
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural basis for substrate discrimination by E. coli repair enzyme, AlkB†
Namrata Jayanth a,
Nirmala Ogiralaa,
Anil Yadavb and
Mrinalini Puranik‡
*a
a,
Nirmala Ogiralaa,
Anil Yadavb and
Mrinalini Puranik‡
*a
aNational Centre for Biological Sciences, Tata Institute of Fundamental Research, GKVK Campus, Bellary Road, Bangalore 560065, India. E-mail: puranik.mrinalini@gmail.com; mrinalini.puranik@unilever.com; Tel: +91-9980633836
bIndian Institute of Science Education and Research (IISER), Pune, Maharashtra 411008, India
First published on 3rd January 2018
Abstract
E. coli AlkB, a repair enzyme of the dioxygenase family, catalyses the removal of mutagenic methylated nucleotides from the genome. Known for substrate promiscuity, AlkB's catalytic mechanism and conformational changes accompanying substrate binding have been extensively dissected. However, the structural parameters of various substrates governing their recognition by AlkB still remain elusive. In this work, through solution-state vibrational spectra of methylated substrates bound to AlkB in combination with computational analysis, we show that the recognition specificity is dictated by the protonation states of the substrates. Specificity is conferred predominantly through hydrogen bonding and cation–π interactions. Furthermore, we report on the interaction of AlkB with normal, unmodified nucleotides, wherein the presence of an exocyclic amino group serves as an essential criterion for the initial process of substrate recognition. Taken together, these results provide a rationale for structural determinants of substrate specificity as well as mode of lesion discrimination employed by AlkB.
Introduction
DNA repair enzymes perform the daunting task of discriminating between normal nucleobases and their close chemical analogues, the results of DNA damage, to efficiently repair and restore DNA. In this, the odds are stacked against the enzymes since there are a few modified bases in several fold higher number of normal bases (∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 base lesions in ∼7 × 109 bp genome).1 An enduring puzzle about DNA repair enzymes is the mechanism through which they screen DNA to find rare lesions. The subsequent steps involving excision or other manner of damage reversal followed by restoration of the DNA are better understood.
000 base lesions in ∼7 × 109 bp genome).1 An enduring puzzle about DNA repair enzymes is the mechanism through which they screen DNA to find rare lesions. The subsequent steps involving excision or other manner of damage reversal followed by restoration of the DNA are better understood.
Several models are proposed for the DNA ‘search’ mechanism.2 In all these models, it is important to know the relative affinities of the repair enzyme for normal and modified bases. The specific contacts that the enzyme makes with normal versus modified bases provide direct information on the mechanism of recognition. Structures of co-complexes of repair enzymes with the damaged DNA inform on the contacts between the lesion and enzyme and are well characterized for several enzymes.2 The challenge has been to understand the interaction between repair enzymes and normal, unmodified DNA because of the very poor binding of repair enzymes to normal bases.
Alkylating agents act on nucleobases to target exocyclic nitrogens and oxygens; in addition to the ring nitrogens that lack hydrogen. These alkyl-modified nucleobases block the process of replication and are genotoxic.3 O6-methyltranferases repair damage inflicted by SN1 agents while α-ketoglutarate-Fe4-dependent oxygenases and glycosylases rectify the damage due to SN2-type alkylating agents. A hallmark feature of repair enzymes of the later two classes is that they recognize and repair a wide variety of modified nucleobases.5,6 AlkB belongs to the dioxygenase family of enzymes which utilizes non-heme mononuclear iron along with 2-OG to demethylate alkylated nucleotides, 1-me-dAMP, 3-me-dCMP, ε-dAMP and 6-me-dAMP.5,7–9
The extraordinary feat of multi-substrate recognition combined with the distinctive ability to specifically recognize methylated substrates versus normal bases by AlkB have initiated a plethora of studies ranging from X-ray crystallography,10–12 NMR,13 computational simulations14 to spectroscopic characterization using absorption15,16 and fluorescence techniques.17 A characteristic feature exhibited by DNA repair enzymes especially the enzymes of the BER pathway are the ‘flipping’ of lesions into their active-site pocket after the process of sampling and recognition.18 Both ALKBH2 and AlkB exhibit the phenomenon of base flipping.9–11 ALKBH2, the human homologue of AlkB, chemically cross-linked with 1-me-dAMP, 3-me-dCMP and ε-dAMP utilizes Phe-102 residue to intercalate the duplex DNA and flip the methylated lesions into its active-site pocket. Conversely, AlkB uses a unique mechanism to flip nucleotides which involves ‘squeezing’ by nucleotides that flank the alkylated substrate.10,11 The nucleotides adjacent to the lesion are ‘squeezed’ such that they stack against each other while enabling the flipping of the lesion. The flipped out nucleotide is then proposed to be stabilized by specific hydrogen bond interaction between Asp135 (Glu 175 in ALKBH2) and the amino group of 1-me-dAMP and 3-me-dCMP based on the proximity of Asp 135 from the methylated nucleotide. An additional key interaction in the active-site of AlkB is the stacking of the substrate by Trp 69 and His 131 residues (Fig. 1).10,12 These remarkable structures chalk out the overall strategy for the stabilization of the damaged DNA-AlkB complex. With this knowledge of the leading players of the active-site interactions, we now wish to understand: (i) how AlkB interacts with normal, unmodified bases and (ii) how the active-site interactions are modulated by the presence of different lesion nucleobases.
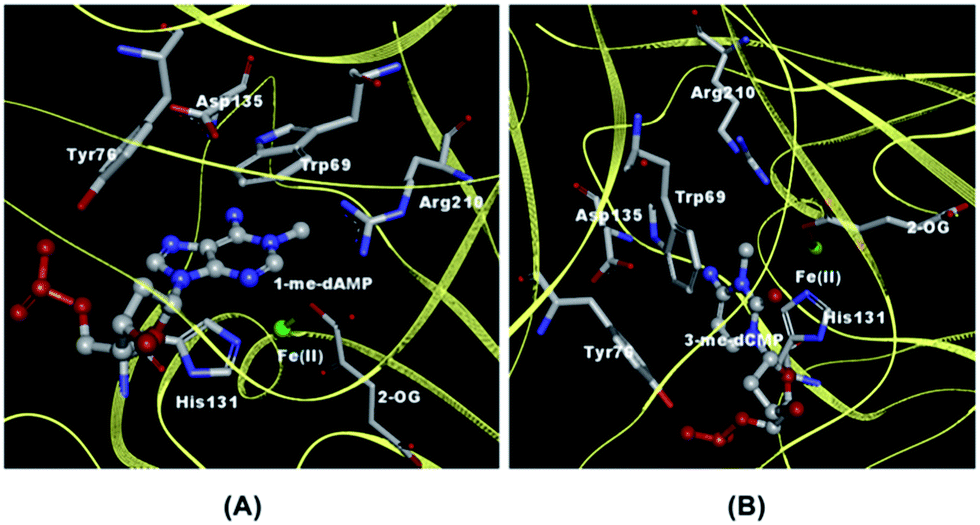 | ||
| Fig. 1 Crystal structures depicting AlkB active-site amino acids in the vicinity of nucleotides. (A) 1-me-dAMP [PDB ID 2FD8] and (B) 3-me-dCMP [PDB ID 3I49]. | ||
Multiple factors are shown to play a role in deciding the substrate specificity of repair enzymes.18 Primary among them is the structural distortion of DNA that results from the presence of a modified nucleotide. This is predominant in DNA duplexes as base pairing properties are affected due to nucleotide modifications leading to disruption of hydrogen bonding interactions between complementary base pairs, in turn affecting the overall stability of the lesion containing duplex.18,19 In fact, the mutagenic potential of most lesions is attributed to discrepancies in the normal Watson–Crick base pairing.19–22 This local alteration could serve as a cue for repair enzymes, which proceed to distort the lesion containing DNA even further by flipping the lesion into their active-site pocket.18
1-me-dAMP incorporated into double-stranded10 DNA does not distort the backbone conformation nor does it disrupt complementary base pairing interaction with dTMP. 1-me-dAMP forms a Hoogsteen base pairing interaction [dTMP(anti):1-meAMP(syn)] which causes only a local change in its sugar pucker and backbone conformation.11,23 3-me-dCMP does not form hydrogen bonds with complementary bases but tends to remain intrahelical in duplex DNA. Hence, recognition of 1-me-dAMP and 3-me-dCMP in duplex DNA by repair enzymes is hypothesized to be from sensing the weakened stability of the non-Watson Crick base pairing exhibited by the alkylated lesions.9 However, AlkB and related human homologues, the ALKBH family, act mostly on single-stranded (ss) DNA and RNA wherein base pairing discrepancies do not exist. Hence, the mechanism of recognition of methylated substrates in ssDNA and RNA by AlkB still remain ambiguous.
The other important criteria that factor in efficient recognition are the inherent structural properties of the modified bases, such as their protonation state, tautomerism and/or syn-anti conformation due to glycosidic bond rotation19. Presence of methyl groups confers a positive charge on the nucleotides which could be the cue recognized by AlkB. The stacking of Trp 69 with the methylated substrates as seen from the crystal structures has been attributed to π–cation interactions.10,12 A recent study by Maciejewska, A. M. et al., reports on the favourable substrate preference displayed by AlkB towards protonated exocyclic adducts.24
However, information on the protonation states of the substrates bound to AlkB and the hydrogen bonding strengths still remain to be deduced. In the current work, we provide finer structural details of the interaction of AlkB with methylated DNA, in solution, with particular emphasis on the structural distortion of substrates upon enzyme binding. We have studied the methylated nucleotides, 1-me-dAMP and 3-me-dCMP containing DNA trimers, present free in solution and when bound to AlkB to understand the nature of the enzyme–substrate interaction. We compare the effect of enzyme active-site on the cognate (methylated) versus normal, unmodified nucleotides to understand the mechanism of discrimination between the two. This is accomplished using UV resonance Raman spectroscopy on the free and enzyme-bound substrates. We utilized the fact that AlkB and methylated nucleotides have complementary electronic absorption spectra in the ultraviolet region (ESI Fig. S1†). As a consequence of this, while methylated and normal nucleotides have a high Raman cross section at 260 nm excitation employed here, contribution from the protein is negligible (ESI Fig. S2A†). In addition, since the Raman excitation is in resonance with the nucleobase, there is minimal contribution from the sugar and phosphate groups present in a nucleotide. Hence, the observed UVRR spectra report exclusively on the changes occurring in the nucleobase upon binding to AlkB.
We show that protonation states of the methylated nucleotides are the deciding criterion for recognition of the methylated nucleotides in ssDNA by AlkB. We have obtained detailed structural information on the protonation states of the substrates when bound in the active-site of AlkB. We present evidence of the predicted non-covalent interactions, namely the stacking of the methylated substrate between His 131 and Trp 69 residues of AlkB. In addition, we report on the complex of AlkB with oligomers containing non-cognate nucleotides, dAMP and dCMP. We find that the discrimination between normal and methylated nucleotides stems from the presence of a positive charge on the molecule along with the requirement for an exocyclic amino group.
Experimental procedures
AlkB purification
E. coli AlkB was purified using E. coli BS127 competent cells containing the pBAR54 plasmid with alkb gene insert. The protein containing amino-terminal His6 tag was purified as described by Sedgwick et al.25 In brief, the E. coli cells were grown at 37 °C until an O.D. of A600 0.9 was reached. The protein was over-expressed using 1 mM IPTG. Cells were harvested, washed with PBSA and resuspended in lysis buffer containing 50 mM Hepes-KOH, pH 8.0 (Himedia), 300 mM NaCl (Himedia), 5% glycerol (Qualigens), 2 mM β-mercaptoethanol3 and 2 mM EDTA3 and subjected to sonication. Cellular debris was pelleted by centrifugation at 12000 rpm at 4 °C. Cell extracts were loaded onto Ni-NTA column (Qiagen) after column equilibration with lysis buffer containing 1 mM imidazole.3 The resin was then washed with lysis buffer containing 40 mM and 60 mM imidazole. The protein eluted out in buffer containing 250 mM imidazole. Purified AlkB was dialysed in storage buffer containing 30 mM potassium phosphate (pH 7.5), 2 mM DTT, 10% glycerol and 300 mM NaCl. Enzyme concentrations were determined from UV absorbance measurements at A280 (Ultraspec 4000 UV/Visible spectrophotometer from Pharmacia Biotech) using an extinction coefficient of 31720 M−1 cm−1.
AlkB enzyme activity assay
The activity of E. coli AlkB was tested by using a standard assay utilising 14C-methylated substrates (American Radiolabeled Chemicals Inc, USA) as described by Sedgwick et al.25 AlkB (1 μM) was added to 14C-poly (dA) substrate (2 μg) in a reaction mixture containing 50 mM Hepes, pH 8.0, 1 mM α-ketoglutarate,3 75 μM ferrous ammonium sulphate (FAS)3 and were incubated at 37 °C for 15 minutes. The reaction was stopped by the addition of 10 mM EDTA.3 The substrate was precipitated by the addition of 100 mM NaCl and calf-thymus DNA (Bangalore Genei, India). 2 volumes of ethanol were added and the mixture was incubated at −70 °C for 45 minutes. The mixture was centrifuged at 12000 rpm for 15 minutes. 80% of the supernatant was monitored for the amount of formaldehyde released using a liquid scintillation counter (Wallac 1409).
Nucleotides and oligonucleotides
The nucleotides, 1-methyl-deoxyadenosine-5′-monophosphate (1-me-dAMP), 3-methyl-deoxycytidine-5′-monophosphate (3-me-dCMP), deoxyadenosine-5′-monophosphate (dAMP) and deoxycytidine-5′-monophosphate (dCMP) were obtained from ChemGenes, USA. Trimers 5′-dT-1-me-dA-dT-3′ and 5′-dT-3-me-dC-dT-3′and pentamer 5′-dC-dT-1-me-dA-dT-dC-3′ were obtained from Biosynthesis, USA. 5′-dT-dA-dT-3′, 5′-dT-dC-dT-3′, 5′-dT-dG-dT-3′ and 5′-dT-dU-dT-3′ were purchased from Sigma.Computational methods
Quantum mechanical calculations were performed on nucleotides used in experiments to assign the experimental observed Raman wavenumber shifts to the corresponding Raman frequencies. Density functional theoretical calculations were performed with Gaussian 09 suite of programs using B3LYP parameterization.26–28 The basis set used was Gaussian 6-31G** with additional polarisation functions on the hydrogen atoms. Geometry optimisation of the nucleotide structures were carried out taking the solvent effect into consideration by using polarizable continuum model (PCM). The potential energy distributions (PEDs) were computed using Vibrational Energy Distribution Analysis (VEDA) 4.0 program.29Since the Raman experiments were carried out at 260 nm laser excitation, wherein only the bands from the nucleobases are resonance enhanced with minimal contribution from the protein in an enzyme–substrate complex, the Raman frequencies were computed exclusively for nucleobases. Detailed vibrational band assignments for the substrates have been reported earlier by us.30 Raman frequencies were calculated from the geometry optimised nucleobase structures. Charge distribution, bond distances, interaction energies and electrostatic potential energy maps were calculated from the geometry optimised structures of nucleobases and nucleotides. The structures of the energy-minimised molecules, charge distribution and bond lengths were visualised using Chemcraft (http://www.chemcraftprog.com).
Electrostatic potential energy surfaces (EPS) of the nucleobases were obtained from the geometry optimised structures of the nucleobases. The EPS was calculated from the electrostatic potential of the nucleobases computed from the total electron density (SCF density matrix). The electrostatic potential maps were visualised using GaussView (version 3.0).31
Sample preparation for UVRR spectroscopy experiments
For the UVRR spectroscopy experiments, the nucleotide and oligomer stocks (10 mM) were prepared in 50 mM Hepes-KOH, pH 8.0. Stocks of the co-factors, FAS and 2-OG were prepared in 50 mM Hepes, pH 8.0. For the UVRR experiments, 0.5 mM FAS, 1 mM 2-OG were added to 200 μM of AlkB in storage buffer followed by the addition of 1 mM nucleotide or 0.5 mM of the trimer or pentamer in reaction buffer containing 50 mM Hepes-KOH, pH 8.0 and 5 mM sodium dithionite. All stock solutions were kept in gas-tight tubes and purged with high purity Argon to remove oxygen. Experiments which required the pH to be maintained at 6.0, the reaction mixture contained 50 mM MES buffer. In order to obtain a stable AlkB-substrate complex, anaerobic conditions were maintained to prevent oxidation of Fe4 leading to catalysis, during sample preparation and data collection. For this, the samples were prepared in an NMR tube which was sealed using rubber septa and purged with ultra pure argon prior to and after mixing the enzyme and substrate components for 20 minutes to ensure complete removal of oxygen before data collection. 30 mM sodium nitrate (NaNO3)3 was added to all the samples just before spectral recording for use as an internal standard reference for further data processing.For the Hydrogen/Deuterium (H/D) exchange experiments, all samples were in buffers prepared in D2O. The nucleotide and oligomer stocks were prepared using 50 mM Hepes-KOH, pH 8.0 in D2O. AlkB in storage buffer was exchanged using Amicon Ultra Centrifugal filter (Millipore) with D2O buffer containing 50 mM Hepes, pH 8.0, 2 mM DTT, 10% glycerol, and 300 mM NaCl.
Data collection and processing
The detailed ultraviolet resonance Raman set up has been described in earlier publications.30,32,33 In brief, resonance Raman spectra were obtained by excitation with 260 nm light. This is the third-harmonic output of a nanosecond-pulsed Nd-YLF laser pumped Ti–S laser (Indigo, Coherent Inc.) operated at 1 KHz. Typical average power at the sample was less than ∼600 μW. Re-absorption of the scattered light was minimized by using back-scattering collection configuration. Light was collected using home-built optics and analyzed with a single grating (3600 grooves per mm) monochromator (Jobin-Yvon). The detector used is a 1024 × 256 pixel, back-illuminated CCD camera (Jobin-Yvon). Spectra were collected by averaging three 14 minute measurements. The spectra were calibrated using standard solvents cyclohexane, acetonitrile, isopropanol, trichloroethylene, and indene. To aid in measurement of the relative wavenumbers changes, the spectra were recorded without changing the spectrometer position. All spectral processing was carried out using the software SynerJY (Jobin-Yvon), a version of Origin 7.0. Lorentzian line shapes were used to fit the calibrated Raman spectra of the samples to obtain wavenumber positions of the observed Raman bands. Band positions were determined by fitting Lorentzian line shapes to the bands in the observed spectra. Equation for Lorentzian line shape:yo is the base line of the peak, A is the area under the curve, w is the width of the peak at half maximum and xc is the centre of the peak.
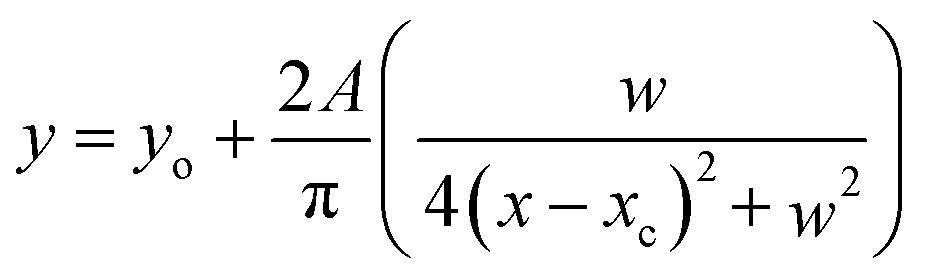
The samples for spectral recording taken in septum sealed NMR tubes (fused silica Suprasil grade, Wilmad Lab Glass, USA) were kept spinning during the entire spectral acquisition to prevent sample degradation. Spectra were collected by averaging three measurements, with each measurement corresponding to an average of 40 frames of 20 seconds laser exposure. The reported spectra are an average of all the 40 frames from the three measurements. In experiments where relative shifts are measured, the Raman spectra were measured without changing the spectrophotometer position and are accurate to ±2 cm−1 for the free nucleotides and ±3 cm−1 for enzyme·substrate complexes.
The laser power impinging on the samples was ∼0.6 mW at 260 nm laser excitation and the power impinging on the samples was 1 mW when the laser excitation wavelength was 225 nm. To ensure that there was no photo-damage of the samples during spectral acquisition, the spectrum obtained from the first frame was compared with the spectrum of the last frame. Relative intensity changes of the Raman bands in the spectrum, appearance of new bands or disappearance of bands during the entire recording period, which are all hallmarks of photo-damage were monitored for during spectral acquisition.
An excitation wavelength of 260 nm was used to study the changes occurring in the amino forms of 1-me-dAMP and 3-me-dCMP in complex with AlkB. There is minimal contribution from the protein to the vibrational spectrum of AlkB in complex with the nucleotides as the protein is not in resonance at 260 nm laser excitation. Spectral contributions from unbound nucleotides, oligomers and AlkB was removed from the UVRR spectrum of the AlkB·nucleotide/oligomer complex to obtain the spectrum of bound nucleotide/oligomer (ESI Fig. S2D†).
Methodology employed for study of UVRR spectra of bound substrates to AlkB
At laser excitation of 260 nm wavelength, the vibrational modes of nucleotides are enhanced in comparison to the protein in an enzyme–substrate complex as nucleotides exhibit absorption maxima at around 260 nm (ESI Fig. S1†).The obtained UVRR spectral data were analysed using the following protocol for complexes of AlkB·substrate complexes:
Spectra of (1) AlkB·substrate complex in the reaction buffer, (2) substrate in reaction buffer, (3) AlkB in reaction buffer and27 reaction buffer with the co-factors (FAS and 2-OG) were recorded. In case of experiments done in D2O, all the above mentioned samples were prepared in D2O. To obtain spectrum of the substrate bound to AlkB, individual contributions from unbound AlkB, substrate and buffer were subtracted from the spectrum of AlkB·substrate complex using the internal standard, NaNO3 as the reference. The subtraction procedure employed is depicted below in ESI Fig. S2.†
The difference spectrum obtained (ESI Fig. S2D†) is the spectrum of the substrate bound to AlkB with all contributions from unbound AlkB and unbound substrate removed. The difference spectrum of a substrate bound to an enzyme were obtained as follows:
Buffer contribution was removed from the spectrum of protein by using the NaNO3 band at 1049 cm−1 as reference. The subtraction of the buffer from the AlkB spectrum was carried out until the NaNO3 band reached baseline, to ensure complete removal of any contribution by the buffer. The difference spectrum of the complex of protein·substrate was obtained by first subtracting the spectrum of protein [AlkB minus buffer] from the protein·substrate complex. Following this, the contribution from unbound substrate (nucleotide or oligomer) was then removed from the protein·substrate minus [AlkB-buffer] spectrum until the bands corresponding to free nucleotide were removed.
The Raman bands of the difference spectrum were fit using Lorentzian line fits to obtain wavenumbers (cm−1) of bands of the substrate bound to AlkB. These wavenumbers were then compared with the wavenumbers obtained from the UVRR spectrum of the free (unbound) substrate to obtain Raman wavenumber shifts. These wavenumber shifts were then used to deduce the specific interactions of AlkB with the substrate.
The band fitting protocol employed for the determining band positions in the difference spectrum of the enzyme·substrate complex was done as follows:
The spectrum of the free (unbound) substrate was fit using the Lorentzian line fitting procedure detailed earlier. Parameters such as the bandwidths, positions and areas of each band were noted down. The Raman bands in the difference spectrum were fit using the Lorentzian line fitting procedure but the width of each band was constrained by fixing the band widths of the Raman bands in the difference spectrum with the band widths obtained from the free substrate. This was followed by relaxing the width constraints of the each of the bands in the difference spectrum, one at a time and checking to ensure the best possible fit that could be obtained for the overall spectrum. The consistency of the fits was checked for experiments done across days using the above mentioned protocol. The reported relative shifts for enzyme·substrate complexes throughout this study were obtained from an average of 3 or more data sets and are accurate to ±3 cm−1.
Results
AlkB selectively binds to the protonated, amino form of 1-me-dAMP
The ideal substrates for AlkB are single-stranded DNA and RNA [kcat/Km 8.6 min−1 μM−1 for methylated poly (dA)].8,34 The minimal substrate for AlkB is the mononucleotide, 1-methyladenosine-5′-monophosphate.8 There are no reported structural studies of the interactions of AlkB with a methylated nucleotide. We examined the complex of AlkB and 1-me-dAMP in solution to infer the interactions of AlkB with its minimal substrate.In a prior study we found that the mononucleotide, 1-me-dAMP with a pKa of 7.2, exists as a mixture of positively charged amino and neutral imino forms at physiological pH.30 AlkB exhibits optimum activity for methylated DNA at pH (8.0).35 At pH 8.0, where the neutral imino form predominates,30 the vibrational spectrum of AlkB·1-me-dAMP shows no perturbations in 1-me-dAMP bands (ESI Fig. S3†). At pH 6.0, wherein the amino form of 1-me-dAMP is the primary tautomer, several changes are observed in the spectrum of 1-me-dAMP in complex with AlkB (Fig. 2). Low intensity bands at 1643 cm−1 and 1594 cm−1 comprising significant contribution from the exocyclic amino group modes all exhibit upshifts upon binding to the enzyme and show increase in Raman band intensities. Downshifts in the wavenumbers of bands at 1565 cm−1 and 1426 cm−1 which correspond to C2–H stretching modes indicate that the substrate is distorted by the overall active-site environment. The two bands at 1330 cm−1 and 1503 cm−1 comprising of the purine ring modes coupled to the methyl modes exhibit downshift of 2 cm−1 in wavenumbers.
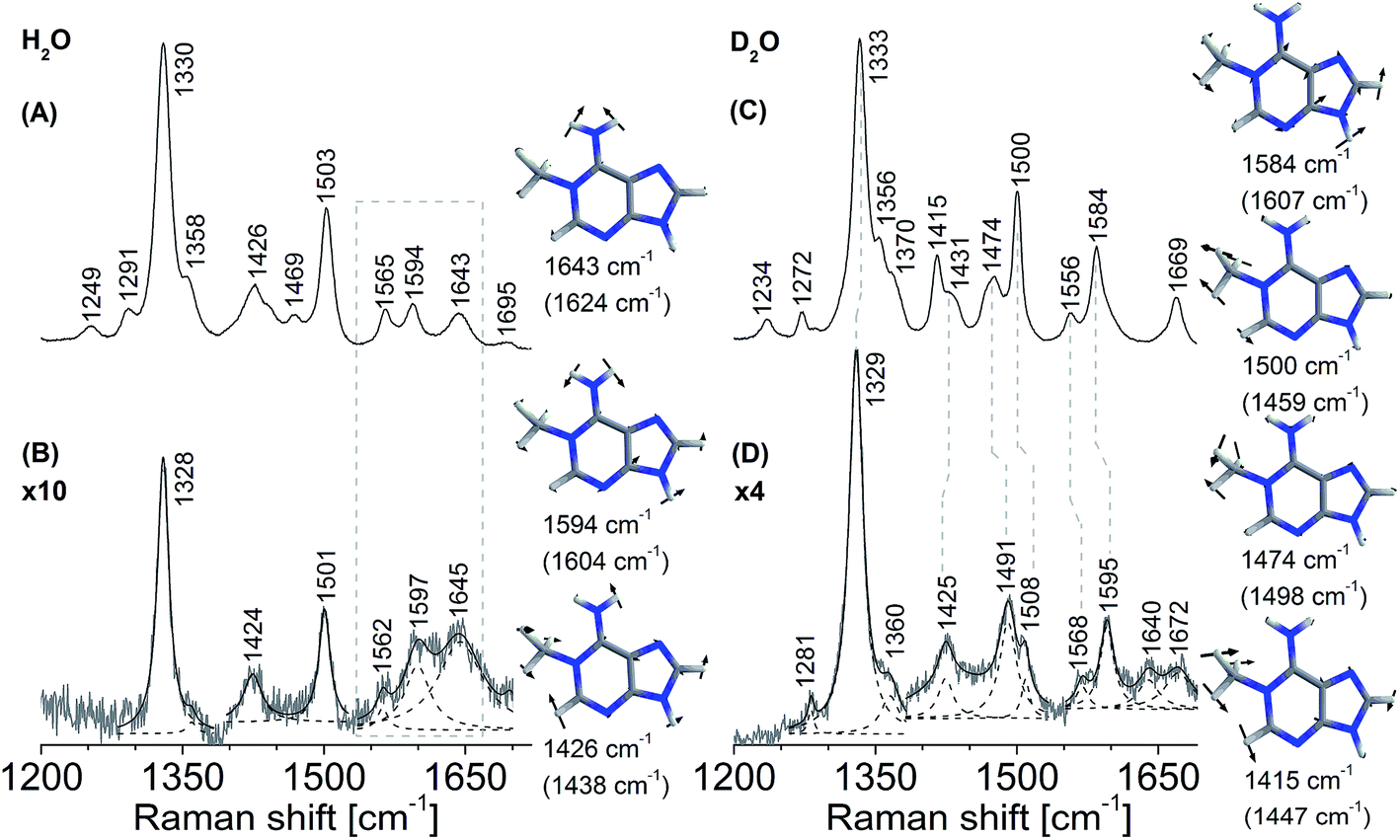 | ||
| Fig. 2 Resonance Raman spectra of AlkB bound to 1-me-dAMP, pH 6.0. (A) 1-me-dAMP in MES buffer (H2O). (B) Difference spectrum of AlkB·1-me-dAMP complex with contributions from unbound AlkB and unbound 1-me-dAMP removed. The result was multiplied by a factor of 10. (C) 1-me-dAMP in MES buffer (D2O). (D) Difference spectrum of AlkB·1-me-dAMP complex with contributions from unbound AlkB and unbound 1-me-dAMP removed. The result was multiplied by a factor of 4. The spectra were obtained using laser excitation of 260 nm. Computed (B3LYP/6-31G**level of theory) wavenumbers are shown in parentheses. Lorentzian band fits are shown as dashed lines. | ||
Deuteration enhances the overall intensities in the 1-me-dAMP spectrum in comparison with the spectrum obtained in water. The positions of bands show significant influence of binding with AlkB. The methyl bending modes coupled to the purine ring stretching modes exhibit substantial changes: bands at 1584 cm−1, 1500 cm−1, and 1474 cm−1 all exhibit upshifts. The methyl modes (1500 cm−1 and 1474 cm−1) show alteration in intensity. The amino group bending mode observed as a band at 1272 cm−1 shows a significant upshift of 9 cm−1 to 1281 cm−1. Thus, these shifts indicate that the protonated amino form of 1-me-dAMP binds to AlkB and the key players in this interaction are the amino and methyl groups.36,37 Detailed descriptions of mode assignments of 1-me-dAMP are given in ESI Table S1.†
TAG I, an E. coli glycosylase involved in base excision repair of methylated bases has been shown to recognize the neutral form of 3-me-dAMP in preference to the positively charged 3-me-dAMP.38 To test whether AlkB binds to the neutral, imino form of 1-me-dAMP as well, we examined the AlkB·1-me-dAMP complex at pH 8.0, wherein the imino form of 1-me-dAMP dominates. For this particular set of experiments, we chose a Raman excitation wavelength of 225 nm. We have shown in previous work that this results in selective enhancement of bands from the imino form of 1-medAMP.30 We find that the presence of AlkB shifts the equilibrium between the two tautomers to favour the amino form. This is manifested as a loss of the intensity of the bands of the imino form. The absence of any perturbations in the positions of the Raman bands of the imino form confirms that it does not bind to AlkB. ESI Fig. S4† and accompanying discussion provide the analysis of these spectra.
Hydrogen-bonding and cation–π interactions stabilize AlkB-trimer complex
In contrast to the nucleotide, 1-me-dAMP in a DNA trimer exists exclusively in the amino form at pH 8.0.30 This phenomenon of change in pKa of a nucleotide when incorporated into an oligomer is known39 and is ascribed to the sequence-specific nearest-neighbor stacking interactions of nucleotides.39 Major changes in the trimer spectra upon AlkB binding map to the amino group, methyl moiety,and the purine ring modes of 1-me-dAMP (Fig. 3). Perturbations are also observed for the C2–H stretching mode seen as a band at cm−1. Shifts are observed for bands at 1582 cm−1 (H2O) and 1584 cm−1 (D2O), corresponding to the amino group bending vibrations, which upshift by 2 cm−1 and 6 cm−1 respectively. Perturbations of modes involving the amino group bending vibrations upon binding to AlkB are indicative of the specific interaction of the exocyclic NH2 group with the active-site residues of AlkB. Crystal structure data shows that Asp 135 is within hydrogen bonding distance from the amino group of 1-me-dAMP.10,12 Extensive biochemical characterisation and computational simulations of AlkB with 3-me-dCMP and exocyclic adducts have deduced the interaction of Asp 135 with the amino group of the substrates of AlkB.24 However, in addition to Asp 135, there is also a water molecule within hydrogen bonding distance of the methylated bases as observed from the crystallographic data (ESI Fig. S10†). Hence, the perturbations observed in the NH2 bending vibrational modes of the nucleotides could also be attributed to the water molecule.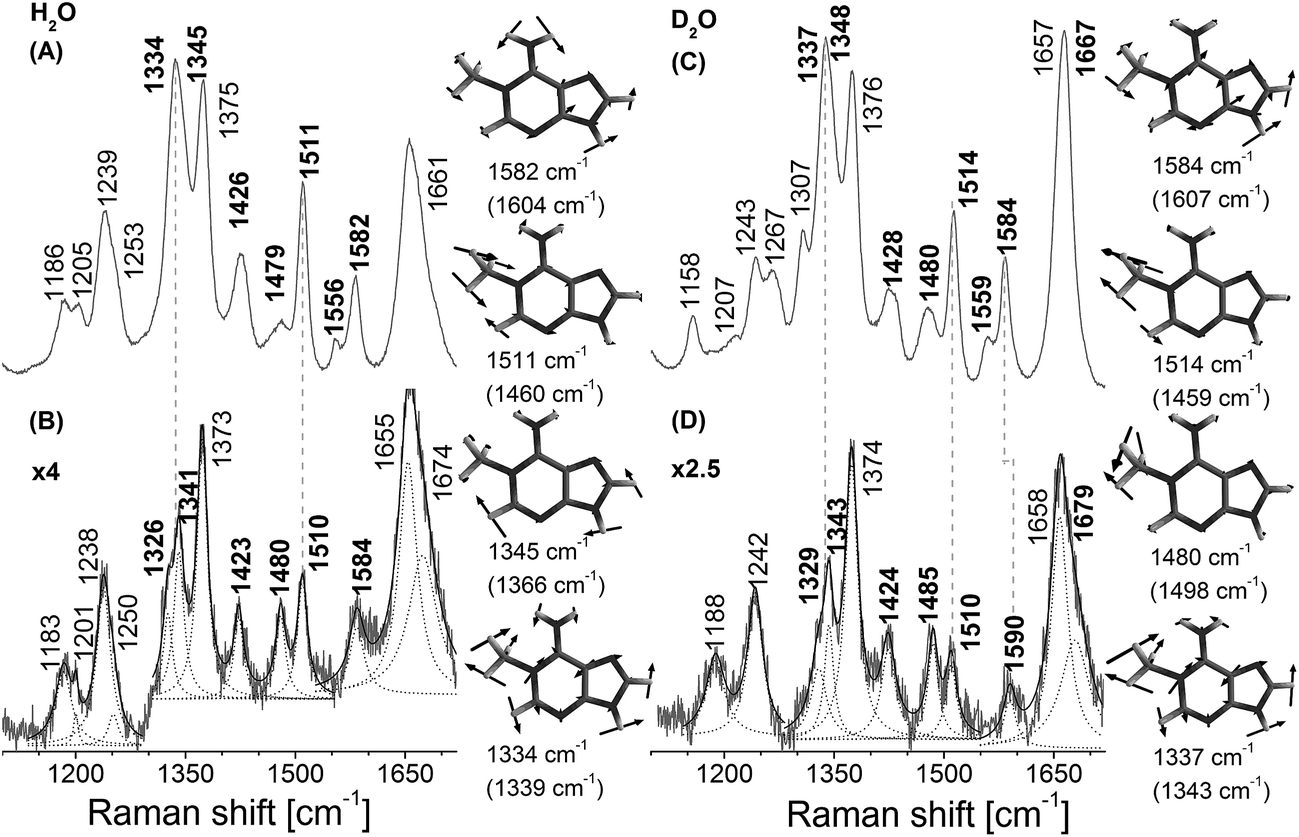 | ||
| Fig. 3 Resonance Raman spectra of AlkB bound to 5′-dT-(1-me-dA)-dT-3′ trinucleotide, pH 8.0. (A) 5′-dT-(1-me-dA)-dT-3′ trinucleotide in Hepes buffer (H2O). (B) Difference spectrum of AlkB·dT-(1-me-dA)-dT complex with contributions from unbound AlkB and unbound dT-(1-me-dA)-dT removed. The result was multiplied by a factor of 4. (C) 5′-dT-(1-me-dA)-dT-3′ trinucleotide in Hepes buffer (D2O). (D) Difference spectrum of AlkB·dT-(1-me-dA)-dT complex with contributions from unbound AlkB and unbound dT-(1-me-dA)-dT (D2O) removed. The result was multiplied by a factor of 2.5. The spectra were obtained using laser excitation of 260 nm. The wavenumbers corresponding to 1-me-dAMP modes are depicted in bold. Lorentzian band fits are shown as dashed lines. Computed (B3LYP/6-31G**level of theory) wavenumbers are shown in parentheses. | ||
The most significant change in the UVRR spectrum of 1-me-dAMP in the trimer upon binding to AlkB is the radical decrease in intensity of bands at 1334 cm−1 and 1345 cm−1, which correspond to the purine ring modes. The origin is accredited to the stacking interactions of 1-me-dAMP and the likely residues are Trp 69 and His 131 (Fig. 1). This stacking effect is more pronounced in the trimer containing 1-me-dAMP than in just the methylated nucleotide. Thus, the flanking nucleobases are important for the appropriate orientation of the methylated nucleotide between the two hydrophobic residues, in addition to the positive charge on 1-me-dAMP, contributing to the cation–π interactions.
The band at 1511 cm−1 corresponding to methyl group bending mode coupled to the purine ring stretching vibrations also shows a reduction in intensity. The change in the methyl group bending mode could be due to the interaction of the CH3 moiety with 2-OG and decrease in intensity from the stacking effect on ring mode vibrations as discussed earlier. In fact, the key contribution of co-factors, 2-OG and Fe2+, to substrate recognition by AlkB is gauged from our experiments wherein AlkB does not bind to the 1-me-dAMP trimer in their absence (ESI Fig. S5†). The decrease in the intensity of the band at 1511 cm−1 comprising of the methyl bending mode and change in the NH2 group bending vibrational modes are well perceived upon isotope exchange of the AlkB·1-me-dAMP complex (seen at 1514 cm−1 in D2O). The decrease in the intensities and changes in the C2–H stretching seen as bands at 1480 cm−1 and 1428 cm−1 are duplicated in the spectrum upon deuteration (Fig. 3D).
3-me-dCMP in the active-site of AlkB
Identical active-site amino acids are observed in the vicinity of both 1-me-dAMP and 3-me-dCMP12 (Fig. 1). Hence, the active-site chemistry of AlkB appears to be similar for both the methylated substrates. From the vibrational spectrum, interaction of the exocyclic amino group of 3-me-dCMP in the active-site of AlkB is evident from the significant shift for NH2 bending vibrations (Fig. 4). This band at 1549 cm−1 comprising of major contribution from the amino group of 3-me-dCMP, shows a downshift of 10 cm−1 (ESI Table S2† and Fig. 4). As mentioned earlier with 1-me-dAMP in the active-site of AlkB, the hydrogen bonding interaction can either be between the NH2 group of 3-me-dCMP and Asp 135 or with the water molecule present in the active-site. The 1270 cm−1 band shows a decrease in intensity with a 3 cm−1 downshift in wavenumber. The primary contributor to this band is the methyl group bending vibrations coupled to pyrimidine ring modes, C2–N3 stretching and C4–N4 stretching vibrations, with minor contribution from the amino group bending vibrations. An interesting result is the perturbation of methyl mode (1455 cm−1) of 3-me-dCMP observed as a downshift of 8 cm−1 upon AlkB addition.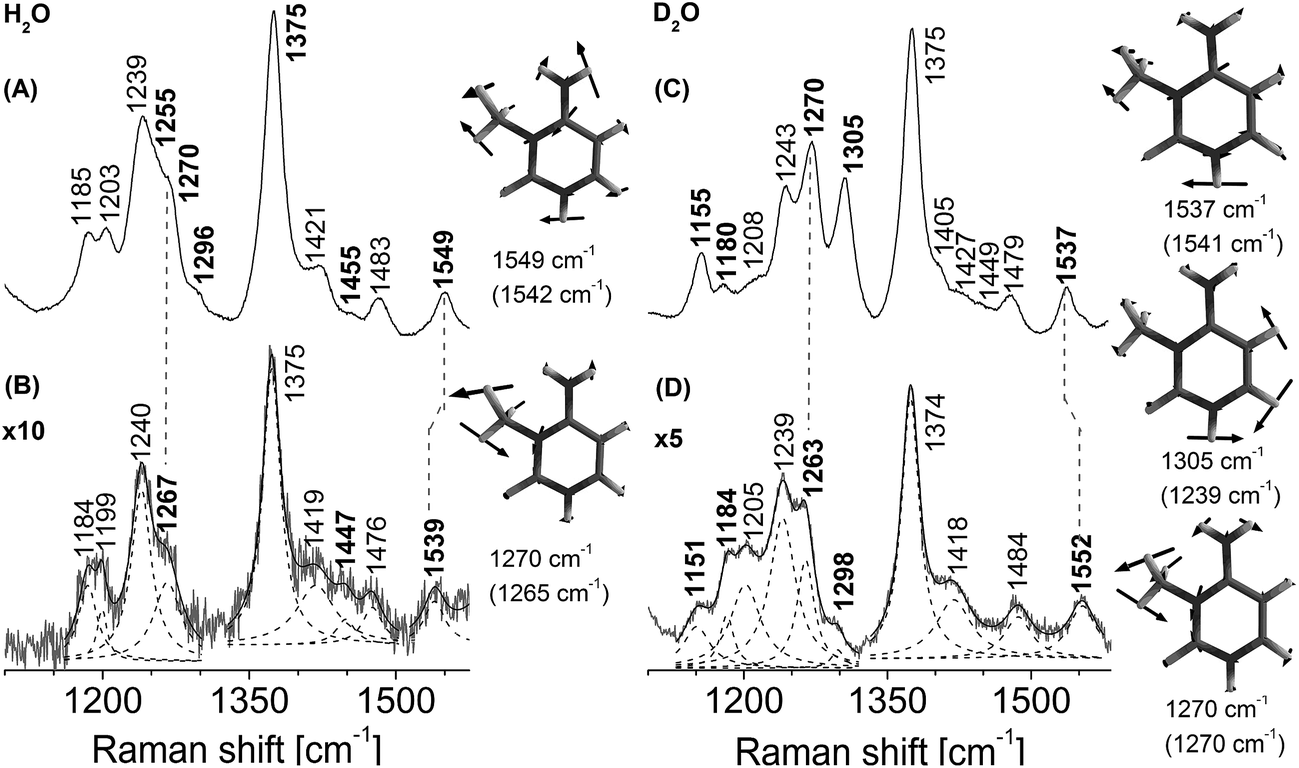 | ||
| Fig. 4 Resonance Raman spectra of AlkB bound to 5′-dT-(3-me-dC)-dT-3′ trinucleotide, pH 8.0. (A) 5′-dT-(3-me-dC)-dT-3′ trinucleotide in Hepes buffer (H2O). (B) Difference spectrum of AlkB·dT-(3-me-dC)-dT complex complex with contributions from unbound AlkB and unbound dT-(3-me-dC)-dT (H2O) removed. The result was multiplied by a factor of 10. (C) 5′-dT-(3-me-dC)-dT-3′ trinucleotide in Hepes buffer (D2O). (D) Difference spectrum of AlkB·dT-(3-me-dC)-dT complex with contributions from unbound AlkB and unbound dT-(3-me-dC)-dT (D2O) removed. The result was multiplied by a factor of 5. The spectra were obtained using laser excitation of 260 nm. The wavenumbers corresponding to 3-me-dCMP modes are depicted in bold. Lorentzian band fits are shown as dashed lines. Computed (B3LYP/6-31G**level of theory) wavenumbers are shown in parentheses. | ||
Upon deuteration, the stacking effect of 3-me-dCMP is evident by the decrease in intensity observed for the 1270 cm−1 band comprising of the ring stretching modes along with a substantial downshift of 7 cm−1. However, the decline in intensity of the ring modes is comparatively lower than that observed for dT-(1-me-dA)-dT. This difference can be explained in terms of the surface area occupied by 1-me-dAMP as compared to 3-me-dCMP. Larger surface area enables better accommodation of bulkier 1-me-dAMP than 3-me-dCMP. The most significant change is in the spectrum of 3-me-dCMP bound to AlkB is in the amino group vibrations seen at 1537 cm−1 which upshift by 15 cm−1 to 1552 cm−1 (Fig. 4D).
Recognition cue of AlkB deduced from studies of AlkB bound to non-cognate nucleotides
Crystal structures of non-cognate enzyme-DNA complexes have provided meaningful insights into mechanisms of enzyme recognition and catalysis. The crystal structures of only a handful of such complexes are currently available. BamHI and EcoRV, both restriction endonucleases which are highly selective in their recognition utilize different mechanisms for recognition of cognate and non-cognate bases.40–42 However there are no studies reporting the interaction of AlkB with non-methylated DNA. This interaction is of relevance as it provides vital clues about the initial process of substrate discrimination by AlkB.It is proposed that AlkB recognizes 1-me-dAMP and 3-me-dCMP in preference to 1-me-dGMP, 3-me-dTMP or dUMP via the position of the NH2 group.12 The Asp 135 residue of AlkB is within hydrogen bond distance from the amino group of the 1-me-dAMP and 3-me-dCMP as seen from the crystal structure data.12 In case of 1-me-dAMP pertaining crystal structures, the distance of exocyclic amino group and Asp 135 residue ranges from 3.2–3.5 Å (ESI Fig. S10†). The Asp135A AlkB mutant protein displays approximately 16% wild-type activity towards ss DNA containing 1-me-dAMP.43 Furthermore, mutation of Asp 135 to residues Asp135I and Asp135N leads to reduction in demethylation activity toward the 1-me-dAMP,44 the Asp135 mutant shows 13% increase in activity towards 1-me-dGMP as compared to the wild-type AlkB,14 highlighting the fact that the presence of carbonyl group at the N6 position deters lesion recognition by AlkB, thus emphasizing the prominence of amino group in recognition. We provide conclusive evidence for this identification cue utilized by AlkB to differentiate between various nucleotides during damage recognition. When AlkB is added to 5′-dT-dA-dT-3′, only the modes arising from the exocyclic amino group are perturbed. Fig. 5A shows a downshift of the 1604 cm−1 band corresponding to the NH2 bending mode of dAMP, by 5 cm−1 to 1599 cm−1 upon AlkB binding. The other mode containing NH2 contribution is seen as a band at 1509 cm−1 which downshifts to 1506 cm−1. Perturbations are not observed for any other bands in the dAMP spectrum implying the interaction of only the amino group of dAMP with AlkB.
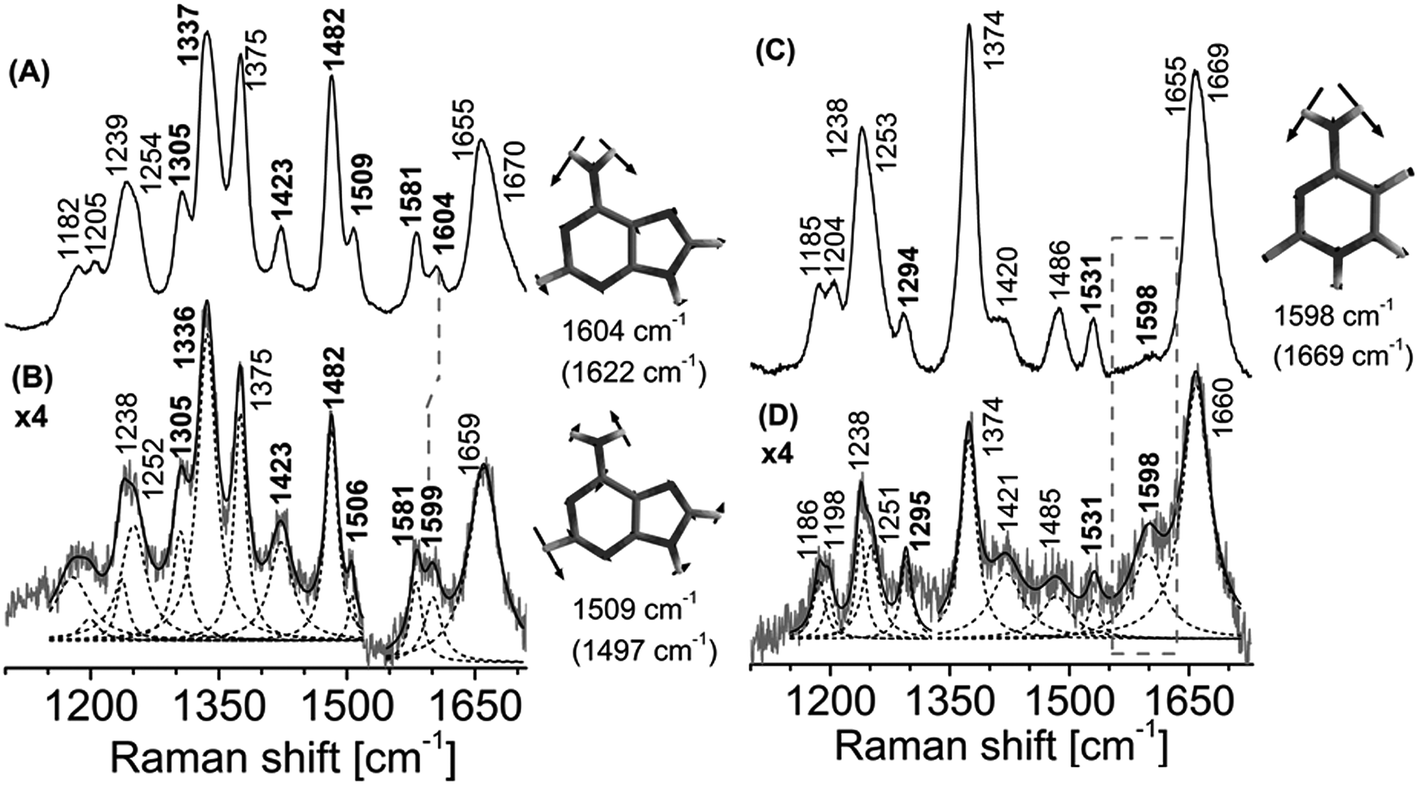 | ||
| Fig. 5 Resonance Raman spectra of AlkB bound to 5′-dT-dA-dT-3′ trinucleotide and AlkB bound to 5′-dT-dC-dT-3′ trinucleotide, pH 8.0. (A) 5′-dT-dA-dT-3′ trinucleotide in Hepes buffer (H2O). (B) Difference spectrum of AlkB·dT-dA-dT complex with contributions from unbound AkB and unbound dT-dA-dT removed. (C) 5′-dT-dC-dT-3′ trinucleotide in Hepes buffer (H2O). (D) Difference spectrum of AlkB·dT-dC-dT complex with contributions from unbound AlkB and unbound dT-dC-dT removed. The result was multiplied by a factor of 4. The spectra were obtained using laser excitation of 260 nm. The wavenumbers corresponding to dAMP and dCMP are depicted in bold. Lorentzian band fits are shown as dashed lines. Computed (B3LYP/6-31 G**) wavenumbers are shown in parentheses. | ||
Similar result is observed in case of the trimer containing dCMP upon AlkB binding (Fig. 5D). In the case of dCMP there is no change in terms of wavenumber of the amino group upon interaction with AlkB. However, there is a significant increase in intensity of the band corresponding to NH2 mode at 1598 cm−1. Changes in the ring vibrational modes along with the NH2 vibrational modes are also observed in complexes of AlkB with 5′-dT-dA-dT-3′and with 5′-dT-dC-dT-3′. The NH2 vibrational modes are coupled to the N3C4 ring vibrational modes. Upon deuteration, the NH2 group which is involved in hydrogen bonding interaction, decouples from the ring modes, providing further evidence for the binding of AlkB to dA and dC containing trimers (ESI Fig. S6, Tables S3 and S4†). In case of trimer, dT bands do not show any shift upon binding to the enzyme, ascribed the limited interaction to dA and dC.
The relevance of NH2 group in recognition by AlkB is further emphasized from the experiments of AlkB with trimers containing dGMP and dUMP. AlkB does not bind either dGMP or dUMP DNA trimers (ESI Fig. S7†), which contain a carbonyl group at the N6 and N4 position respectively, in place of the amino group.
Criterion for substrate recognition – charge distribution in nucleotides, deduced from computational prediction
From our results, we find that AlkB binds to the positively charged methylated substrates and there is a substantial decrease in the ring modes of the methylated nucleotides upon AlkB binding. We attribute the binding of AlkB to the positively charged nucleotides and stacking of the methylated bases in the active-site of AlkB to cation–π interactions.Cation–π interactions are important non-covalent bonding interactions contributing to protein stability, receptor-ligand recognition and in protein–DNA interactions.45 A number of experiments and theoretical data have established that it is a strong interaction, with several studies demonstrating it to be nearly comparable in energy to a hydrogen bond.45 The N-alkylated heterocyclic compounds are effective cations in a cation–π interaction.46 In addition, tryptophan possessing the highest electrostatic potential for binding cations, is reported to be the most favorable amino acid for cation–π interactions.47 Previous studies suggest that AlkB enzyme preferentially binds to cationic base over neutral base to perform repair mechanism.24
We have used ab initio quantum mechanical calculations to substantiate our experimental results of the presence of positive charge on the nucleotides, thus providing basis for the cation–π interaction. Computed charge distribution shows that the positive charge on 1-me-dAMP and 3-me-dCMP is largely localized on the pyrimidine ring (ESI Fig. S8†). Our results from the Raman experiments show a substantial decrease in the intensity of ring modes of 1-me-dAMP in comparison to 3-me-dCMP. This is in agreement with the crystal structure data that show a distance of 3.790 Å between 1-me-dAMP and Trp 69 and a distance of 4.365 Å between 3-me-dCMP and Trp 69 (ref. 12). Furthermore, the electrostatic potential energy surface (EPS) maps reporting on the charges of molecules were calculated (Fig. 6). These depict the distribution of positive charge on the overall surface of 1-methyladenine and 3-methylcytosine whereas the non-cognate bases, adenine and cytosine, display a trend towards the negative charge.
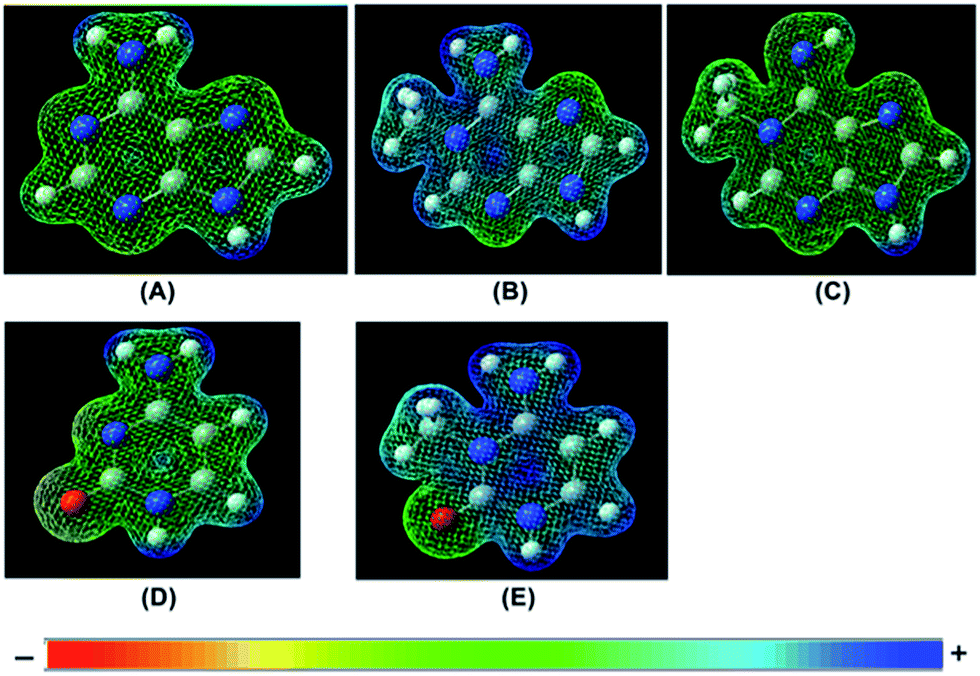 | ||
| Fig. 6 Computed (B3LYP/6-31G** level of theory) electrostatic potential energy surfaces. (A) Adenine. (B) 1-Methyl-adenine, amino form. (C) 1-Methyl-adenine, imino form. (D) Cytosine. (E) 3-Methyl-cytosine, amino form. The color bar at the bottom depicts the color gradient representing the charge. Blue represents the most positive charge and red depicts the most negative charge. | ||
Thus the presence of a positive charge on the nucleotides is one of the deciding factors utilized by AlkB to single out methylated bases amongst other normal nucleotides.
Discussion
AlkB recognizes methylated purines and pyrimidines in addition to bulky etheno adducts like ethenodA, making it one of the most promiscuous repair enzymes of the dioxygenase family. In order to deduce the structural changes occurring in the positively charged methylated substrates rather than the conformational changes in AlkB, we have employed UV resonance Raman spectroscopy to study the complex of AlkB with its substrates in solution. The specific excitation wavelength of 260 nm enables us to monitor the changes occurring exclusively in the methylated nucleotides in the protein–DNA complex.Our results show that AlkB binds the protonated amino form of 1-me-dAMP in preference to the neutral imino form. Our data emphasizes the role of protonation state of the substrates as a crucial factor governing their affinity to AlkB. We show that hydrogen bond formed between the amino group of the methylated substrates with the active-site residues of AlkB, distorts the NH2 bending mode of 1-me-dAMP (Fig. 2–4), manifested as shifts in Raman wavenumber of bands corresponding to the NH2 group vibrations. Stacking interactions between the substrates and active-site amino acids of AlkB are inferred by the significant decrease in intensities of bands corresponding to the purine ring modes. Positively charged 1-me-dAMP and 3-me-dCMP are accommodated suitably in the active-site pocket of AlkB between the amino acids, Trp 69 and His 131.
We ascribe the affinity of AlkB to protonated substrates with an overall positive charge, to cation–π interactions. Many enzymes involved in the repair of methylated substrates appear to acquire their substrate specificity through favorable stacking interactions between their active-site residues and substrates. ABH2, the human homologue of AlkB which recognizes 1-me-dAMP has Phe 124 and His 171 stacked against the charged nucleotide.
AlkB binds to the protonated methylated nucleotides with higher affinity than to neutral ε-dAMP, most likely due to the absence of cation–π interaction in case of ε-dAMP.12 π–π stacking interactions are weaker than cation–π interactions and enzymes utilizing π–π stacking compensate for the weak stacking interaction by forming multiple hydrogen bonds with the substrate, thus stabilizing the substrate in the active-site pocket. These enzymes, such as AAG, usually prefer the bulkier substrates like ε-dAMP48 or in case of FTO49 and TAG38 that recognize neutral 3-me-dTMP and 3-me-dAMP, there are 3 and 4 hydrogen bonding partners, respectively, conferring specificity as compared to only one hydrogen bond in case of AlkB and its methylated substrates. Yi et al.14 have calculated the natural bond orbital (NBO) charge for 3-me-dCMP and have ascribed the AlkB recognition factor to be the positive charge on the two methylated substrates (1-me-dAMP and 3-me-dCMP). The exocyclic amino group holds a positive change and shows interaction with anionic Asp135 (ref. 14). Protonation of purine has been shown to enhance the π-orbital overlap between the methylated base and aromatic amino acid leading to favorable stacking interaction.4,50–52
Energetic evaluation of favorable cation–π interactions predicted from ab initio calculations of structures of various proteins, have shown tryptophan to be the most likely aromatic amino acid to be involved in cation-π interaction.53 The importance of Trp in the cation–π interaction predicted for AlkB with methylated substrates has also been established from mutagenesis experiments of Trp 69 (W69A), that abolishes the activity of AlkB.43 In addition, the stacking interactions between the nucleotides and aromatic amino acids decrease in the order of tryptophan, tyrosine and phenylalanine.51 Furthermore, from our data, the absence of stacking interactions in non-methylated substrates exemplifies the presence of cation–π interactions in the active-site of AlkB with methylated nucleotides.
To our knowledge we provide the first report of the stable complex of AlkB with non-methylated oligomer containing dAMP and oligomer with dCMP. We find that the only contacts established by AlkB with these normal, unmodified nucleotides are with the amino group. dUMP and dGMP that possess carbonyl group instead of an amino group are not recognized by AlkB. The presence of Asp 135 residue renders the presence of the carbonyl group at the N6/N4 position unfavorable for binding. Hence, the exocyclic amino group is a preliminary criterion for recognition of potential substrates by AlkB. Through our experiments of AlkB with normal, unmodified oligomers, dT-dA-dT, and dT-dC-dT, we provide a rationale for the DNA sampling and recognition by AlkB. In addition, we demonstrate the presence of a carbonyl group at N6 or N4 positions for purines (dGMP) or pyrimidines (dUMP) respectively, acts a deterring factor for recognition by AlkB.
The presence of neighboring nucleotides further contributes to the proper positioning of the nucleotide in the active-site as observed from the spectra of nucleotide versus the trimer; wherein the decrease in intensities of bands corresponding to the ring modes of the methylated substrates are less prominent in case of the nucleotide versus the trimer, suggestive of higher stacking interactions between amino acids of AlkB with 1-me-dAMP containing trimer (ESI Fig. S9†).
Our experiments show that the combination of positive charge coupled with increased surface area along with the presence of amino group provide additional stability along with specificity and render 1-me-dAMP followed by 3-me-dCMP suitable substrates for AlkB.
Conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article.Abbreviations
| 1-me-dAMP | 1-Methyl-deoxyadenosine-5′-monophosphate |
| 3-me-dCMP | 3-Methyl-deoxycytidine-5′-monophosphate |
| dAMP | Deoxyadenosine-5′-monophosphate |
| dCMP | Deoxycytidine-5′-monophosphate |
| 2-OG | 2-Oxoglutarate |
| UVRR | Ultraviolet Resonance Raman |
| D2O | Deuterium oxide |
| EPS | Electrostatic potential energy surface |
| NBO | Natural bond orbital |
Acknowledgements
The authors thank Dr Barbara Sedgwick (London Cancer Research Institute, UK) for kindly providing us with the E. coli strain with pBAR plasmid expressing AlkB. This work was supported by grants from the Department of Biotechnology, Government of India to M. P. M. P. is a recipient of the Innovative Young Biotechnologist Award, Department of Biotechnology, The Government of India. N. J. was supported by a PhD fellowship by the Council for Scientific and Industrial Research, India.References
- T. Lindahl, Nature, 1993, 362, 709–715 CrossRef CAS PubMed.
- J. I. Friedman and J. T. Stivers, Biochemistry, 2010, 49, 4957–4967 CrossRef CAS PubMed.
- J. C. Delaney and J. M. Essigmann, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14051–14056 CrossRef CAS PubMed.
- T. Ishida, M. Shibata, K. Fujii and M. Inoue, Biochemistry, 1983, 22, 3571–3581 CrossRef CAS PubMed.
- F. Drablos, E. Feyzi, P. A. Aas, C. B. Vaagbo, B. Kavli, M. S. Bratlie, J. Pena-Diaz, M. Otterlei, G. Slupphaug and H. E. Krokan, DNA Repair, 2004, 3, 1389–1407 CrossRef CAS PubMed.
- B. Sedgwick, Nat. Rev. Mol. Cell Biol., 2004, 5, 148–157 CrossRef CAS PubMed.
- L. Aravind and E. V. Koonin, Genome Biol., 2001, 2(3), research0007.1–research.8 CrossRef.
- P. Koivisto, T. Duncan, T. Lindahl and B. Sedgwick, J. Biol. Chem., 2003, 278, 44348–44354 CrossRef CAS PubMed.
- C. Yi, B. Chen, B. Qi, W. Zhang, G. Jia and L. Zhang, et al., Nat. Struct. Mol. Biol., 2012, 19(7), 671–676 CAS.
- B. Yu, W. C. Edstrom, J. Benach, Y. Hamuro, P. C. Weber, B. R. Gibney and J. F. Hunt, Nature, 2006, 439, 879–884 CrossRef CAS PubMed.
- C. G. Yang, C. Q. Yi, E. M. Duguid, C. T. Sullivan, X. Jian, P. A. Rice and C. He, Nature, 2008, 452, 961–964 CrossRef CAS PubMed.
- B. Yu and J. F. Hunt, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14315–14320 CrossRef CAS PubMed.
- B. Bleijlevens, T. Shivarattan, E. Flashman, Y. Yang, P. J. Simpson, P. Koivisto, B. Sedgwick, C. J. Schofield and S. J. Matthews, EMBO Rep., 2008, 9, 872–877 CrossRef CAS PubMed.
- C. Q. Yi, G. F. Jia, G. H. Hou, Q. Dai, W. Zhang and G. Q. Zheng, et al., Nature, 2010, 468(7321), 330–333 CrossRef CAS PubMed.
- Y. Mishina, L. X. Chen and C. He, J. Am. Chem. Soc., 2004, 126, 16930–16936 CrossRef CAS PubMed.
- T. F. Henshaw, M. Feig and R. P. Hausinger, J. Inorg. Biochem., 2004, 98, 856–861 CrossRef CAS PubMed.
- T. W. Roy and A. S. Bhagwat, Nucleic Acids Res., 2007, 35(21), e147 CrossRef PubMed.
- W. Yang, Cell Res., 2008, 18, 184–197 CrossRef CAS PubMed.
- N. R. Jena, J. Biosci., 2012, 37, 503–517 CrossRef CAS PubMed.
- M. Kouchakdjian, Biochemistry, 1991, 30, 1403–1412 CrossRef CAS PubMed.
- C. D. M. Churchill, L. A. Eriksson and S. D. Wetmore, Chem. Res. Toxicol., 2011, 24, 2189–2199 CrossRef CAS PubMed.
- M. S. Cooke, M. D. Evans, M. Dizdaroglu and J. Lunec, FASEB J., 2003, 17, 1195–1214 CrossRef CAS PubMed.
- H. Yang, Y. Q. Zhan, D. Fenn, L. M. Chi and S. L. Lam, FEBS Lett., 2008, 582, 1629–1633 CrossRef CAS PubMed.
- A. M. Maciejewska, J. Poznanski, Z. Kaczmarska, B. Krowisz, J. Nieminuszczy, A. Polkowska-Nowakowska, E. Grzesiuk and J. T. Kusmierek, J. Biol. Chem., 2013, 288, 432–441 CrossRef CAS PubMed.
- B. Sedgwick, P. Robins and T. Lindahl, in DNA Repair, Pt A, 2006, vol. 408, pp. 108–120 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- M. H. Jamroz, Vibrational Energy Distribution Analysis VEDA 4.0, Warsaw, Poland, 2004 Search PubMed.
- N. Jayanth and M. Puranik, J. Phys. Chem. B, 2011, 115, 6234–6242 CrossRef CAS PubMed.
- P. Commercial Molecular Graphics Software, 2005, GaussView 2005. Gaussian Inc, PA Search PubMed.
- S. Gogia, H. Balaram and M. Puranik, Biochemistry, 2011, 50, 4184–4193 CrossRef CAS PubMed.
- N. Jayanth, S. Ramachandran and M. Puranik, J. Phys. Chem. A, 2009, 113, 1459–1471 CrossRef CAS PubMed.
- S. Dinglay, S. C. Trewick, T. Lindahl and B. Sedgwick, Genes Dev., 2000, 14, 2097–2105 CAS.
- A. M. Maciejewska, K. P. Ruszel, J. Nieminuszczy, J. Lewicka, B. Sokolowska, E. Grzesiuk and J. T. Kusmierek, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2010, 684, 24–34 CrossRef CAS PubMed.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, Introduction to Infrared and Raman Spectroscopy, Academic Press, New York, 2nd edn, 1975 Search PubMed.
- A. Barth and C. Zscherp, Q. Rev. Biophys., 2002, 35, 369–430 CrossRef CAS PubMed.
- C. Y. Cao, K. Kwon, Y. L. Jiang, A. C. Drohat and J. T. Stivers, J. Biol. Chem., 2003, 278, 48012–48020 CrossRef CAS PubMed.
- S. Acharya, J. Barman, P. Cheruku, S. Chatterjee, P. Acharya, J. Isaksson and J. Chattopadhyaya, J. Am. Chem. Soc., 2004, 126, 8674–8681 CrossRef CAS PubMed.
- D. A. Hiller, A. M. Rodriguez and J. J. Perona, J. Mol. Biol., 2005, 354, 121–136 CrossRef CAS PubMed.
- H. Viadiu, R. Kucera, I. Schildkraut and A. K. Aggarwal, J. Struct. Biol., 2000, 130, 81–85 CrossRef CAS PubMed.
- F. K. Winkler, D. W. Banner, C. Oefner, D. Tsernoglou, R. S. Brown, S. P. Heathman, R. K. Bryan, P. D. Martin, K. Petratos and K. S. Wilson, EMBO J., 1993, 12, 1781–1795 CAS.
- P. J. Holland and T. Hollis, PLoS One, 2010, 5(1), e8680 Search PubMed.
- C. Zhu and C. Yi, Angew. Chem., Int. Ed. Engl., 2014, 53, 3659–3662 CrossRef CAS PubMed.
- N. Zacharias and D. A. Dougherty, Trends Pharmacol. Sci., 2002, 23, 281–287 CrossRef CAS PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS PubMed.
- D. E. Schlamadinger, M. M. Daschbach, G. W. Gokelb and J. E. Kim, J. Raman Spectrosc., 2010, 42(4), 633–638 CrossRef PubMed.
- A. Y. Lau, M. D. Wyatt, B. J. Glassner, L. D. Samson and T. Ellenberger, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13573–13578 CrossRef CAS PubMed.
- Z. F. Han, T. H. Niu, J. B. Chang, X. G. Lei, M. Y. Zhao, Q. Wang, W. Cheng, J. J. Wang, Y. Feng and J. J. Chai, Nature, 2010, 464, 1205–1209 CrossRef CAS PubMed.
- B. F. Eichman, E. J. O'Rourke, J. P. Radicella and T. Ellenberger, EMBO J., 2003, 22, 4898–4909 CrossRef CAS PubMed.
- T. Ishida, M. Doi and M. Inoue, Nucleic Acids Res., 1988, 16, 6175–6190 CrossRef CAS PubMed.
- T. Ishida, M. Doi, H. Ueda, M. Inoue and G. M. Scheldrick, J. Am. Chem. Soc., 1988, 110, 2286–2294 CrossRef CAS.
- J. P. Gallivan and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: This article contains supplemental Fig. S1–S10, supplemental Tables S1–S4 and additional references. See DOI: 10.1039/c7ra11333a. |
| ‡ Present address: Hindustan Unilever Research Centre, Whitefield, Bangalore 560066, India |
| This journal is © The Royal Society of Chemistry 2018 |