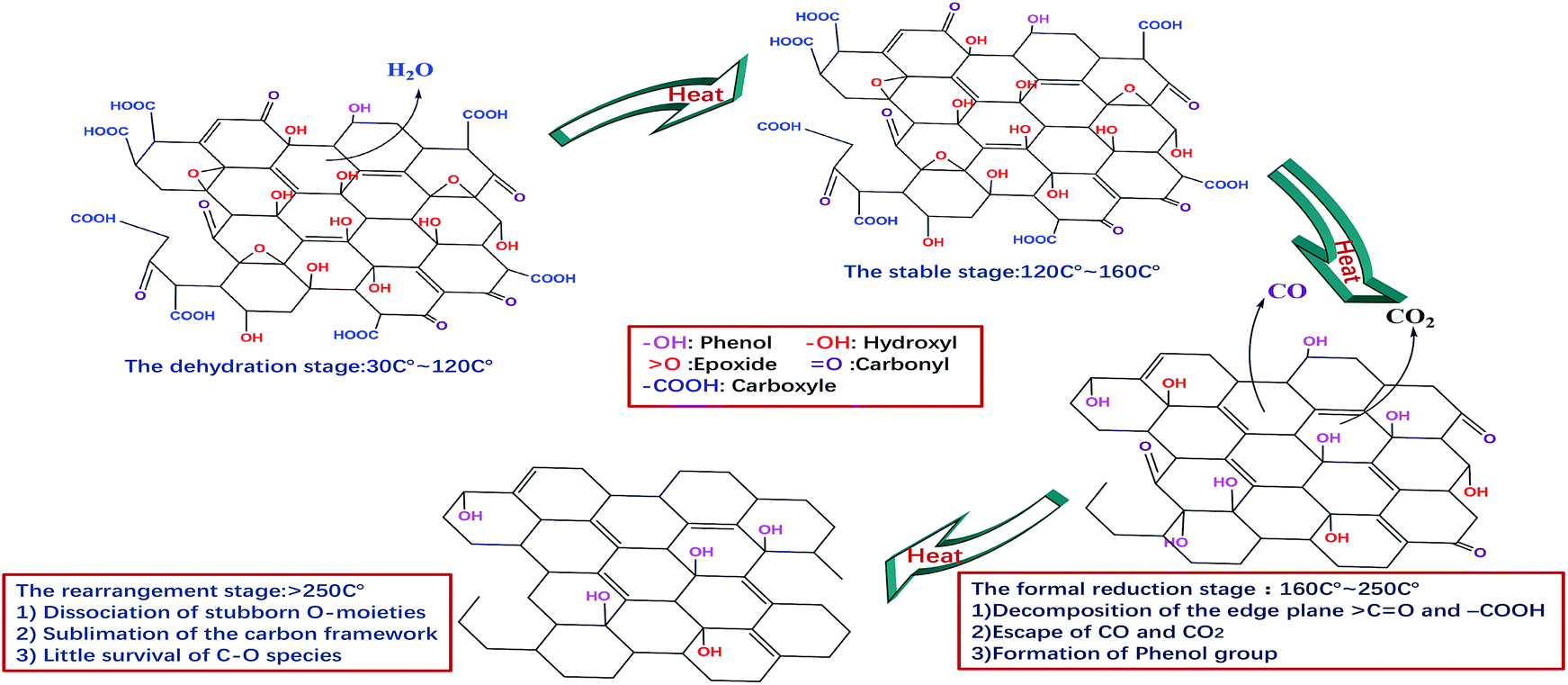 | ||
| Fig. 8 Schematic diagram of the temperature evolution of EGO. | ||
CO, COOH) will resolve. While under extreme heat, permanent defects are generated that include missing carbon atoms or stable oxygen-containing structures.39,42 Meanwhile, the generated carbon oxides and water vapor may react with the carbon atoms at high temperature, which causes more defects (vacancies) than the case at low temperature.9,10Apart from temperature, heating rate is regarded as another key factor in thermal reducing oxidized graphite. At low external heating rates, graphite oxide undergoes a smooth, non-explosive decomposition and yields products with low surface area and low degrees of exfoliation. Above a critical heating rate, the decomposition switches to the explosive mode and produces much higher internal porosity and larger surface area. So, it is significant to select a modest heat rate and annealing temperature.9,10,43
Fundamentally, the very nature of EGO primarily determines the internal disciplines of reduction. The activation energy of deoxygenation of EGO in our experiment is lower than that of GO prepared by traditional methods. This phenomenon is intimately linked to the degree of oxidation of graphite material and the relative content of oxygen functionalities. EGO exhibits a thermolabile behavior that is because thermally-unstable CO bond types are dominant among the oxygen functionalities of EGO. Thus, the minimum energy required to start the deoxygenation reaction is relatively small.
The thermal deoxygenation of oxidized graphite is analogous to the ‘elimination reaction’ in organic chemistry, in which the small molecules are detached from the parent macromolecular. And the mechanisms of thermal decarbonylation and decarboxylation have been investigated by several research groups.40 Based on the previous researches' experience, the process of detachment of CO from CO in aldehyde form can be described as follow:
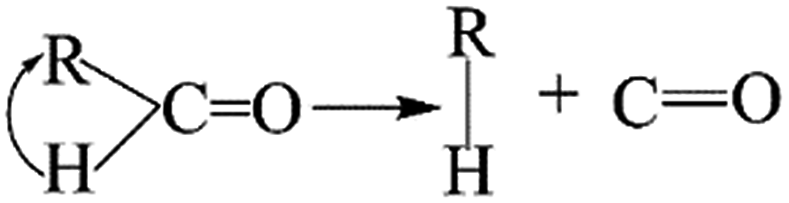
The elimination of CO2 from the carboxyl group can also follow the same route:40
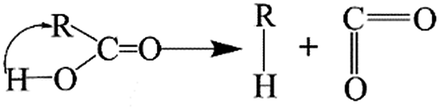
As described above, the H atom of the carboxyl group is attached to the nearest carbon atom of the radical R, with the separation of CO2 from the main parent carbon framework.
The removal of the carbon dioxide molecule from COOH group requires little energy, as the carboxylic compounds cannot take a coplanar geometry with the graphite sheet. That makes the carboxylic group become the most susceptible to thermal treatment. In certain situations, CO also can be detached due to the rupture of C–O of carboxyl group:
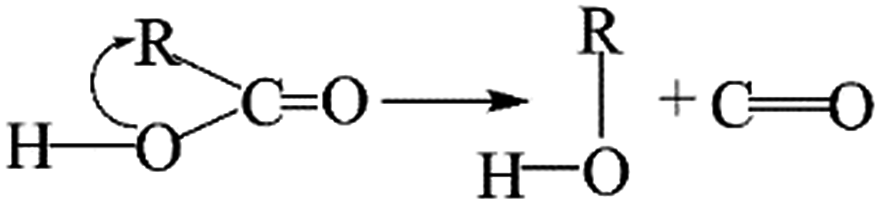
The cleavage of C–O makes the whole –OH transfer to the closest C atom of radical R with the separation of CO molecule from the parent groups. Consequently, the carboxylic was actually converted into hydroxyl functionality. This conversion of carboxylic to hydroxyl can be regarded as one of origins of the rise of C–O bond types between 200 °C and 300 °C. And, the C–OH functional groups can also be transformed from C–O–C groups. The existed C–H species in oxidized graphite transfer the H atom to the closest O atom of epoxy group and prompt the rupture of C–O, leading to the transformation of epoxy to hydroxyl. Besides, C–H interacts with the adjacent hydroxyl group via atom migration, eliciting the dehydration reaction.40
In addition, a high-temperature thermal annealing can directly eliminate CO from CO in ketone form through a one-step deoxygenation step:42
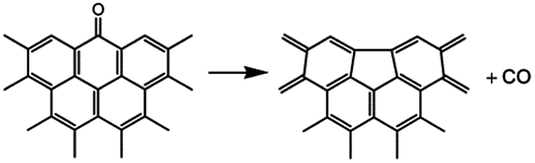
The dissociation of terminal carbonyl, carboxyl, and hydroxyl groups is feasible at low temperature (160–250 °C). However, the removal of basal plane groups requires more energy, and can only be achieved at high temperature. And simultaneously more defects will be induced into the carbon backbone of REGO.
For example, the basal plane hydroxyl can be thermally reduced in two steps process and yields a monoxide (CO) with a defective graphite sheet:42
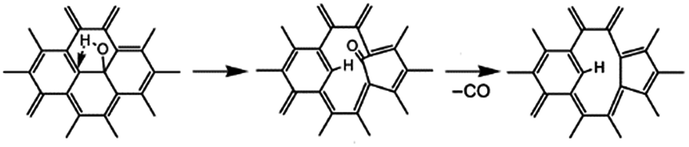
Interestingly, some research reported that hydroxyls linked to the inner aromatic domains of oxidized graphite can be expected to migrate to the edges of graphite sheets under the heat treatment and ultimately reduced at the edges domains.42
However, our XPS analysis illustrates the OH-moieties on basal plane that was identified as phenolic group were the most thermally stable species in our work. The temperature of 1000 °C is inadequate to remove all the C–O of the EGO. This indicates that there are high requirements for temperature to migrate and dissociate the basal plane C–O.
Nevertheless, the carbon framework becomes unstable and degrades at elevated temperatures.39 Meanwhile, other complex functionalities such as lactones, semiquinones, and ethers are manufactured with the evolution of volatile organic molecules.31,40 The separation of parent EGO molecules, which requires the presence of unsaturated C–C bonds involves a more complex series of reactions, and consequently causes vacancies and topological defects on the carbon backbone that have been thoroughly reported in the literatures.40–42
The above mechanism we proposed match well with our experimental data, and can preliminarily explain the evolution of structure of oxidized graphite and the observed emission of CO2 and CO during thermal process. However, the deep mechanisms of these reactions are muddled and need further exploration.
4. Conclusions
In conclusion, thermochemistry of annealing process of oxidized graphite and the evolution of electronic structure of EGO during thermal process is systematically researched. Using these results, we have clarified some important issues concerning the deoxygenation of EGO upon the thermal treatment.From the results above, it is apparent that the oxygen-containing functional groups of EGO are sensitive to thermal treatment. They will decompose upon heating and generate gases including H2O, CO2, and CO. Among these functionalities, the edge-plane COOH group is the most unstable one, and becomes the first to be removed. Additionally, the CO bond types decomposes with the annealing temperature rising, and thoroughly disappear at 1000 °C. As the heat-sensitive CO and COOH groups are dominant among the O-moieties of EGO, most of the oxygen species can be removed at a temperature lower than 300 °C.
However, there is a significant amount of oxygen species persists in the reduced products, and they can't be removed even at high temperatures. The thermoresistant groups that residue in the structure of reduced EGO is identified to be C–OH group. These types of species can be assured from two sources: the original O-species of pristine nuclear graphite, and the newly-formed C–O moieties in phenolic form during thermal annealing process. The good news is that the remnant oxygen species are stable enough to resist the effect of high-heat environment. Thus, it seems to be unnecessary to entirely eliminate these stubborn species as it can meet the requirements for further safety storage.
In addition, we synthetically analyze the continuous thermal process of EGO, and then expound the influence of temperature and heating rate on the reduction process, and lastly a plausible reduction mechanism has been discussed based on our detailed experimental data and the results from previous researches. However, the graphitic materials we used in these experiments hadn't been irradiated in the nuclear reactor. The chemical and physical properties of these materials are different from the irradiated graphitic material that generated from the decommissioning process of the HTGR. Thus, these results can only be essential references for optimizing the conditions for the large-scale reduction of the EGO. Because such reduced products from nuclear industry must be stored or processed discreetly under ambient conditions or even in extreme environments, the influences of irradiation effect on the carbon framework of nuclear graphitic material is highly important for the applications of thermal processing method and should be considered in future researches.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the National Science and Technology Major Project (2014ZX06901-016) and Program for Changjiang Scholars and Innovative Research Team in University (IRT13026) are gratefully acknowledged.Notes and references
- C. H. Tang, Y. P. Tang, J. G. Zhu, Y. W. Zou, J. H. Li and X. J. Ni, Nucl. Eng. Des., 2002, 218, 91–102 CrossRef CAS.
- S. Kazuhiro and U. Shohei, Nucl. Eng. Des., 2004, 233, 163–172 CrossRef.
- M. Masson, S. Grandjean, J. Lacquement, S. BourgJean, M. Delauzun and J. Lacombe, Nucl. Eng. Des., 2006, 236, 516–525 CrossRef CAS.
- M. IroIshihara, J. Sumita, T. Shibata, T. Iyoku and T. Oku, Nucl. Eng. Des., 2004, 233, 251–260 CrossRef.
- G. Y. Zhang, M. F. Wen, S. W. Wang, J. Chen and J. C. Wang, J. Appl. Electrochem., 2016, 46, 1163–1176 CrossRef CAS.
- L. F. Tian, M. F. Wen and J. Chen, Electrochim. Acta, 2010, 56, 985–989 CrossRef CAS.
- L. F. Tian, M. F. Wen and J. Chen, J. Nucl. Mater., 2013, 432, 113–119 CrossRef CAS.
- Y. Qiu, S. Moore, R. Hurt and I. Külaots, Carbon, 2017, 111, 651–657 CrossRef CAS PubMed.
- Y. Qiu, F. Collin, R. H. Hurt and I. Külaots, Carbon, 2016, 96, 20–28 CrossRef CAS PubMed.
- Y. Qiu, F. Guo, R. Hurt and I. Külaots, Carbon, 2014, 72, 215–223 CrossRef CAS PubMed.
- H. K. Jeong, Y. P. Lee, M. H. Jin, E. S. Kim, J. J. Bae and Y. H. Lee, Chem. Phys. Lett., 2009, 470, 255–258 CrossRef CAS.
- C. Zhang, W. Lv, X. Y. Xie, D. M. Tang, C. Liu and Q. H. Yang, Carbon, 2013, 62, 11–24 CrossRef CAS.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291–312 RSC.
- V. G. Plotnikov, V. A. Smirnov, M. V. Alfimov and Y. M. Shul'Ga, High Energy Chem., 2011, 45, 411–415 CrossRef CAS.
- V. A. Smirnov, Y. M. Shul Ga, N. N. Denisov, E. I. Kresova and N. Y. Shul Ga, Nanotechnol. Russ., 2012, 7, 81–86 Search PubMed.
- K. Yin, H. T. Li, Y. D. Xia, H. C. Bi, J. Sun, Z. G. Liu and L. Sun, Nano-Micro Lett., 2011, 3, 51–55 CrossRef CAS.
- H. R. Thomas, S. P. Day, W. E. Woodruff, C. Vallés, R. J. Young, I. A. Kinloch, G. W. Morley, J. V. Hanna, N. R. Wilson and J. P. Rourke, Chem. Mater., 2013, 25, 3580–3588 CrossRef CAS.
- W. Lv, D. M. Tang, Y. B. He, C. H. You, Z. Q. Shi, X. C. Chen, C. M. Chen, P. X. Hou, C. Liu and Q. H. Yang, ACS Nano, 2009, 3, 3730–3736 CrossRef CAS PubMed.
- M. Z. Cai, D. Thorpe, D. H. Adamsonb and A. H. C. Schniepp, J. Mater. Chem., 2012, 22, 24992–25002 RSC.
- C. Botas, P. Álvarez, C. Blanco, R. Santamaría, M. Granda, M. D. Gutiérrez, F. R. Reinoso and R. Menéndez, Carbon, 2013, 52, 476–485 CrossRef CAS.
- S. Eigler, C. Dotzer, A. Hirsch, M. Enzelberger and P. Müller, Chem. Mater., 2012, 24, 1276–1282 CrossRef CAS.
- W. K. Choi, B. J. Kim, E. Kim, S. H. Chi and S. J. Park, Nucl. Eng. Des., 2011, 241, 82–87 CrossRef CAS.
- E. S. Kim and H. C. No, J. Nucl. Mater., 2006, 349, 182–194 CrossRef CAS.
- L. Payne, P. J. Heard and T. B. Scott, PLoS One, 2015, 10, e143041 Search PubMed.
- A. Ganguly, S. Sharma, P. Papakonstantinou and J. Hamilton, J. Phys. Chem. C, 2011, 115, 17009–17019 CAS.
- A. Lerp, H. Y. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef.
- H. Y. He, T. Riedl, A. Lerf and J. Klinowski, J. Phys. Chem., 1996, 100, 19954–19958 CrossRef CAS.
- D. W. Boukhvalov and M. I. Katsnelson, J. Am. Chem. Soc., 2008, 130, 10697–10701 CrossRef CAS PubMed.
- L. B. Casabianca, M. A. Shaibat, W. W. Cai, S. J. Park, R. Piner, R. S. Ruoff and Y. Ishii, J. Am. Chem. Soc., 2010, 132, 5672–5676 CrossRef CAS PubMed.
- X. F. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2010, 114, 832–842 CAS.
- R. Larciprete, P. Lacovig, S. Gardonio, A. Baraldi and S. Lizzit, J. Phys. Chem. C, 2012, 116, 9900–9908 CAS.
- O. Akhavan, Carbon, 2010, 48, 509–519 CrossRef CAS.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS.
- A. Bagri, C. Mattevi, M. Acik, Y. J. Chabal, M. Chhowalla and V. B. Shenoy, Nat. Chem., 2010, 2, 581–587 CrossRef CAS PubMed.
- H. C. Schniepp, J. L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'Homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS PubMed.
- D. Zhan, Z. H. Ni, W. Chen, L. Sun, Z. Q. Luo, L. F. Lai, T. Yu, A. T. S. Wee and Z. Shen, Carbon, 2011, 49, 1362–1366 CrossRef CAS.
- A. Ambrosi, G. K. S. Wong, R. D. Webster, Z. Sofer and M. Pumera, Chem.–Eur. J., 2013, 19, 14446–14450 CrossRef CAS PubMed.
- Z. Sofer, P. Simek and M. Pumera, Phys. Chem. Chem. Phys., 2013, 15, 9257–9264 RSC.
- S. Eigler, S. Grimm and A. Hirsch, Chem.–Eur. J., 2014, 20, 984–989 CrossRef CAS PubMed.
- Z. Sofer, O. Jankovský, P. Šimek, D. Sedmidubský, J. Šturala, J. Kosina, R. Mikšová, A. Macková, M. Mikulics and M. Pumera, ACS Nano, 2015, 9, 5478–5485 CrossRef CAS PubMed.
- C. G. Navarro, J. C. Meyer, R. S. Sundaram, A. Chuvilin, S. Kurasch, M. Burghard, K. Kern and U. Kaiser, Nano Lett., 2010, 10, 1144–1148 CrossRef PubMed.
- X. F. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2010, 114, 832–842 CAS.
- X. J. Chen, D. L. Meng, B. Wang, B. W. Li, W. Li, C. W. Bielawski and R. S. Ruoff, Carbon, 2016, 101, 71–76 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11578d |
| This journal is © The Royal Society of Chemistry 2018 |