
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel insensitive energetic-cocrystal-based BTO with good comprehensive properties†
Jingjing Taoa,
Bo Jin *a,
Shijin Chua,
Rufang Peng*a,
Yu Shanga and
Bisheng Tanb
*a,
Shijin Chua,
Rufang Peng*a,
Yu Shanga and
Bisheng Tanb
aState Key Laboratory Cultivation Base for Nonmetal Composites and Functional Materials, Southwest University of Science and Technology, Mianyang 621010, China
bInstitute of Chemical Materials, Chinese Academy of Engineering Physics, Mianyang 621010, China
First published on 8th January 2018
Abstract
Combining a layer construction strategy with cocrystallization techniques, we designed and prepared a structurally unusual 1H,1′H-5,5′-bistetrazole-1,1′-diolate (BTO) based energetic cocrystal, which we also confirmed by single-crystal X-ray diffraction and powder-crystal X-ray diffraction. The obtained cocrystal crystallizes in a triclinic system, P-1 space group, with a density of 1.72 g cm−3. The properties including the thermal stability, sensitivity and detonation performance of the cocrystal were analyzed in detail. In addition, the thermal decomposition behavior of the cocrystal was studied by differential calorimetry and thermogravimetry tandem infrared spectroscopy. The results indicated that the cocrystal exhibits strong resistance to thermal decomposition up to 535.6 K. The cocrystal also demonstrates a sensitivity of >50 J. Moreover, its formation enthalpy was estimated to be 2312.0 kJ mol−1, whereas its detonation velocity and detonation pressure were predicted to be 8.213 km s−1 and 29.1 GPa, respectively, by applying K–J equations. Therefore, as expected, the obtained cocrystal shows a good comprehensive performance, which proves that a high degree of layer-by-layer stacking is essential for the structural density, thermal stability and sensitivity.
Introduction
The development of high-performance insensitive energetic materials has been of continued interest due to the increasing demands for military and civilian applications in the energetic materials field.1–3 Currently, the relationship between layer packing strategy and energetic materials with augmented density, decreased sensitivity to mechanical stimuli, and increased performance is paid much attention.4–6 The layer-by-layer assembly of energetic molecules greatly affects the physicochemical and detonation properties. To our best knowledge, such face-to-face arrangements are present in the crystal structure of some cocrystal explosives.7,8 Besides the rational design and screening of energetic backbones and building blocks, cocrystallization is a powerful method to improve the density and ameliorate sensitivity at the molecular level.9 The most common definition of cocrystallization is the generation of neutral complexes consisting of two or more components through non-covalent interactions, such as hydrogen bonding, π-stacking, and van der Waals forces.10 Meanwhile, hydrogen bonding has been reported as essential for almost all cocrystals.11–13 These two strategies illuminate the above-mentioned issue and effectively combine the layer packing strategy and cocrystallization to generate compounds with unique properties.In our design concept, rationally selecting H-bond donor–acceptor units is the key to forming a layer-by-layer structure in cocrystal molecule. 1H,1′H-5,5′-bistetrazole-1,1′-diolate (BTO) was selected as an H-acceptor group that includes electronegative atoms. This group is a nearly flat building block that features an excellent detonation performance, low production cost, environmental friendliness, and favorable thermal stability. However, the group also displays a comparative density and sensitivity because of the lack of H-bond donor and acceptor units in the crystal structure of some energetic-salts-based BTO. Therefore, the high crystal packing cannot be achieved, and interlayer sliding is prevented. Additionally, for most energetic compound, there is an inverse relationship between energy density and molecular stability. In the quest for energetic compound with better detonation behavior, there have been reports of CHON-based high explosives. Unfortunately, the majority of them suffer similar problems, for instance, low molecular stabilities, complex synthetic routes and poor resistance to hydrolysis. Since Thomas M. Klapötke reported on BTO salts, numerous ionic salts based on BTO salts have been developed as potential energetic explosives.14–17 Nevertheless, introducing chain cations or cyclic ring cations, including triazole, furazan, and tetrazole, decreases the density and sensitivity of title energetic salts, such as 1,3-diamino-1,2,3-triazolium 5,5′-bis(1-oxidotetrazolate) (ρ = 1.711 g cm−3, 2 J),18 1H,1′H-5,5′-bistetrazole-1,1′-diolate·2-(5-amino-2-methyl-2H-tetrazole) (ρ = 1.608 g cm−3, 8 J),19 and 1,3-diamino-1,2,3-triazolium nitrotetrazolate-2-oxide (ρ = 1.716 g cm−3, 3 J).18 Furthermore, density is a major factor affecting explosive performance, which limits the salt's practical application despite a very high nitrogen content reaching 66.7%, and even more importantly, the detonation pressure increases with the square of the density.
Consequently, we considered introducing another H2BTO neutral molecule in the imidazolium 5,5′-bis(1-oxidotetrazolate) (IMBTO) to tune the packing column and increase density via cocrystallization. The assumption was that the IMBTO was chosen as H-bond acceptor, and the H2BTO molecule provides both H-bond acceptor and H-bond donor units for pairing. The combination of the IMBTO and H2BTO molecules was expected to promote increased packing, augmented nitrogen content, enhanced energetic properties, and remarkable sensitivities.
Given the above-mentioned points as guidelines, we present the facile synthesis of a unique energetic cocrystal composed of H2BTO and IMBTO. As a curious-type energetic cocrystal, the cocrystal was fully characterized through X-ray diffraction and powder X-ray diffraction and proved the feasibility of our strategy. The expected cocrystal imidazolium BTO (CIMBTO) displayed a desirable crystal density of 1.720 g cm−3, which is significantly higher than that of the non-cocrystal IMBTO (1.588 g cm−3) recently published.20 Meanwhile, multinuclear nuclear magnetic resonance (NMR) spectroscopy, infrared (IR) spectroscopy, elemental analysis (EA), differential scanning calorimetry (DSC), and thermogravimetric analysis (TGA) were employed to characterize the prepared cocrystal. The resultant cocrystal also exhibited potentially decent thermal stability, good detonation properties, and extremely low sensitivity toward accidental stimuli; these characteristics highlight the cocrystal's potential energetic application.
Results and discussion
Synthesis
H2BTO was easily prepared in accordance with a previously reported procedure (Scheme 1).19 H2BTO was then synthesized from glyoxal, which was treated with hydroxylamine to form glyoxime. Glyoxime was then chlorinated with Cl2 in ethanol. Azido exchange occurred and yielded diazidoglyoxime, which was cyclized under acidic conditions (HCl gas in diethyl ether) to produce BTO. The metathesis reaction is analogous to that of non-cocrystal IMBTO.20 Thereafter, a cocrystallization experiment was conducted by dissolving the same equivalent of H2BTO and IMBTO in a minimum amount of distilled water, and the mixtures were stirred slowly to dissolve. Then, the solution was filtered and the solvent was evaporated over a period of several days at room temperature. A new colorless CIMBTO was then obtained.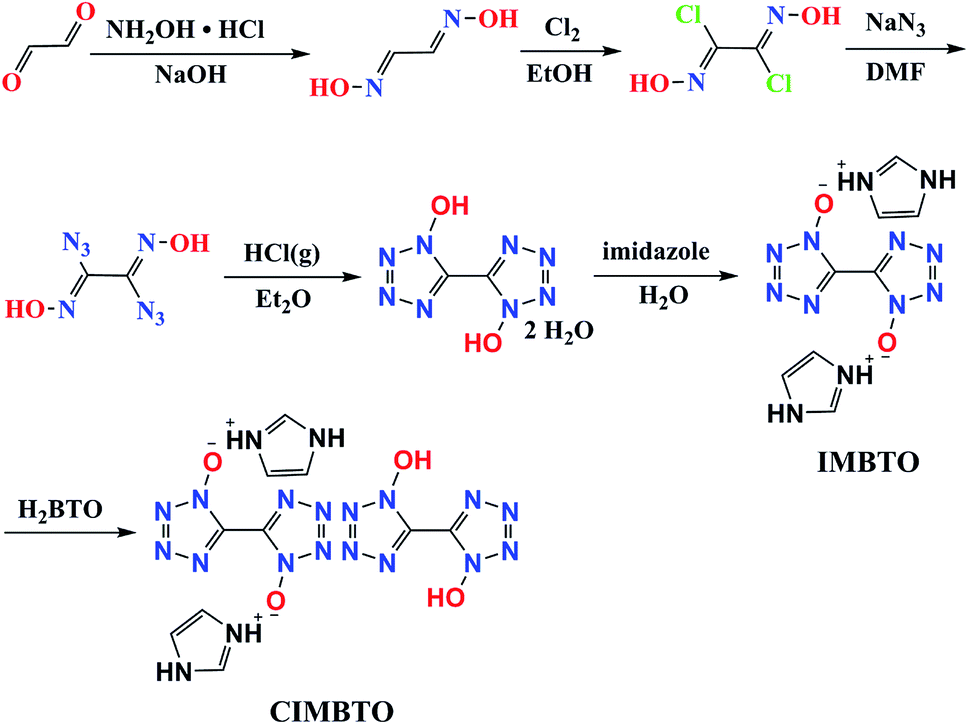 | ||
| Scheme 1 Synthesis route of CIMBTO. | ||
Crystal structure
A high-quality cocrystal was formed by slowly growing from distilled water at room temperature and then assessed by single-crystal X-ray analysis. The results concerning crystallographic data collection and structural refinement are summarized in Table 1, and the molecular moiety of the CIMBTO is displayed in Fig. 1. The selected bond lengths and angles are given in Table 2, and the hydrogen bond lengths and bond angles of CIMBTO are listed in Table 3.| Parameter | CIMBTO |
|---|---|
| a GOF = Goodness of Fit.b R1 = ∑||FO − |FC||/∑||FO.c ωR2 = [(ω(FO2 − FC2)2/ω(FO2)2)]1/2. | |
| Empirical formula | C10H12N20O4 |
| Formula weight | 476.40 |
| CCDC number | 1487781 |
| Crystal size [mm3] | 0.19 × 0.17 × 0.16 |
| Crystal system | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a [Å] | 3.6752(10) |
| b [Å] | 9.158(2) |
| c [Å] | 13.799(4) |
| α[°] | 92.786(3) |
| β[°] | 97.372(3) |
| γ[°] | 90.753(3) |
| V [Å3] | 460.0(2) |
| Z | 1 |
| ρcalcd [g cm−3] | 1.720 |
| T [K] | 150(2) |
| F(000) | 244 |
| Rint. | 0.0505 |
| Data | 1616 |
| Restraints | 1 |
| Parameters | 155 |
| GOFa on F2 | 1.046 |
| R1b (I > 2σ (I)) | 0.0449 |
| ωR2 (I > 2σ (I)) | 0.1209 |
| R1 (all data) | 0.0478 |
| ωR2c (all data) | 0.1235 |
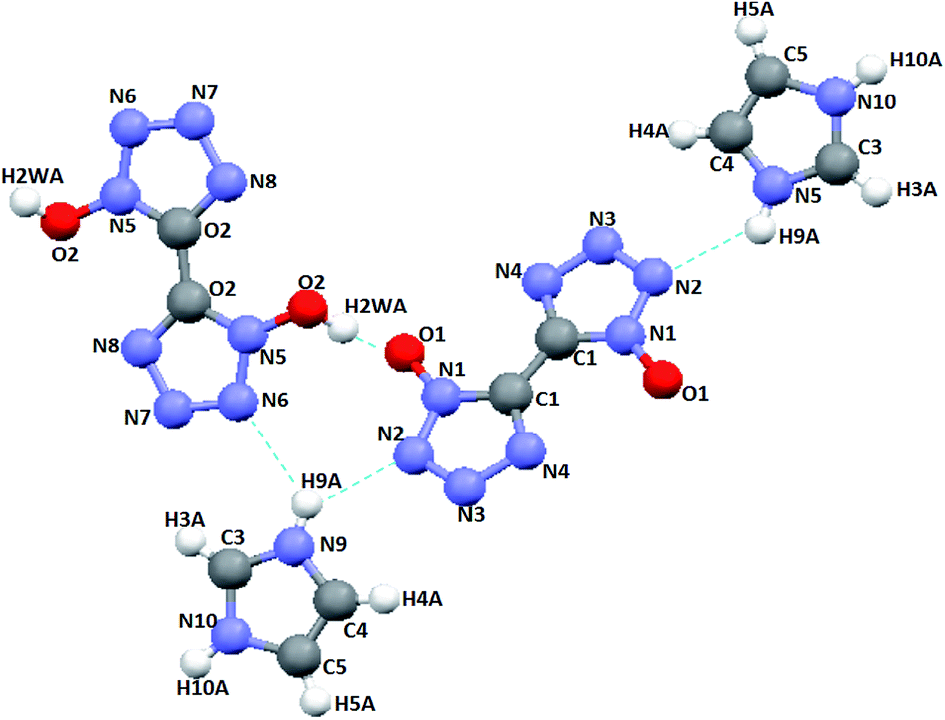 | ||
| Fig. 1 Molecular structure of CIMBTO. | ||
| Bond | Length/Å | Bond | Angle/° |
|---|---|---|---|
| N(1)–N(2) | 1.329(3) | N(2)–N(1)–O(1) | 121.94(16) |
| N(1)–O(1) | 1.331(2) | N(2)–N(1)–C(1) | 109.24(16) |
| N(2)–N(3) | 1.296(3) | N(3)–N(2)–N(1) | 106.12(16) |
| N(3)–N(4) | 1.345(3) | N(2)–N(3)–N(4) | 110.94(18) |
| N(5)–O(2) | 1.327(2) | O(2)–N(5)–C(2) | 128.36(17) |
| N(5)–N(6) | 1.333(3) | C(2)–N(5)–N(6) | 109.14(16) |
| N(6)–N(7) | 1.299(3) | N(7)–N(6)–N(5) | 105.93(16) |
| N(7)–N(8) | 1.342(2) | N(6)–N(7)–N(8) | 110.93(18) |
| D–H | d(D–H) | d(H⋯A) | <DHA | d(D⋯A) | A |
|---|---|---|---|---|---|
| a [−x + 1, −y + 2, −z + 1].b [−x, −y + 1, −z + 1]. | |||||
| N9–H9Aa | 0.880 | 2.390 | 129.44 | 3.026 | N2 |
| N9–H9Aa | 0.880 | 2.395 | 131.01 | 3.046 | N6 |
| N10–H10A | 0.880 | 2.241 | 139.21 | 2.964 | N8 |
| N10–H10Ab | 0.880 | 2.436 | 142.63 | 3.180 | O2 |
| O2–H2WA | 0.873 | 1.555 | 170.12 | 2.420 | O1 |
| O2–H2WA | 0.873 | 2.314 | 155.73 | 3.130 | N1 |
As expected, the test result is consistent with our previous design idea. The developed cocrystal crystallizes in a triclinic system, P space group, with a density of 1.72 g cm−3 at 150 K, which is fairly lower than that of H2BTO (1.811 g cm−3) but much superior to that of imidazole (1.030 g cm−3). More importantly, the cocrystal is also much superior to the non-cocrystal IMBTO (1.588 g cm−3) as recently published.20 The graphite-like structure of CIMBTO in the solid state was the same as that of the predicted mode and highly contributed to the high density in Fig. 3. By contrast, the favorable density evidently revealed that the cocrystal was certainly a new substance and was partially attributed to the good detonation properties. Additionally, in this work, the energetic cocrystal was formed by combining an energetic salt with a neutral co-former, which is different from many of other cocrystals. It is noted that the imidazole salt of BTO was first prepared, where proton transfer has occurred. Afterward, the salt was co-crystallized with H2BTO. This would mean that the proton transfer happened before the cocrystallization. Analogously, the similar example can be found in the U.S. patent which present a novel cocrystal composed of HMX and ammonium perchlorate.21
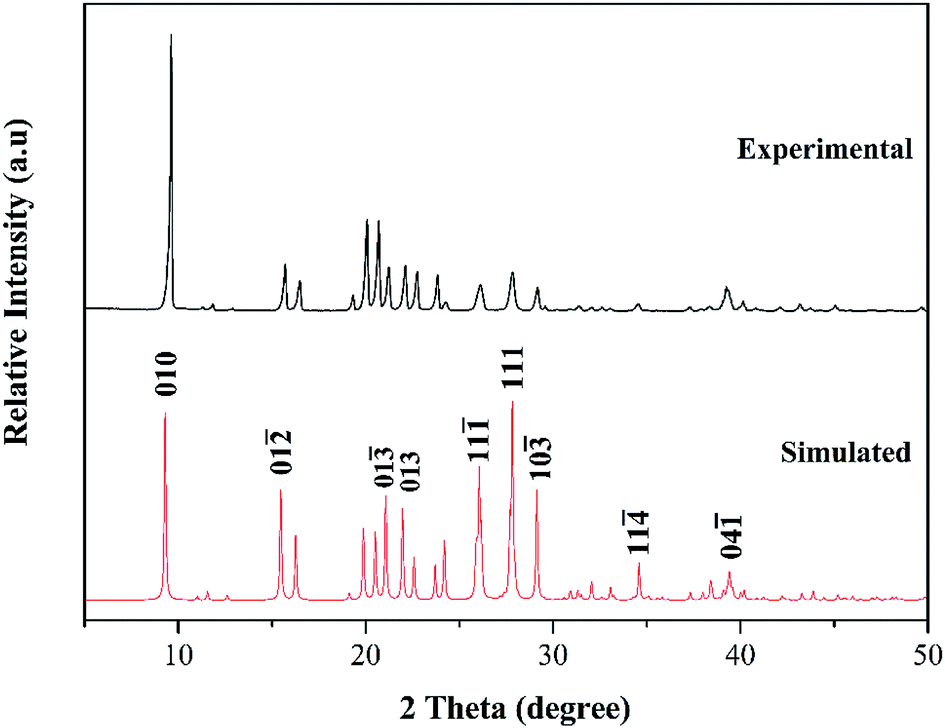
Additionally, the phase purity of the cocrystal was verified by performing the powder X-ray diffraction experiment, and then compare it with the simulated data from the SC-XRD. As shown in Fig. 4, the powder X-ray diffraction (PXRD) pattern of the as-prepared cocrystal is in agreement with the numerical calculation results converted from the single crystal data, which identifies the cocrystal as a new material with high phase purity. The characteristic peaks are at 9.66°, 26.28°, and 28.17°, which can be attributed to the (010), (11), and (111) respectively. Due to the simulated growth conditions and test conditions of the single crystal are different from the experimental one, variance was found to some degree in the intensity.
To verify our assumption further, we studied the packing coefficient, which was calculated as the ratio of the total molecular volume to the unit cell volume, where 73.64% for CIMBTO was closer to the value for H2BTO itself (74.35%) and higher than that of IMBTO (71.53%).20 This result signifies that the volume and amount of the internal cavities were largely reduced in the cocrystal and that the cocrystal was a new compound.
In the tetrazolate of BTO anion (Table 2), the N–N bond lengths varied from 1.296(3) Å for N2–N3 to 1.345(3) Å for N3–N4. These lengths were between those of the N–N single bonds (1.454 Å) and N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds (1.245 Å).22 However, the C–C bond connecting the two tetrazole moieties was 1.446 Å long. Such length was located within the normal range of 1.32 Å for the CC bond and 1.53 Å for the C–C bond.23 Additionally, the N–O bonds are found between a N–O single bond and a NO double bond (d(O001–N001) = 1.331(2) Å).24 These observations indicate the existence of a multiple bond and the conjugation of the negative charge throughout the aromatic rings caused by the presence of oxygen atoms. All atoms of the BTO anion were almost coplanar, and the largest torsional angle was 2.58(0.36)°, which was located at O001–N001–C001–C001.
N double bonds (1.245 Å).22 However, the C–C bond connecting the two tetrazole moieties was 1.446 Å long. Such length was located within the normal range of 1.32 Å for the CC bond and 1.53 Å for the C–C bond.23 Additionally, the N–O bonds are found between a N–O single bond and a NO double bond (d(O001–N001) = 1.331(2) Å).24 These observations indicate the existence of a multiple bond and the conjugation of the negative charge throughout the aromatic rings caused by the presence of oxygen atoms. All atoms of the BTO anion were almost coplanar, and the largest torsional angle was 2.58(0.36)°, which was located at O001–N001–C001–C001.
A series of strong intermolecular N–H⋯N and O–H⋯O hydrogen bonds formed (Table 3 and Fig. 2). The BTO anion was a H-bond acceptor, and the imidazole cation was a H-bond donor. However, the H2BTO molecule was a donor and an acceptor of the hydrogen bonds. The packing structure was held not only by electrostatic attraction through cations and anions but also by hydrogen bonds and π–π interactions. Strong hydrogen bonds were formed in the crystal structure of CIMBTO (N10–H10A⋯N008 [2.964 Å], N9–H9A⋯N2 [3.026 Å], N4–H4⋯N2 [2.433 Å], N10–H10A⋯O2 [3.180 Å]) (Fig. 1). Its packing structure of CIMBTO was built and linked to a 2D layer by various hydrogen bonds. Apart from the hydrogen bonds, the π-stacking interactions were also significant in the cocrystal packing. The structures of CIMBTO and imidazolium BTO salt along the b axis is depicted in Fig. 3. After the layer was assembled, further interlinking of the 2D layers into an intricate 3D supermolecule was stimulated by hydrogen bonding and π stacking. Remarkably, these 2D layers in the b axis direction were arranged in a face-to-face arrangement horizontally. As such, the arrangement led to a decent thermal stability and insensitivity.
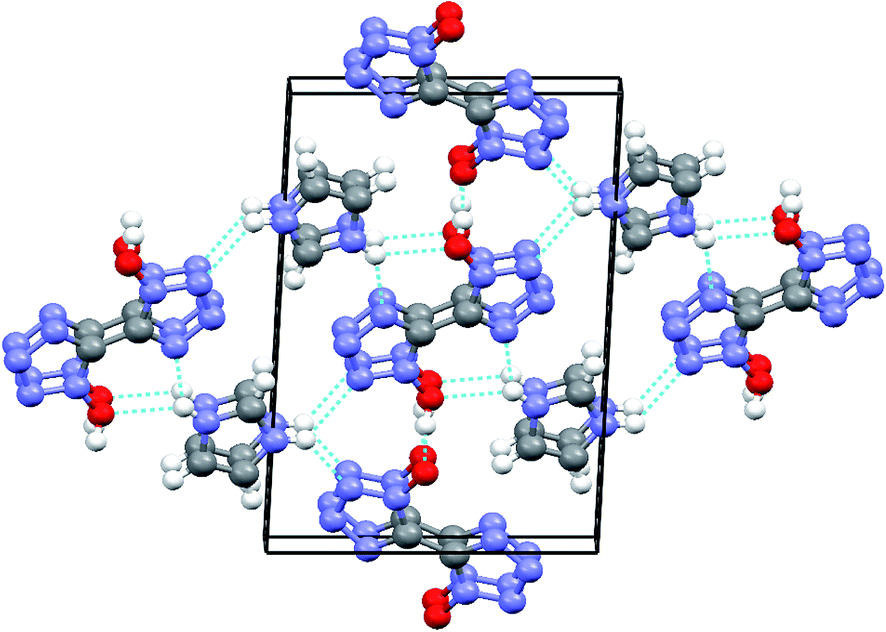 | ||
| Fig. 2 Packing diagram of CIMBTO along the a axis. | ||
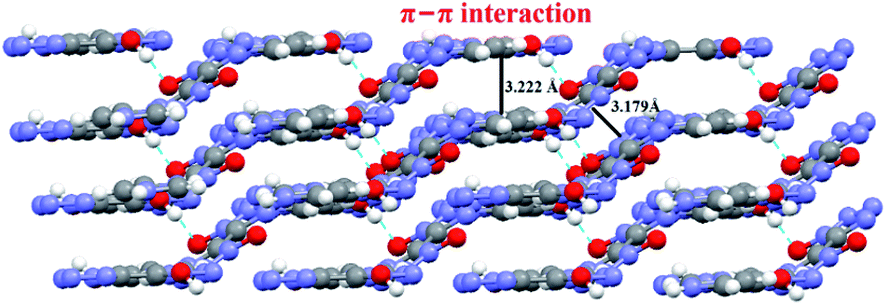 | ||
| Fig. 3 Packing diagram of CIMBTO along the b axis. | ||
 | ||
| Fig. 4 PXRD patterns of CIMBTO and simulated data from SC-XRD. | ||
Spectroscopy
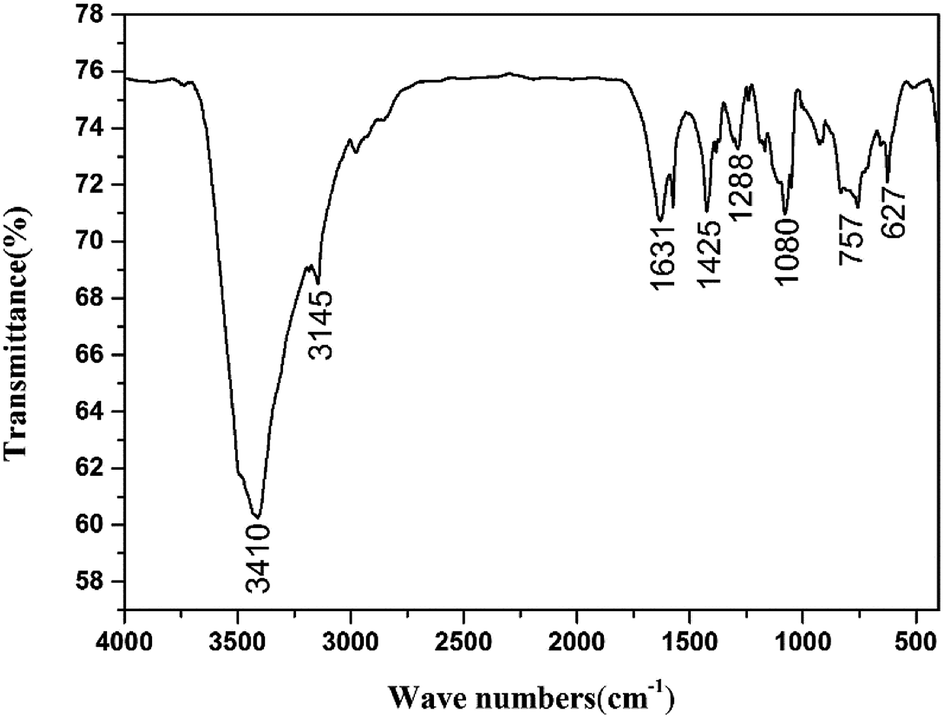
The structure of CIMBTO was supported further by its corresponding FT-IR, 1H NMR, 13C NMR, and elemental analysis (Experimental section). As shown, in the FT-IR spectrum (Fig. 5), several sharp characteristic bands at 1631, 1425, and 1288 cm−1 were ascribed to the stretching vibration absorption peaks of CN, NN, and N–O, respectively.19 The intense broad band at 3410 cm−1 was attributed to the N–H stretching vibrations of CIMBTO, and the peak at 3145 cm−1 corresponded to the stretching vibrations of C–H.25 In addition, the peak at 757 cm−1 was caused by the out-plane flexural vibration absorption of C–H.25 Consequently, the FT-IR spectrum was consistent with the structural features of the compound.
 | ||
| Fig. 5 FT-IR spectrum of CIMBTO. | ||
The 1H NMR and 13C NMR spectra also verified the correct structure of CIMBTO. In the 1H NMR spectrum, the signal for the amino group of salt occurred as a broad single band at δ/ppm = 9.09 ppm [s, CH, NH+] and δ/ppm = 7.70 [s, CHCH–NH+].25 Three signals in the 13C NMR [DMSO-d6] spectrum of the cocrystal were found at δ/ppm = 134.86 [CNH+], 119.77 [CH–NH, CH–NH+], and 135.62 [CN4O], respectively.19,25
Energetic performance
HOF is an important parameter that represents the energy content of an energetic material. Positive values correspond to enhanced detonation properties. The computations were performed with the Gaussian 09 program.26Isodesmic reactions were used to obtain the formation enthalpy of CIMBTO at 298 K. Thus, the empirical Kamlet–Jacobs equations27 were employed to estimate the values of detonation velocity (D) and detonation pressure (P) for CIMBTO, as shown in the following equations:
D = 1.01(N![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) 1/2Q1/2)1/2(1 + 1.3ρ) 1/2Q1/2)1/2(1 + 1.3ρ)
| (1) |
|
P = 1.558ρ2N1/2Q1/2
| (2) |
is the gaseous product of the average molecular weight (g mol−1), Q is the explosive detonation chemical energy per gram (kJ g−1), and ρ is the density (g cm−3). Q should be calculated before D and P are determined. Moreover, Q is also attained by the ΔHf of the detonation reactant and product.
Given the theoretical HOFs and the experimental room temperature density, the detonation properties for IMBTO were predicted by K–J equations27 (Table 4). CIMBTO showed a positive HOF of 2312.0 kJ mol−1, which was higher than those of HMX (104.8 kJ mol−1), TNT (95.3 kJ mol−1), and RDX (83.8 kJ mol−1) (Table 4).
| Sample | Tdeca [K] | ρb [g cm−3] | N + Oc [%] | ΔfHd [kJ mol−1] | Pe [GPa] | Df [m s−1] | ISg [J] |
|---|---|---|---|---|---|---|---|
| a Decomposition peak temperature.b Calculated density.c Nitrogen and oxygen content.d Calculated molar formation enthalpy of the salts.e Detonation pressure.f Detonation velocity.g Impact sensitivity.h Ref. 28.i Ref. 29. | |||||||
| CIMBTO | 535.6 | 1.720 | 72.27 | 2312.0 | 29.1 | 8213 | >50 |
| IMBTO | 545.9 | 1.588 | 65.36 | 717.6 | 19.0 | 6812 | >40 |
| TNTh | 568.2 | 1.648 | 18.50 | 95.3 | 19.5 | 6881 | 15 |
| RDXh | 503.2 | 1.806 | 37.84 | 83.8 | 34.9 | 8748 | 7.5 |
| TATBi | 597.2 | 1.930 | 69.74 | −139.7 | 31.15 | 8114 | 50 |
The performance characteristics are presented in Table 4. As shown, the theoretically computed detonation velocity (D) and detonation pressure (P) were 8213 m s−1 and 23 GPa, respectively, which were superior to those of TNT. By contrast, the detonation velocity was roughly at the same level as that of RDX, but the cocrystal was more insensitive than the so-called wood explosive TATB. These properties demonstrated that the cocrystal may be used as an insensitive and nitrogen-rich energetic material.
Considering our cocrystal's practical application, the impact sensitivity was determined using a standard BAM Fall hammer apparatus with a 10 kg drop weight. Fortunately, the attained impact sensitivity of >50 J fulfilled our expectations (Table 4). The result shows the low sensitivity of the cocrystal to external stimuli because of a special molecular stacking, including the extensive intermolecular interactions and graphite-like stacking. The majority of explosives, such as TATB, LLM-105, and FOX-7, involve graphite-like stacking, which allows interlayer sliding within a crystal lattice and contributes to low sensitivity.30 The cocrystal exhibited a lower sensitivity to external stimuli than those in the traditional energetic materials of RDX (7.4 J), HMX (7 J), and TNT (15 J).31
Thermal behavior
Thermal stability is a crucial factor in energetic materials because an unacceptable thermal stability considerably restricts these materials' applications. DSC and TG-DTG were employed with a linear heating rate of 10 K min−1 under flowing high-purity nitrogen to determine the thermal decomposition behaviors of the cocrystal. As shown by the DSC curve (Fig. 6), the cocrystal melted at 531.5 K similar to that in RDX. Such temperature is significantly higher than that of pure H2BTO (372.95 K) and implies that the cocrystallization is effective in increasing the melting point of low-melting-point explosives to broaden their applications.32 After the melting process was finished, the cocrystal soon exhibited an intense endothermic process with a peak temperature of 535.6 K, which was higher than that of RDX (503.15 K). This temperature level indicates that CIMBTO is a promising energetic material with an excellent thermal stability.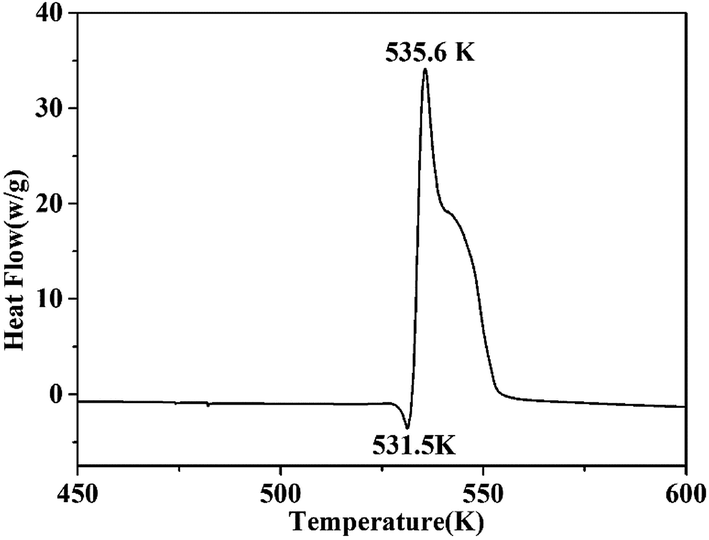 | ||
| Fig. 6 DSC curve of CIMBTO at the heating rate of 10 K min−1. | ||
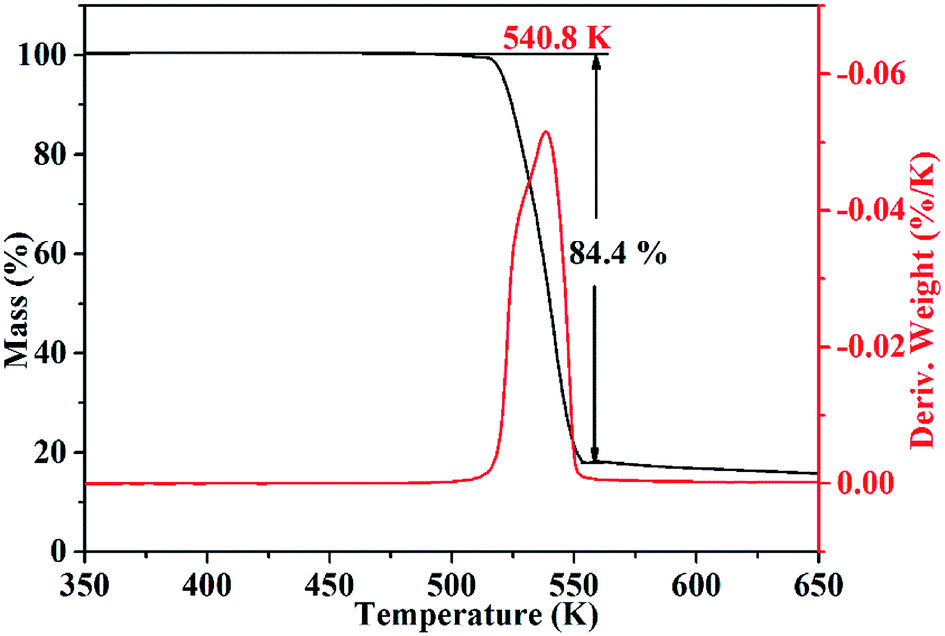
Corresponding to the decomposition processes of the DSC curve, the TG–DTG curves (Fig. 7) show a severe weight loss step of 84.4% in the range of 509–554 K, and the main exothermic temperature range of CIMBTO was consistent with that of the weight loss. The decomposition behavior of the cocrystal may be explained by the destruction of the crystal structure with rising temperature.
 | ||
| Fig. 7 TG and DTG curves of IMBTO. | ||
To gain further insight, we also investigated TGA tandem IR spectrum to rapidly identify the constituents of the thermal decomposition gas. Fig. 8 depicts the FT-IR spectra of the thermal decomposition gas during decomposition at their different temperatures. The figure displays that the thermal decomposition of CIMBTO also mainly includes a single stage, which is consistent with the TG–DTG results. The intense decomposition occurred at approximately 540.8 K. From Fig. 8, we deduced that the decomposition products were mainly CO2 (2360 cm−1) and N2O (2192–2330 cm−1 and 748 cm−1), NH3 (3456, 930, and 965 cm−1), and NO (1786 cm−1).33,34 When the heating temperature was increased to 598.6 K, IR signals nearly disappeared, which proved that CIMBTO was completely decomposed.
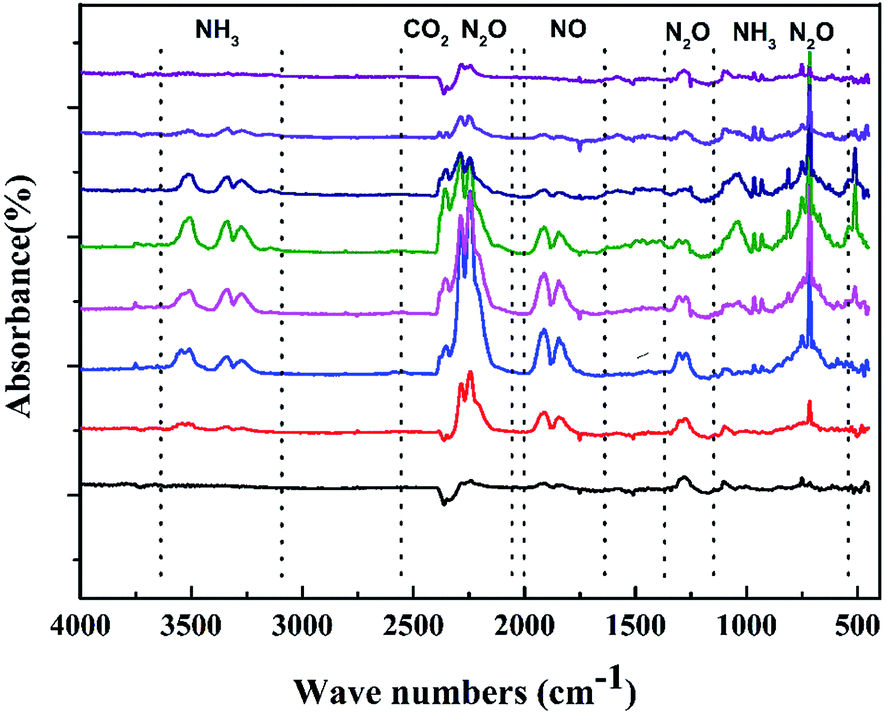 | ||
| Fig. 8 FT-IR spectra of gas products of CIMBTO during decomposition at individual temperatures. | ||
Conclusions
An unusual nitrogen-rich energetic cocrystal was discovered and fully characterized through multinuclear NMR spectroscopy, IR spectroscopy, EA, DSC, and TGA. The structure of CIMBTO was further confirmed through single-crystal X-ray diffraction and powder X-ray diffraction. DSC results revealed that CIMBTO possesses an excellent thermal stability at a decomposition temperature of 535.6 K and an extremely high melting point of 531.5 K. CIMBTO is insensitive to impact (>50 J) and can thus be safely handled and applied to specific areas. The superior sensitivity and stability may be explained by the strong hydrogen bond network and graphite-like stacking. Furthermore, the new energetic compound was calculated to yield a detonation pressure of 29.1 GPa and velocity of 8213 m s−1. Given its reasonable detonation property, excellent impact sensitivity, and decent stability, CIMBTO can be used as a potential insensitive energetic material for commercial applications.Experimental section
Safety precautions!
Although we experienced no difficulties in the synthesis and characterization of CIMBTO, small-scale syntheses is strongly encouraged. All of the compounds should be handled with extreme care, and eye protection and gloves must be worn at all times.General methods
Chemical reagents and solvents were purchased from Aladdin (Shanghai, China) and used as supplied without further purification. 1H NMR and 13C NMR spectra were obtained using a JEOL GSX 600 MHz nuclear magnetic resonance (NMR) spectrometer in a d6-DMSO solution by using tetramethylsilane as internal standard. Fourier transform infrared (FT-IR) spectra were recorded on a Nicolet-5700 FTIR spectrometer using pressed KBr pellets in the wavelength range of 4000 cm−1 to 400 cm−1. Elemental analyses (C, H, and N) were conducted using a Vario EL CUBE device. DSC was performed by a Q200 DSC instrument (TA Instruments, United States) at a heating rate of 10 K min−1 under flowing high-purity nitrogen. TGA was performed with an SDT Q600 TGA instrument (TA Instruments, United States) at a heating rate of 10 K min−1 under flowing high-purity nitrogen.Synthesis of 1H,1′H-5,5′-bistetrazole-1,1′-diolate dihydrate (H2BTO)
1H,1′H-5,5′-bistetrazole-1,1′-diolate dihydrate was synthesized according to a procedure reported in the literature.19 H2BTO·2H2O was synthesized starting from glyoxal, which was treated with hydroxylamine to form glyoxime. Glyoxime was then chlorinated with Cl2 gas in ethanol; chloro/azido exchange occurred to yield diazidoglyoxime, which was cyclized under acidic conditions (HCl gas in diethyl ether) to produce H2BTO.Synthesis of bis-imidazolium 1H,1′H-5,5′-bistetrazole-1,1′-diolate (IMBTO)
1H,1′H-5,5′-bistetrazole-1,1′-diolate dihydrate (206 mg, 1 mmol) was dissolved in 3 mL distilled water, and then imidazole (136.2 mg, 2 mmol), previously dissolved in 4 mL water, was added. The mixture was briefly heated to reflux and filtered. After cooling to room temperature, IMBTO crystallized as colorless needles, yield 233 mg (0.76 mmol, 76%). IR (KBr) v/cm−1: 3468, 3184, 3151, 3085, 1427, 1234, 1163, 1097, 927, 747, 627. 1H NMR (600 MHz, [D6]DMSO, 25 °C) δ/ppm: 8.62, 7.37. 13C NMR (150 MHz, [D6]DMSO, 25 °C) δ/ppm: 162.17, 132.18, 121.46, 114.55. Elemental analysis (%) calcd for C8H10N12O2 (306.20): C, 31.38; H, 3.29; N, 54.88; found: C, 31.30; H, 3.32; N, 54.79.Synthesis of the cocrystal CIMBTO
After confirmed the structure of IMBTO, the same equivalent of H2BTO (156.56 mg, 0.76 mmol) and IMBTO (233 mg, 0.76 mmol) was dissolved in a minimum amount of distilled water, and the mixtures were stirred slowly to dissolve. Then, the solution was filtered and the solvent was evaporated over a period of several days at room temperature. A new colorless CIMBTO was then obtained, yield of 267 mg (0.56 mmol, 74%). IR (KBr) v/cm−1: 3410, 3145, 1631, 1574, 1425, 1288, 1169, 1080, 927, 757, 627. 1H NMR (600 MHz, [D6]DMSO, 25 °C) δ/ppm: 9.09, 7.70. 13C NMR (150 MHz, [D6]DMSO, 25 °C) δ/ppm: 135.62, 134.86, 119.77. Elemental analysis (%) calcd for C8H10N12O2 (306.20): C, 25.22; H, 2.54; N, 58.81; found: C, 25.10; H, 2.56; N, 58.90.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (project no. 51372211) and Open Project of State Key Laboratory Cultivation Base for Nonmetal Composites and Functional (project no. 14tdfk05). Moreover, we also gratefully acknowledge the support of China Academy of Engineering Physics Research Institute of Safety and Ammunition Research and Development Center (project no. RMC2015B01) and the Institute of Chemical Materials, China Academy of Engineering Physics (project no. 16zh0101).References
- D. Fischer, T. M. Klapotke and J. Stierstorfer, Angew. Chem., 2015, 54, 10299–10302 CrossRef CAS PubMed.
- A. A. Dippold and T. M. Klapotke, J. Am. Chem. Soc., 2013, 135, 9931–9938 CrossRef CAS PubMed.
- A. A. Gidaspov, V. V. Bakharev, K. Y. Suponitsky, V. G. Nikitin and A. B. Sheremetev, RSC Adv., 2016, 6, 104325–104329 RSC.
- W. Zhang, J. Zhang, M. Deng, X. Qi, F. Nie and Q. Zhang, Nat. Commun., 2017, 8, 181 CrossRef PubMed.
- J. Zhang, Q. Zhang, T. T. Vo, D. A. Parrish and J. M. Shreeve, J. Am. Chem. Soc., 2015, 137, 1697–1704 CrossRef CAS PubMed.
- J. Zhang, L. A. Mitchell, D. A. Parrish and J. M. Shreeve, J. Am. Chem. Soc., 2015, 137, 10532–10535 CrossRef CAS PubMed.
- Y. Wang, Z. Yang, H. Li, X. Zhou, Q. Zhang, J. Wang and Y. Liu, Propellants, Explos., Pyrotech., 2014, 39, 590–596 CrossRef CAS.
- Z.-B. Zhang, T. Li, L. Yin, X. Yin and J.-G. Zhang, RSC Adv., 2016, 6, 76075–76083 RSC.
- O. Bolton and A. J. Matzger, Angew. Chem., 2011, 50, 8960–8963 CrossRef CAS PubMed.
- C. B. Aakeröy and D. J. Salmon, CrystEngComm, 2005, 7, 439 RSC.
- O. Bolton, L. R. Simke, P. F. Pagoria and A. J. Matzger, Cryst. Growth Des., 2012, 12, 4311–4314 CAS.
- J.-T. Wu, J.-G. Zhang, T. Li, Z.-M. Li and T.-L. Zhang, RSC Adv., 2015, 5, 28354–28359 RSC.
- J. Zhang, D. A. Parrish and J. M. Shreeve, Chem. Commun., 2015, 51, 7337–7340 RSC.
- N. Fischer, D. Fischer, T. M. Klapötke, D. G. Piercey and J. Stierstorfer, J. Mater. Chem., 2012, 22, 20418 RSC.
- T. M. Klapötke, P. C. Schmid, S. Schnell and J. Stierstorfer, J. Mater. Chem. A, 2015, 3, 2658–2668 Search PubMed.
- Y. Shang, B. Jin, R. Peng, Z. Guo, Q. Liu, J. Zhao and Q. Zhang, RSC Adv., 2016, 6, 48590–48598 RSC.
- Z. B. Zhang, C. X. Xu, X. Yin and J. G. Zhang, Dalton Trans., 2016, 45, 19045–19052 RSC.
- T. M. Klapötke, D. G. Piercey and J. Stierstorfer, Eur. J. Inorg. Chem., 2013, 2013, 1509–1517 CrossRef.
- N. Fischer, T. M. Klapötke, M. Reymann and J. Stierstorfer, Eur. J. Inorg. Chem., 2013, 2013, 2167–2180 CrossRef CAS.
- T. Yang, Z.-B. Zhang and J.-G. Zhang, Z. Anorg. Allg. Chem., 2017, 643, 413–419 CrossRef CAS.
- U. S. P., 086, 110.
- D. Fischer, T. M. Klapötke and J. Stierstorfer, Propellants, Explos., Pyrotech., 2012, 37, 156–166 CrossRef CAS.
- L. Liang, H. Huang, K. Wang, C. Bian, J. Song, L. Ling, F. Zhao and Z. Zhou, J. Mater. Chem., 2012, 22, 10 Search PubMed.
- H. Huang, Z. Zhou, L. Liang, J. Song, K. Wang, D. Cao, C. Bian, W. Sun and M. Xue, Z. Anorg. Allg. Chem., 2012, 638, 392–400 CrossRef CAS.
- J. Xu, J. Wei, F. Li, Q. Ma and X. Peng, New J. Chem., 2014, 38, 5303–5311 RSC.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, J. Montgomery, J. T. Vreven, K. Kudin and J. Burant, Revision A.02, Gaussian Inc, 2009 Search PubMed.
- H. D. B. Jenkins, H. K. Roobottom, J. Passmore and L. Glasser, Inorg. Chem., 1999, 38, 3609–3620 CrossRef CAS PubMed.
- Y. Tang, H. Gao, D. A. Parrish and J. M. Shreeve, Chemistry, 2015, 21, 11401–11407 CrossRef CAS PubMed.
- H. Gao and J. M. Shreeve, Chem. Rev., 2011, 111, 7377–7436 CrossRef CAS PubMed.
- Y. Ma, A. Zhang, C. Zhang, D. Jiang, Y. Zhu and C. Zhang, Cryst. Growth Des., 2014, 14, 4703–4713 CAS.
- R.-H. Wang, H.-Y. Xu, Y. Guo, R. J. Sa and J. M. Shreeve, J. Am. Chem. Soc., 2010, 132, 11904–11905 CrossRef CAS PubMed.
- C. Guo, H. Zhang, X. Wang, J. Xu, Y. Liu, X. Liu, H. Huang and J. Sun, J. Mol. Struct., 2013, 1048, 267–273 CrossRef CAS.
- Z. Zhang, G. Wang, N. Luo, M. Huang, M. Jin and Y. Luo, J. Appl. Polym. Sci., 2014, 131, 40965 Search PubMed.
- J. Zhao, B. Jin, R. Peng, N. Deng, W. Gong, Q. Liu and S. Chu, RSC Adv., 2015, 5, 90422–90427 RSC.
Footnote |
| † CCDC 1487781. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra11428a |
| This journal is © The Royal Society of Chemistry 2018 |