
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure and magnetism of two chair-shaped hexanuclear dysprosium(III) complexes exhibiting slow magnetic relaxation†
Zi-Yuan Liua,
Hua-Hong Zou*a,
Rong Wanga,
Man-Sheng Chena and
Fu-Pei Liang *ab
*ab
aState Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, School of Chemistry & Pharmacy of Guangxi Normal University, Guilin 541004, P. R. China. E-mail: gxnuchem@foxmail.com
bGuangxi Key Laboratory of Electrochemical and Magnetochemical Functional Materials, College of Chemistry and Bioengineering, Guilin University of Technology, Guilin 541004, P. R. China. E-mail: fliangoffice@yahoo.com
First published on 3rd January 2018
Abstract
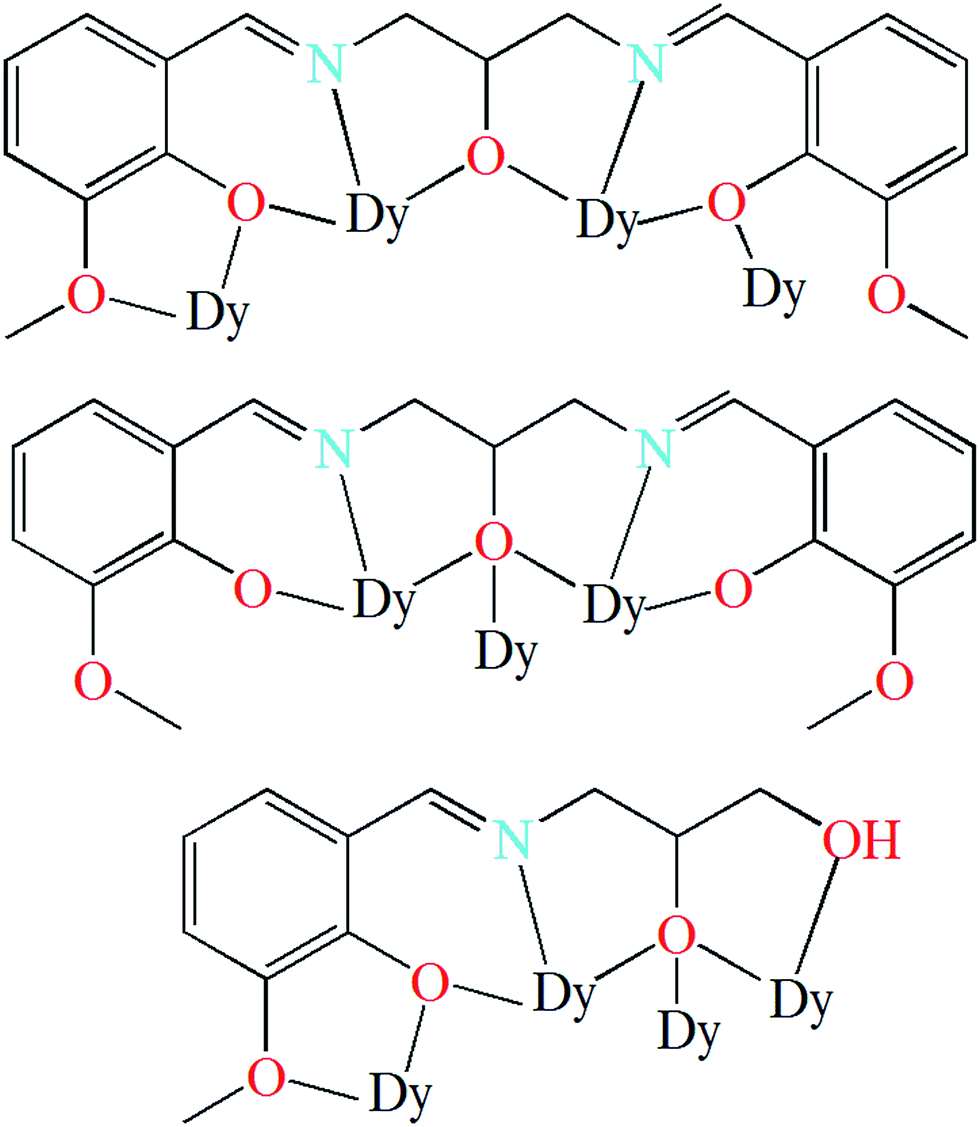
Two novel hexanuclear DyIII complexes with polyhydroxy Schiff-base ligands, [Dy6(L1)4(μ3-OH)4(MeOH)4]Cl2·2MeOH·2MeCN (1) and [Dy6(HL2)2(μ3-OH)2(μ3-OCH3)2(piv)10(MeOH)2] (2) (H3L1 = N,N′-bis(3-methoxysalicylidene)(propylene-2-ol)-1,3-diamine, H3L2 = 2,3-dihydroxypropylimino)methyl)-6-methoxyphenol, piv = pivalate), have been prepared under solvothermal conditions and structurally characterized by single-crystal X-ray diffraction, elemental analyses, thermal analyses, and IR spectroscopy. Each of the hexanuclear complexes is constructed with Dy3 triangular motifs as building blocks, and the six DyIII ions are arranged in a chair-shaped conformation. Variable-temperature magnetic susceptibility measurements in the temperature range of 2–300 K indicate dominant ferromagnetic exchange interactions between the DyIII ions in the complexes. Both complexes exhibit slow magnetic relaxation behavior.
Introduction
The synthesis of new single-molecule magnets (SMMs) containing lanthanide metals continues to grow because of their fascinating magnetic behaviours and potential applications in many fields such as high-density information storage, quantum computing, molecular spintronics etc.1–6 Most of the 4f-based SMMs reported in the literature are derived from DyIII ion due to its unquenched orbital angular momentum and large intrinsic magnetoanisotropy.7–11 In the past decade, a number of mono- and polynuclear DyIII complexes have been synthesized and characterized,12 and many of them have been demonstrated to exhibit remarkable properties of single-molecule magnets. For example, mononuclear DyIII complexes with pentagonal bipyramidal coordination geometry featuring strong axial ligand field and weak equatorial donors exhibit very high effective energy barriers for magnetic relaxation.13 A dinuclear DyIII complex bridged by an N23− radical ligand was observed to show magnetic hysteresis up to 8.3 K.14 A tetranuclear DyIII alkoxide cage complex and a square-based pyramid iso-propoxide-bridged pentanuclear DyIII complex have been reported to possess large thermal energy barrier for the reversal of magnetization.1b,15Among the various DyIII-based SMMs reported, the Dy3 triangular molecules have been found to be a magnetically interesting system. This system has an essentially nonmagnetic spin ground state, but exhibits SMM behavior of thermally populated excited states, as well as toroidal magnetic moments which are useful in molecule-based multiferroics.16 Stimulated by the fascinating magnetic behaviors of these intriguing Dy3 triangles, research on the utilization of theirs as building blocks to construct larger SMMs with enhanced magnetic properties has attracted much attention. It was reported that linking of two such highly anisotropic Dy3 triangles in different forms led to the creation of hexanuclear dysprosium SMMs,17 of which enhanced slow magnetic relaxation was observed,17b and enhanced toroidal magnetisms were obtained by fine-tuning the arrangements of the DyIII ions or by modifying the local ligand-field around the DyIII ions.17d,e Combination of two independent Dy3 SMM-building blocks by a paramagnetic [Dy(μ2-CH3O)2Dy]4+ linker afforded an octanuclear DyIII complexes with SMM behavior inherited from its Dy3 precursor.18 An even larger cluster of decanuclear DyIII based on the peculiar Dy3 triangles was obtained by incorporating two sets of vertex-sharing Dy3 triangular motifs.19 These successful examples represent a promising strategy for preparation of novel Dy-based SMMs by using highly anisotropic Dy3 triangles as building blocks.
Herein, we report the synthesis, structure, and magnetic properties of two analogous hexanuclear DyIII complexes, [Dy6(L1)4(μ3-OH)4(MeOH)4]Cl2·2MeOH·2MeCN (1) and [Dy6(HL2)2(μ3-OH)2(μ3-OCH3)2(piv)10(MeOH)2] (2), (H3L1 = N,N′-bis(3-methoxysalicylidene)(propylene-2-ol)-1,3-diamine, H3L2 = 2,3-dihydroxypropylimino)methyl)-6-methoxyphenol, and piv = pivalate, Scheme 1). The hexanuclear complexes can be viewed as being constructed with Dy3 triangular motifs as building blocks. The six DyIII ions are arranged in a chair-shaped conformation that has never been shown for the hexanuclear lanthanide complexes. In addition, intramolecular ferromagnetic interactions are observed, which is rarely reported for the lanthanide SMMs.17a,20 Magnetic investigations indicate that both of complexes exhibit single-molecule magnets behavior.
 | ||
| Scheme 1 Schiff-base ligands of H3L1 (left) and H3L2 (right). | ||
Experimental section
Materials and methods
Metal salts and solvents were purchased from commercially available and used directly without further purification in the preparation of the free ligands and complexes. The Schiff-base ligands H3L1 and H3L2 were prepared as previously described.21 IR spectra were recorded in the range of 4000–400 cm−1 on Perkin-Elmer Spectrum Two FT/IR spectrometer using a KBr pellet. Elemental analysis (C, H, N) was performed on a Elementar Micro cube CHN elemental analyzer. The thermal analysis was performed on Labsys Evo TG-DTG/DSC. The crushed single-crystal sample was heated up to 1000 °C in N2 (99.99%) at a heating rate of 10 °C min−1. Magnetic susceptibility measurements were performed in the temperature range of 2–300 K, using a Quantum Design MPMS SQUID-XL-5 magnetometer equipped with a 5 T magnet. The diamagnetic corrections for these complexes were estimated using Pascal's constants, and magnetic data were corrected for diamagnetic contributions of the sample holder.22 Alternating current susceptibility measurements were taken of powdered samples to determine the in-phase and out-of-phase components of the magnetic susceptibility. The data were collected by increasing temperature from 2 K to 10 K, with no applied external dc field and a drive frequency of 2.5 Oe, with frequencies between 10 and 1000 Hz. In the samples where free movement of crystallites were prevented, silicone grease was employed for the embedding.Single-crystal X-ray crystallography
Diffraction data for these complexes were collected on a Bruker SMART CCD diffractometer (Mo Kα radiation and λ = 0.71073 Å) in Φ and ω scan modes. The structures were solved by direct methods followed by difference Fourier syntheses, and then refined by full-matrix least-squares techniques on F2 using SHELXL-97.23 All other non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen atoms were placed in calculated positions and refined isotropically using a riding model. X-ray crystallographic data and refinement details for the complexes are summarized in Table S1.† Full details can be found in the CIF files provided in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was stirred at room temperature for 30 min (pH = 7.6), and then the final mixture was sealed in a 25 mL Teflon-lined autoclave and heated at 80 °C for five days. After cooling to room temperature slowly, yellow block crystals of 1 were isolated (the yield based on DyCl3·6H2O: 42%). Elemental analysis calcd (%) for 1, C86H110Dy6N10O30Cl2: C, 56.30; H, 6.04; N, 7.63. Found: C, 56.51; H, 6.12; N, 7.73. IR (KBr disk, cm−1): 3408 (s), 3056 (w), 2967 (m), 1642 (m), 1558 (s), 1482 (s), 1421 (s), 1376 (m), 1231 (s), 1018 (m), 896 (w), 736 (w), 599 (w).:1) was stirred at room temperature for 60 min (pH = 6.7), and then the final mixture was sealed in a 25 mL Teflon-lined autoclave and heated at 120 °C for six days. After cooling to room temperature slowly, yellow block crystals of 2 were isolated (the yield based on Dy(NO3)3·6H2O: 31%). Elemental analysis calcd (%) for 2, C76H124Dy6N2O34: C, 35.31; H, 4.84; N, 1.08. Found: C, 35.46; H, 4.97; N, 1.13. IR (KBr disk, cm−1): 3406 (s), 2940 (w), 1642 (m), 1630 (w), 1482 (w), 1467 (m), 1385 (s), 1295 (m), 1218 (m), 1158 (w), 1073 (m), 896 (w), 742 (w), 599 (w).
:1) was stirred at room temperature for 30 min (pH = 7.6), and then the final mixture was sealed in a 25 mL Teflon-lined autoclave and heated at 80 °C for five days. After cooling to room temperature slowly, yellow block crystals of 1 were isolated (the yield based on DyCl3·6H2O: 42%). Elemental analysis calcd (%) for 1, C86H110Dy6N10O30Cl2: C, 56.30; H, 6.04; N, 7.63. Found: C, 56.51; H, 6.12; N, 7.73. IR (KBr disk, cm−1): 3408 (s), 3056 (w), 2967 (m), 1642 (m), 1558 (s), 1482 (s), 1421 (s), 1376 (m), 1231 (s), 1018 (m), 896 (w), 736 (w), 599 (w).:1) was stirred at room temperature for 60 min (pH = 6.7), and then the final mixture was sealed in a 25 mL Teflon-lined autoclave and heated at 120 °C for six days. After cooling to room temperature slowly, yellow block crystals of 2 were isolated (the yield based on Dy(NO3)3·6H2O: 31%). Elemental analysis calcd (%) for 2, C76H124Dy6N2O34: C, 35.31; H, 4.84; N, 1.08. Found: C, 35.46; H, 4.97; N, 1.13. IR (KBr disk, cm−1): 3406 (s), 2940 (w), 1642 (m), 1630 (w), 1482 (w), 1467 (m), 1385 (s), 1295 (m), 1218 (m), 1158 (w), 1073 (m), 896 (w), 742 (w), 599 (w).Results and discussions
Syntheses
Both complexes were synthesized under solvothermal conditions. Reactions of DyCl3·6H2O with H3L1 in the presence of Et3N in 10:5:2 molar ratio in a mixed solvent of CH3OH and CH3CN (1:1) in Teflon-lined autoclave at 80 °C for five days produced yellow crystals of 1 in 42% yield. Utilization of single solvent could not yield the compound. The Schiff base ligand in the complex has a −3 charge with all hydroxyl groups deprotonated. Reactions of Dy(NO3)3·6H2O, H3L2, pivalic acid, and Et3N in 5:5:30:2 molar ratio in similar solvent as 1 in Teflon-lined autoclave at 120 °C for six days produced yellow crystals of 2 in 31% yield. The ligand in complex 2 has a −2 charge with the phenolic hydroxyl group and one alcoholic hydroxyl group deprotonated. Reactions of H3L1 or H3L2 with other Dy(III) sources, such as Dy(NO3)3·6H2O for H3L1 and DyCl3·6H2O for H3L2, have been tested, but no crystals of 1 and 2 were obtained.
Crystal structures
The solid state structures of 1–2 were determined by single crystal X-ray crystallographic studies. Crystallographic data for complexes 1–2 are presented in Table S1,† and selected bond lengths and angles are given in Tables S3–S5† in the ESI.†Single-crystal X-ray studies revealed that complex 1 crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with the asymmetric unit containing the entire molecule as well as a chloride counterion and MeOH and MeCN solvent molecules. A view of the cationic complex 1 is shown in Fig. 1. The X-ray structure of 1 reveals a centrosymmetric core with two equivalent Dy3 moieties linked by two μ3-OH− anions. Each μ3-OH− anion links three Dy ions, and the Dy–Dy separations are similar at 3.521 Å, 3.507 Å and 3.639 Å for Dy1–Dy2, Dy1–Dy3 and Dy2–Dy3, respectively. Dy1 is coordinated with six oxygen atoms and two nitrogen atoms, which originating from two different L1 ligands, and two μ3-OH− anions. The Dy2 center is ligated with seven oxygen atoms (Dy–O = 2.047(6)–2.235(5) Å) and one nitrogen atom (Dy–N = 2.235(5) Å) from three different Schiff-base ligands, one μ3-OH− anion, and one MeOH molecule. Dy3 is coordinated with seven oxygen atoms and one nitrogen atom from two Schiff-base ligands, three μ3-OH− anions, one MeOH molecule. All Dy ions are 8-coordinate and display distorted bicapped trigonal prism geometry. The hexanuclear Dy(III) complex contain four Dy3 triangles. The Dy1, Dy3, Dy1A, and Dy3A atoms are in the same plane, forming a parallel quadrilateral, which containing two Dy3 triangles (Dy1, Dy3, Dy1A, and Dy1A, Dy3A, Dy1). Each triangle link by one μ3-OH− anion. Dy2 and Dy2A are located at both ends of the parallel quadrilateral, Dy2A lying above the plane and Dy2 below. Therefore, two other Dy3 triangles (Dy1, Dy2, Dy3, and Dy1A, Dy2A, Dy3A) are formed. The latter two Dy3 triangles are linked via one μ3-bridging hydroxyl group of the ligand L1 and one μ3-OH− (located on the front and back of the triangle). Obviously, the layout of the six Dy atoms forms a chair-shaped. Notably, such a hexanuclear Dy(III) complex is quite different from other known examples.24 First, there exit a parallel quadrilateral containing two Dy3 triangles. Second, all Dy atoms are 8-coordinate with distorted bicapped trigonal prism geometry. Furthermore, the Schiff-base ligand has a −3 charge with two phenolic hydroxyl groups and one backbone hydroxyl group deprotonated in 1. And the ligands adopt two different coordination fashions (Scheme 2). Adjacent molecules are segregated by these ligands, the shortest intermolecular Dy–Dy separation is 8.478 Å, indicating that the molecules are quite well isolated. Hydrogen atoms were determined by analyzing the coordination environment of oxygen atoms and hydrogen bonds were found in 1. There is a triply hydrogen-bonded among the chloride ion (Cl1) and the solvent methanol molecule (O15), the coordinated methanol (O13), μ3-OH (O1) (Fig. S1†). Meanwhile, there is a strong hydrogen bonds interaction between the solvent methanol molecule (O15) and coordinated oxygen atoms (O2) with the O2–H2⋯O15 distances of 2.853 Å.
, with the asymmetric unit containing the entire molecule as well as a chloride counterion and MeOH and MeCN solvent molecules. A view of the cationic complex 1 is shown in Fig. 1. The X-ray structure of 1 reveals a centrosymmetric core with two equivalent Dy3 moieties linked by two μ3-OH− anions. Each μ3-OH− anion links three Dy ions, and the Dy–Dy separations are similar at 3.521 Å, 3.507 Å and 3.639 Å for Dy1–Dy2, Dy1–Dy3 and Dy2–Dy3, respectively. Dy1 is coordinated with six oxygen atoms and two nitrogen atoms, which originating from two different L1 ligands, and two μ3-OH− anions. The Dy2 center is ligated with seven oxygen atoms (Dy–O = 2.047(6)–2.235(5) Å) and one nitrogen atom (Dy–N = 2.235(5) Å) from three different Schiff-base ligands, one μ3-OH− anion, and one MeOH molecule. Dy3 is coordinated with seven oxygen atoms and one nitrogen atom from two Schiff-base ligands, three μ3-OH− anions, one MeOH molecule. All Dy ions are 8-coordinate and display distorted bicapped trigonal prism geometry. The hexanuclear Dy(III) complex contain four Dy3 triangles. The Dy1, Dy3, Dy1A, and Dy3A atoms are in the same plane, forming a parallel quadrilateral, which containing two Dy3 triangles (Dy1, Dy3, Dy1A, and Dy1A, Dy3A, Dy1). Each triangle link by one μ3-OH− anion. Dy2 and Dy2A are located at both ends of the parallel quadrilateral, Dy2A lying above the plane and Dy2 below. Therefore, two other Dy3 triangles (Dy1, Dy2, Dy3, and Dy1A, Dy2A, Dy3A) are formed. The latter two Dy3 triangles are linked via one μ3-bridging hydroxyl group of the ligand L1 and one μ3-OH− (located on the front and back of the triangle). Obviously, the layout of the six Dy atoms forms a chair-shaped. Notably, such a hexanuclear Dy(III) complex is quite different from other known examples.24 First, there exit a parallel quadrilateral containing two Dy3 triangles. Second, all Dy atoms are 8-coordinate with distorted bicapped trigonal prism geometry. Furthermore, the Schiff-base ligand has a −3 charge with two phenolic hydroxyl groups and one backbone hydroxyl group deprotonated in 1. And the ligands adopt two different coordination fashions (Scheme 2). Adjacent molecules are segregated by these ligands, the shortest intermolecular Dy–Dy separation is 8.478 Å, indicating that the molecules are quite well isolated. Hydrogen atoms were determined by analyzing the coordination environment of oxygen atoms and hydrogen bonds were found in 1. There is a triply hydrogen-bonded among the chloride ion (Cl1) and the solvent methanol molecule (O15), the coordinated methanol (O13), μ3-OH (O1) (Fig. S1†). Meanwhile, there is a strong hydrogen bonds interaction between the solvent methanol molecule (O15) and coordinated oxygen atoms (O2) with the O2–H2⋯O15 distances of 2.853 Å.
 | ||
| Fig. 1 Crystal structure and chair-shaped of complex 1 (left) and 2 (right); hydrogen atoms and solvents are omitted for clarity. Color scheme: Dy, purple; O, red; N, light blue; C, green. | ||
 | ||
| Scheme 2 The coordination modes of (L1)3− in 1 (top (μ4-L1-κ9O1,O2:O2,N1,O3:O3,N2,O4:O4) and middle (μ3-L1-κ7O2,N1,O3:O3:O3,N2,O4)) and (HL2)2− in 2 (down (μ4-L2-κ8O1,O2:O2,N1,O3:O3:O3,O4)). | ||
A perspective view of 2 is depicted in Fig. 1. Complex 2 crystallises in the monoclinic space group P21/c. The hexanuclear complex surrounded by two Schiff-base ligands and ten pivalate ligands. Only one coordination and bridging mode can be observed for the unique polydentate Schiff-base HL2 ligand (Scheme 2). Three different coordination modes of pivalate ligands coexist in the crystal structure (Scheme 3). Six of the ten pivalate anions acts as a bridging ligand with two oxygen atoms coordinating separately to two Dy ions. Two pivalate anions coordinate to two Dy ions respectively in μ2-pivalate-κ3O1O2:O2 fashion. The other two pivalate anions coordinate respectively to Dy ions in monodentate mode. Two methanol molecules occupy the remaining coordination sites of the Dy2 and Dy2A metal centres. The Dy1 is nine-coordinate with distorted capped square antiprism geometries (Fig. S2†), Dy2 and Dy3 are eight-coordinate, displaying triangular dodecahedron and square antiprism geometry, respectively. In the hexanuclear unit, Dy1, Dy2, Dy1A and Dy2A ions are present in the same plane, forming a parallel quadrilateral, whereas the Dy3 and Dy3A ions are arranged in a trans geometry with respect to each other and are displaced by 1.618 Å from the plane. Thus, the hexanuclear core adopts the chair conformation. This Dy6 complex also contain four Dy3 triangles. The two Dy3 triangles (Dy1, Dy2, Dy1A, and Dy1, Dy2A, Dy1A) are linked by one μ3-OH− anion, respectively. Two other Dy3 triangles (Dy1, Dy2, Dy3, and Dy1A, Dy2A, Dy3A) are linked via one μ3-bridging hydroxyl group of the ligand L2 and one μ3-OCH3. The closest intercluster Dy–Dy distances is 9.49 Å, also indicating that the molecules are quite well isolated.
 | ||
| Scheme 3 Three coordination modes of pivalate ligands in complex 2 (pivalate-κO, left; μ2-pivalate-κ2O1:O2, middle; μ2-pivalate-κ3O1O2:O2, right). Hydrogen atoms are omitted for clarity. Color scheme: Dy, purple; O, red; C, green. | ||
Up to now, many hexanuclear DyIII complexes with different topologies have been reported (Table S2†). The arrangements of six DyIII ions in the complex with ring (or wheel),25 hollow,26 hemi-cubane,27 trigonal prism,28 edge-to-edge,17c,29 etc. have been described. The chair-shaped arrangement described here is newly-reported, and contributes an interesting example to the topology catalogues of the Dy6 complex.
Metal complexes of ligands H3L1 and H3L2 have been reported in literatures.24,30–32 For ligand H3L1, most of reported complexes, with 3d, 4f, 3d–4f, and others as metal centers, are finite multinuclear compounds.24,30,31a–f Only two examples containing Cu ions are coordination polymers with one-dimensional structure which was built by the bridging of alcoholic hydroxyl group of the ligand.31g,h The coordination modes of the ligand H3L1 in these reported complexes are summarized in Scheme S1.† For ligand H3L2, 3d and 4f metal complexes are reported, which are all finite multinuclear.32 The ligand in the complexes is found to show a variety of coordination modes that are listed in Scheme S2.† It is interesting to note that the coordination modes of the ligands H3L1 and H3L2 exhibited in this work (Scheme 1) have never been reported previously.
Spectroscopic and thermal analyses of complexes 1–2
The infrared (IR) spectra of complexes 1–2 have been recorded between 4000 and 400 cm−1 with KBr pellets (Fig. S3–4†). In the low-wavenumber region, the spectra exhibit the characteristic Dy–O and Dy–N vibration bands. The characteristic strong absorptions at ∼1642 cm−1 were ascribed to the vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, indicating the formation of the Schiff-base ligand.33 Furthermore, the resonances at 2967 and 2940 cm−1 are assigned to the ν(CH2) stretching vibrations, and the signals at 1482–1421 cm−1 and 1467–1385 cm−1 correspond to the δ(CH2) bending vibrations, respectively.34 The peaks appearing in the range of 1640–1390 cm−1 are assigned to the stretch of benzene ring from Schiff-base ligands. The ν(O–H) vibration at around 3400 cm−1 can also be observed in complexes 1–2, respectively. In summary, the results of the IR spectras are consistent with the single-crystal structural analyses.
N bond, indicating the formation of the Schiff-base ligand.33 Furthermore, the resonances at 2967 and 2940 cm−1 are assigned to the ν(CH2) stretching vibrations, and the signals at 1482–1421 cm−1 and 1467–1385 cm−1 correspond to the δ(CH2) bending vibrations, respectively.34 The peaks appearing in the range of 1640–1390 cm−1 are assigned to the stretch of benzene ring from Schiff-base ligands. The ν(O–H) vibration at around 3400 cm−1 can also be observed in complexes 1–2, respectively. In summary, the results of the IR spectras are consistent with the single-crystal structural analyses.
Thermal analyses have been carried out to examine the thermal stability of the complexes 1–2. The crushed single-crystal samples were heated up to 1000 °C in N2 at a heating rate of 10 °C min−1. The TG curves of 1 (Fig. S5†) show that the first weight loss of 7.6% between 30 and 155 °C corresponds to the loss of solvent MeCN, MeOH and chloride counterion (calc. 7.8%). The weight gradually decrease 4.3% with temperature increasing from 155 and 203 °C corresponds to the loss of terminal MeOH molecules (calc. 4.5%). The residue is stable up to 213 °C, where after pyrolysis of Schiff-base ligand and then ends at 710 °C (weight loss: 45.3%; calc. 45.7%). The final residual weight of 42.5% (calc. 46.7%) corresponds to that of Dy2O3. The above thermal analysis results basically agree with the formula of 1. The thermal properties of the complex 2 was also investigated. The weight loss of 4.9% is observed at 200 °C which maybe corresponding to the coordinated MeOH molecules and part of pivalate ligands. When the temperature continues rising, the Schiff-base ligands and other pivalate ligands begin to decompose in the 200 to 600 °C temperature range. Finally, the remaining weight of 43.8% is in agreement with the proportional weight (calc. 43.3%) of Dy2O3.
Magnetic properties
The direct-current (dc) magnetic susceptibility of 1–2 were measured in the range of 300–2 K with an applied magnetic field of 1000 Oe and can be seen plotted as χMT vs. T in Fig. 2. The room temperature χMT values of 84.95 and 84.57 cm3 K mol−1 for 1–2, respectively, which is in agreement with the expected value of 85.02 cm3 K mol−1 for six uncoupled DyIII ions (DyIII: 6H15/2, S = 5/2, L = 5, J = 15/2, g = 4/3). The χMT values of 1–2 gradually increase with decreasing temperature to reach a maximum of 92.39 cm3 K mol−1 at 50 K and 87.81 cm3 K mol−1 at 60 K, before decreasing to 39.12 cm3 K mol−1 and 49.01 cm3 K mol−1 at 2 K, respectively. The first increase of the χMT values suggests the presence of a dominant intramolecular ferromagnetic interactions between the DyIII ions. The low temperature decrease may be due to a combination of intermolecular antiferromagnetic interactions, large magnetic anisotropy and thermal population of the excited states of the DyIII ions (Stark sublevels of the 6H15/2 state).35 | ||
| Fig. 2 Plots of χMT vs. T for 1–2 under applied dc magnetic field of 1000 Oe. | ||
The field dependence of the magnetization (M) for 1–2 were performed in the field (H) range of 0–5 T at 2–5 K. As expected for ferromagnetically coupled spins, the M vs. H data below 5 K reveal a rapid increase of the magnetization at low magnetic fields (Fig. S6†). At high fields, M increases rapidly to reach 45.76 Nβ and 36.38 Nβ at 2 K and under 5 T, respectively. These values are all lower than the corresponding theoretical saturated values. The non-superposition of the M vs. H/T data (Fig. 3) on a single master curve and the highfield non-saturation suggests the presence of significant magnetic anisotropy and/or low-lying excited states in complexes 1–2.36,37
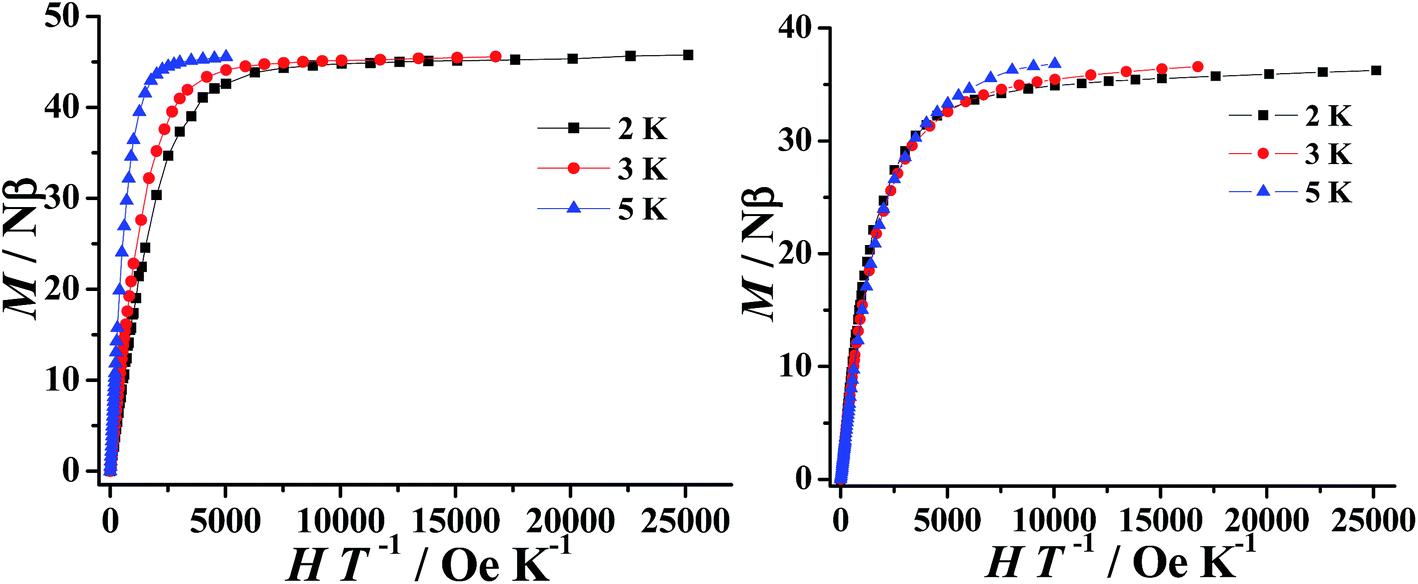 | ||
| Fig. 3 M vs. H/T plot at various temperatures for complexes 1 (left) and 2 (right). | ||
The temperature-dependent alternating-current (ac) susceptibility of complexes 1–2 were measured under zero-dc field and 2.5 Oe ac field (Fig. 4 and S7†). Both the in-phase (χ′) and out-of-phase (χ′′) signals of 1–2 show frequency dependence at low temperatures 2–10 K. The shape and the frequency dependence of the ac susceptibility signal indicate the slow magnetic relaxation behavior of 1–2. Although such ac signals were observed above 2 K, no obvious hysteresis was detected in the M vs. H data obtained using a traditional SQUID magnetometer (Fig. S8–9†).
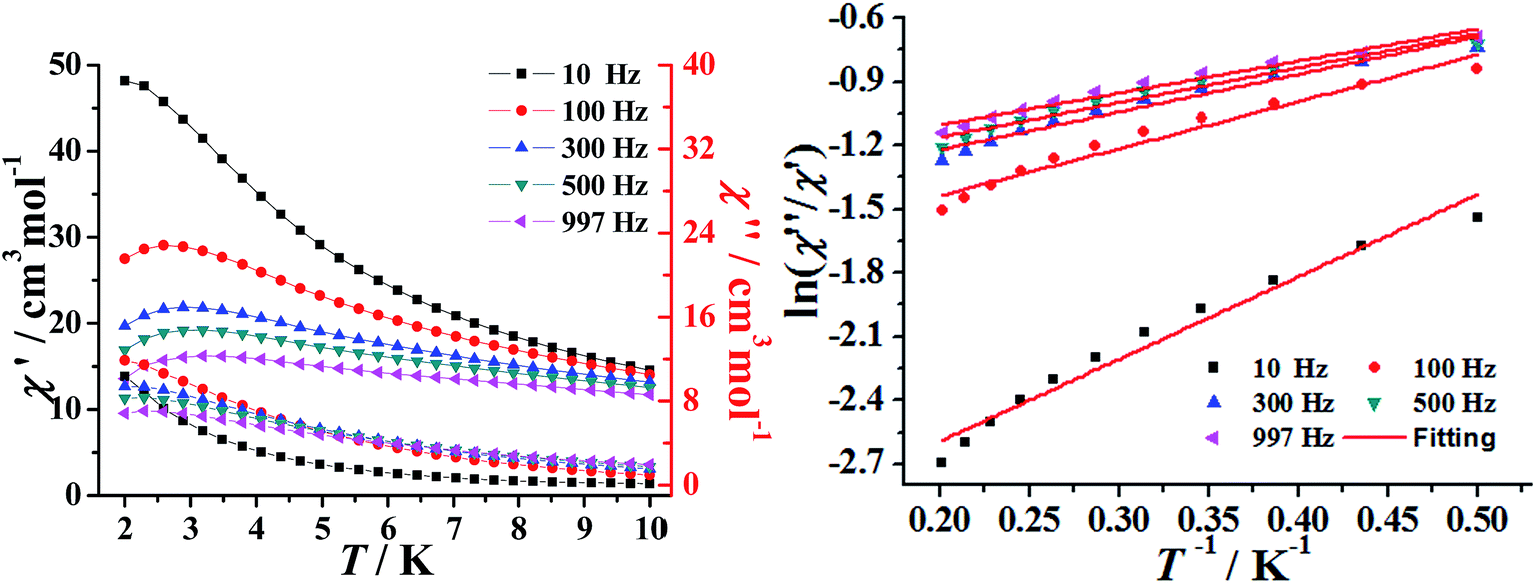 | ||
| Fig. 4 Temperature dependent ac susceptibility for 1 in the absence of a dc field (Hac = 2.5 Oe) (left); plots of ln(χ′′/χ′) vs. 1/T for 1. The solid lines represent the fitting results over the temperature range of 2–5 K (right). | ||
The above magnetic determination indicates that complexes 1–2 display a clear signature of slow magnetic relaxation behavior. Unfortunately, the expected maximum due to the blocking could not be observed above 2 K, even by applying a dc magnetic field up to 1000 and 2000 Oe (Fig. S10†). Under the assumption that the SMM relaxation has just one characteristic time, we could obtain the energy barriers and τ0 values by fitting the ac susceptibility data from adopt the Debye model.37 This gave τ0 = 9.03 × 10−5 s and Ueff = 1.03 K for 1 and τ0 = 2.36 × 10−5 s and Ueff = 4.21 K for 2 (Fig. 4 and S7†). The Cole–Cole diagram can be used to study the distribution of the relaxation process, which is frequently characterized and discussed for SMMs. The data of 1–2 plotted as Cole–Cole diagrams are shown in Fig. S11.† The shape of the Cole–Cole plot of 1–2 are relatively symmetrical and can be fitted to the generalized Debye model with a parameter α ranging from 0.11 to 0.15 for 1, 0.07–0.13 for 2, respectively, the relatively small α value indicates that a single relaxation time is mainly involved in the present relaxation process independently of the temperature.
Conclusions
In summary, we have synthesized two hexanuclear DyIII complexes based on two polyhydroxy Schiff-base ligands. It is interested that the complexes can be regarded as the result of the construction with Dy3 triangular motifs as building blocks, and the six DyIII ions in the complex are arranged in a newly-reported chair-shaped geometry. The measurements of the dc susceptibility and of the field dependence of magnetization indicated the existence of the intramolecular ferromagnetic interactions. The frequency-dependent ac susceptibility signals revealed by the ac susceptibility investigations evidenced the SMM behaviors under zero dc field for two compounds. The lanthanide SMMs exhibiting intramolecular ferromagnetic interactions are rarely reported, and the synthesis of such a lanthanide SMM is believed to be of great challenges. The results of this work might imply an effective approach for the rational design and construction of lanthanide SMMs with novel magnetic behaviors by means of the lanthanide building blocks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51572050, 21601038 and 21461004), the Guangxi Natural Science Foundation (No. 2015GXNSFDA139007 and 2016GXNSFAA380085), and Guangxi Key Laboratory of Electrochemical and Magnetochemical Functional Materials (EMFM20162107).References
-
(a) M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef CAS PubMed
; (b) R. J. Blagg, L. Ungur, F. Tuna, J. Speak, P. Comar, D. Collison, W. Wernsdorfer, E. J. L. McInnesl, L. F. Chibotaru and R. E. P. Winpenny, Nat. Chem., 2013, 5, 673–678 CrossRef CAS PubMed
-
(a) Y. Z. Zheng, M. Evangelisti, F. Tuna and R. E. P. Winpenny, J. Am. Chem. Soc., 2012, 134, 1057–1065 CrossRef CAS PubMed
- A. Candini, S. Klyatskaya, M. Ruben, W. Wernsdorfer and M. Affronte, Nano Lett., 2011, 11, 2634–2639 CrossRef CAS PubMed
- S. K. Langley, N. F. Chilton, B. Moubaraki and K. S. Murray, Inorg. Chem., 2013, 52, 7183–7192 CrossRef CAS PubMed
- J. Rinck, G. Novitchi, W. V. Heuvel, L. Ungur, Y. Lan, W. Wernsdorfer, C. E. Anson, L. F. Chibotaru and A. K. Powell, Angew. Chem., Int. Ed., 2010, 49, 7583–7587 CrossRef CAS PubMed
- S. K. Langley, D. P. Wielechowski, V. Vieru, N. F. Chilton, B. Moubaraki, B. F. Abrahams, L. F. Chibotaru and K. S. Murray, Angew. Chem., Int. Ed., 2013, 52, 12014–12019 CrossRef CAS PubMed
-
(a) J. L. Liu, J. Y. Wu, Y. C. Chen, V. Mereacre, A. K. Powell, L. Ungur, L. F. Chibotaru, X. M. Chen and M. L. Tong, Angew. Chem., Int. Ed., 2014, 53, 12966–12970 CrossRef CAS PubMed
- S. Y. Lin, J. F. Wu and Z. K. Xu, RSC Adv., 2017, 7, 47520–47526 RSC
- C. Papatriantafyllopoulou, T. C. Stamatatos, C. G. Efthymiou, L. Cunha-Silva, F. A. A. Paz, S. P. Perlepes and G. Christou, Inorg. Chem., 2010, 49, 9743–9745 CrossRef CAS PubMed
- A. Yamashita, A. Watanabe, S. Akine, T. Nabeshima, M. Nakano, T. Yamamura and T. Kajiwara, Angew. Chem., Int. Ed., 2011, 50, 4016–4019 CrossRef CAS PubMed
-
(a) S. K. Langley, D. P. Wielechowski, V. Vieru, N. F. Chilton, B. Moubaraki, L. F. Chibotaru and K. S. Murray, Chem. Sci., 2014, 5, 3246–3256 RSC
-
(a) R. Sessoli and A. K. Powell, Coord. Chem. Rev., 2009, 253, 2328–2341 CrossRef CAS
-
(a) Y. C. Chen, J. L. Liu, L. Ungur, J. Liu, Q. W. Li, L. F. Wang, Z. P. Ni, L. F. Chibotaru, X. M. Chen and M. L. Tong, J. Am. Chem. Soc., 2016, 138, 2829–2837 CrossRef CAS PubMed
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538–542 CrossRef CAS PubMed
- R. J. Blagg, C. A. Muryn, E. J. L. McInnes, F. Tuna and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2011, 50, 6530–6533 CrossRef CAS PubMed
-
(a) J. Tang, I. J. Hewitt, N. T. Madhu, G. Chastanet, W. Wernsdorfer, C. E. Anson, C. Benelli, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 1729–1733 CrossRef CAS PubMed
-
(a) B. Hussain, D. Savard, T. J. Burchell, W. Wernsdorfer and M. Murugesu, Chem. Commun., 2009, 1100–1102 RSC
- L. Zhang, P. Zhang, L. Zhao, J. F. Wu, M. Guo and J. Tang, Dalton Trans., 2016, 45, 10556–10562 RSC
- H. Ke, G. F. Xu, L. Zhao, J. Tang, X. Y. Zhang and H. J. Zhang, Chem.–Eur. J., 2009, 15, 10335–10338 CrossRef CAS PubMed
-
(a) S. F. Xue, X. H. Chen, L. Zhao, Y. N. Guo and J. Tang, Inorg. Chem., 2012, 51, 13264–13270 CrossRef CAS PubMed
-
(a) F. Lam, J. Xu and K. Chan, J. Org. Chem., 1996, 61, 8414–8418 CrossRef CAS
- E. A. Boudreaux and J. N. Mulay, Theory and Application of Molecular Diamagnetism, John Wiley and Sons, New York, 1976 Search PubMed
-
(a) G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed
- S. Liao, X. P. Yang and R. A. Jones, Cryst. Growth Des., 2012, 12, 970–974 CAS
-
(a) S. K. Langley, B. Moubaraki, C. M. Forsyth, I. A. Gass and K. S. Murray, Dalton Trans., 2010, 39, 1705–1708 RSC
- T. T. da Cunha, F. Pointillart, B. Le Guennic, C. L. M. Pereira, S. Golhen, O. Cador and L. Ouahab, Inorg. Chem., 2013, 52, 9711–9713 CrossRef CAS PubMed
- A. Adhikary, J. A. Sheikh, S. Biswas and S. Konar, Dalton Trans., 2014, 43, 9334–9343 RSC
-
(a) H. Q. Tian, M. Wang, L. Zhao, Y. Guo, J. Tang and Z. L. Liu, Chem.–Eur. J., 2012, 18, 442–445 CrossRef CAS PubMed
- S. Das, S. Hossain, A. Dey, S. Biswas, J. P. Sutter and V. Chandrasekhar, Inorg. Chem., 2014, 53, 5020–5028 CrossRef CAS PubMed
-
(a) L. Jiang, D. Y. Zhang, J. J. Suo, W. Gu, J. L. Tian, X. Liu and S. P. Yan, Dalton Trans., 2016, 45, 10233–10248 RSC
-
(a) V. Chandrasekhar, A. Dey, S. Das, M. Rouzières and R. Clérac, Inorg. Chem., 2013, 52, 2588–2598 CrossRef CAS PubMed
-
(a) H. H. Zou, L. B. Sheng, F. P. Liang, Z. L. Chen and Y. Q. Zhang, Dalton Trans., 2015, 44, 18544–18552 RSC
-
(a) W. J. Wang, Q. H. Chen, Q. Li, Y. Sheng, X. J. Zhang and K. Uvdal, Cryst. Growth Des., 2012, 12, 2707–2713 CrossRef CAS
-
(a) W. Z. Li, P. F. Yan, G. F. Hou, H. F. Li and G. M. Li, Dalton Trans., 2013, 42, 11537–11547 RSC
- P. H. Lin, T. J. Burchell, R. Clérac and M. Murugesu, Angew. Chem., Int. Ed., 2008, 47, 8848–8851 CrossRef CAS PubMed
-
(a) P. H. Lin, W. B. Sun, M. F. Yu, G. M. Li, P. F. Yan and M. Murugesu, Chem. Commun., 2011, 47, 10993–10995 RSC
-
(a) J. Ferrando-Soria, D. Cangussu, M. Eslava, Y. Journaux, R. Lescouëzec, M. Julve, F. Lloret, J. Pasán, C. Ruiz-Pérez, E. Lhotel, C. Paulsen and E. Pardo, Chem.–Eur. J., 2011, 17, 12482–12494 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1042130 and 1042131. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra11378a |
| This journal is © The Royal Society of Chemistry 2018 |