
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImprovement of O2 adsorption for α-MnO2 as an oxygen reduction catalyst by Zr4+ doping
Yicheng Wang a,
Yaozong Lia,
Zhenghang Lub and
Wei Wang*a
a,
Yaozong Lia,
Zhenghang Lub and
Wei Wang*a
aDepartment of Applied Chemistry, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, PR China. E-mail: wangweipaper@163.com; Fax: +86-22-27403389; Tel: +86-13512958953
bKimball Union Academy, NH 03770, USA
First published on 15th January 2018
Abstract
Zr4+ doped α-MnO2 nanowires were successfully synthesized by a hydrothermal method. XRD, SEM, TEM and XPS analyses indicated that Mn3+ ions, Mn4+ ions, Mn4+δ ions and Zr4+ ions co-existed in the crystal structure of synthesized Zr4+ doped α-MnO2 nanowires. Zr4+ ions occupied the positions originally belonging to elemental manganese in the crystal structure and resulted in a mutual action between Zr4+ ions and Mn3+ ions. The mutual action made Mn3+ ions tend to lose their electrons and Zr4+ ions tend to get electrons. Cathodic polarization analyses showed that the electrocatalytic activity of α-MnO2 for oxygen reduction reaction (ORR) was remarkably improved by Zr4+ doping and the Zr/Mn molar ratio notably affected the ORR performance of the air electrodes prepared by Zr4+ doped α-MnO2 nanowires. The highest ORR current density of the air electrodes prepared by Zr4+ doped α-MnO2 nanowires in alkaline solution appeared at Zr/Mn molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :110, which was 23% higher than those prepared by α-MnO2 nanowires. EIS analyses indicated that the adsorption process of O2 molecules on the surface of the air electrodes prepared by Zr4+ doped α-MnO2 nanowires was the rate-controlling step for ORR. The DFT calculations revealed that the mutual action between Zr4+ and Mn3+ in Zr4+ doped α-MnO2 nanowires enhanced the adsorption process of O2 molecules.
:110, which was 23% higher than those prepared by α-MnO2 nanowires. EIS analyses indicated that the adsorption process of O2 molecules on the surface of the air electrodes prepared by Zr4+ doped α-MnO2 nanowires was the rate-controlling step for ORR. The DFT calculations revealed that the mutual action between Zr4+ and Mn3+ in Zr4+ doped α-MnO2 nanowires enhanced the adsorption process of O2 molecules.
1. Introduction
Oxygen reduction reaction (ORR), as the main kinetically limiting cathode reaction in clean energy related technologies, is the primary cause of voltage loss in fuel cells and metal–air batteries.1,2 Developing catalysts for ORR is of significant importance. At present, the most extensively used ORR catalysts are still Pt-based materials due to their relatively high activity and stability.3,4 However, the low abundance of Pt in the earth's crust together with its high price limits its wide and practical applications in fuel cells. In recent years, various efforts have been made to explore non-Pt-based catalysts for ORR, such as Ag-based catalysts,5,6 Pd-based materials,7,8 N-doped carbon,9–11 M–N4-macrocycles,12 Fe–N–C hybrids13–15 as well as some transition metal oxides.16–19 Nevertheless, exploiting inexpensive and highly active electrocatalysts for ORR remains a great challenge.20,21Manganese dioxide (MnO2) has been utilized as an electrocatalyst in metal–air batteries owing to its unique electrochemical performance, low cost and environmentally friendly properties.22–26 MnO2 is a polymorphic material with three different crystal phases including α, β, and γ phases.27 The catalytic activity of MnO2 depends strongly on the crystallographic structure, following an order of α > β > γ as reported in the literature.28 The structural variations of manganese dioxides result from the different bonding ways of the basic [MnO6] octahedral units. Among them, α-MnO2 possesses both (2 × 2) and (1 × 1) tunnels surrounded by double binding octahedral chains, β-MnO2 consists of only (1 × 1) tunnels separated by single chains, and γ-MnO2 displays (1 × 1) and (1 × 2) tunnels enveloped in double chains.29 To the best of our knowledge, morphology is another important influential factor for the electrochemical properties. It was reported that α-MnO2 nanowires possess enhanced electrocatalytic activity compared to the other two shapes namely nanotubes and nanoparticles despite the nanotubes showing a much higher specific surface area.30
Previous research on manganese dioxide as the catalysts toward the ORR mainly focused on the effect of the morphology, crystallographic structure and chemical composition on the catalytic activity.31–34 Recently, most research has been concentrated on further improving the ORR activity by hybridizing or doping manganese oxides with other transition metals, such as Ag,35–37 Pd38 and Ni.39,40 Zr4+ doped cathode materials for metal-ion batteries could improve electronic structure and electrochemical performance.41,42 Up to now, related studies on Zr4+ doped α-MnO2 have not been reported yet.
In the present work, Zr4+ doped α-MnO2 nanowires were prepared via an in situ hydrothermal method. The structures and properties were characterized using X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS). The electrocatalytic activity of Zr4+ doped α-MnO2 nanowires for ORR was evaluated by cathodic polarization curve (CPC) and electrochemical impedance spectroscopy (EIS) measurements. The enhancement mechanism of Zr4+ doping was further explored by density functional theory (DFT) calculations.
2. Experimental
2.1. Catalyst synthesis
Manganese dioxide (α-MnO2) nanowires were synthesized via a hydrothermal method as reported in the literatures.43,44 Typically, 30 mL of 0.1 M KMnO4 aqueous solution was dropped into 30 mL of the MnSO4 solution (0.1 M) at room temperature with vigorous stirring for 30 min. Then the mixture was transferred into a 100 mL Teflon-lined autoclave reactor to perform a hydrothermal reaction at 140 °C for 24 h in a hot air oven. After that, the reactor was cooled naturally to room temperature. The obtained precipitate was washed several times with distilled water and ethanol. Finally, the product was dried overnight in an oven at 60 °C and used for further characterization.For the synthesis of Zr4+ doped α-MnO2 nanowire catalysts, a certain amount of Zr(NO3)4·5H2O was added into 30 mL of 0.1 M MnSO4 solution under vigorous stirring until the formation of a homogeneous solution. The dosage of Zr(NO3)4·5H2O changed depending on the Zr/Mn molar ratio desired 1:70, 1:90, 1:110, 1:140 and 1:150. The other preparation processes were exactly the same with the preparation of α-MnO2 nanowires.
2.2. Physical characterization
The X-ray diffraction (XRD) patterns of the prepared α-MnO2 and Zr4+ doped α-MnO2 catalysts were recorded with a Rigaku MiniFlex II X-ray diffractometer using Cu Kα radiation with a Ni filter. The tube current was 30 mA with a tube voltage of 40 kV. The 2θ angular regions between 20° and 75° were explored at a scan rate of 4° min−1. The morphologies and surfaces of the prepared catalysts were investigated using a Nanosem 430 field-emission scanning electron microscope (FE-SEM, FEI Company) with an acceleration voltage range of 10–30 kV and a Philips Tecnai G2 F20 field-emission transmission electron microscope (TEM, FEI Company) operated at 200 kV. The binding energy of the elements in the catalysts was analyzed by X-ray photoelectron spectroscopy (XPS, PHI1600 ESCA System, PERKIN ELMER, USA) with Al Kα radiation (hv = 1486.6 eV).2.3. Preparation of the air electrode
Briefly, the air electrode was prepared by dispensing 2.2 g of the synthesized catalyst, 1.1 g of acetylene black and 2.1 g of polytetrafluoroethylene (PTFE, 60 wt%) into 20 mL of ethanol. The mixture was stirred at room temperature for 1 hour and then stirred continually at 60 °C for 15 minutes until the mixture getting together. Afterwards, the mixture was rolled to form a film with thickness 0.6 mm. Finally, the film was coated onto a stainless steel net with area 2 cm × 2 cm.2.4. Electrochemical measurements
The electrochemical measurements were carried out in a three-electrode and two-circuit electrochemical cell. One side of the working electrode was immersed in a 3 M KOH solution and the other side was exposed to the air. A ruthenium–titanium mesh was used as a counter electrode. An Hg/HgO (3 M KOH) reference electrode was used through a salt bridge, and all the potentials in this work were referred to Hg/HgO (3 M KOH) reference electrode. The cathodic polarization curves were recorded from the open circuit potentials and the scan rate of the potential was 0.5 mV s−1. Electrochemical impedance spectroscopy (EIS) measurements were performed with an applied potential amplitude ± 5 mV in a frequency range from 10 kHz to 10 mHz. All the electrochemical measurements were conducted in 3 M KOH aqueous solution at 25 ± 0.1 °C. Distilled water and analytical grade reagents were used to prepare the solutions.The electrochemical tests for charge transfer and durability analyses were carried out on a CHI660D electrochemical workstation (CH Instruments, China) with a rotation disk electrode (Pine instrument), using a conventional three-electrode electrochemical cell at 25 ± 0.1 °C. A glassy carbon electrode (GCE) with an area of 0.196 cm2 was used as the substrate for the working electrode, a ruthenium–titanium mesh and Hg/HgO (1 M KOH) were employed as the counter and reference electrodes, respectively. The catalyst ink was fabricated as follows: 2.67 mg as-prepared Zr4+ doped α-MnO2 and 1.33 mg acetylene black was ultrasonically dispersed in a 1.0 mL solution (0.9 mL of ethanol and 0.1 mL of 5 wt% Nafion solution) for 30 min. Then 10 μL of the ink was pipetted onto the GCE surface, dried in air naturally. Linear sweep voltammetry (LSV) curves were obtained in O2-saturated 0.1 M KOH solution at different electrode rotated speeds with a potential scan rate of 5 mV s−1. The current–time durability measurement at −0.4 V (vs. Hg/HgO) was conducted by a rotating disk electrode (RDE) in O2-saturated 0.1 M KOH solution with a rotation rate of 1600 rpm. Koutecky–Levich plots (K–L plots) was fitted by a line and the slopes was used to analyse the number of electrons transferred (n) during the ORR on the basis of Koutecky–Levich equation (K–L equation)45
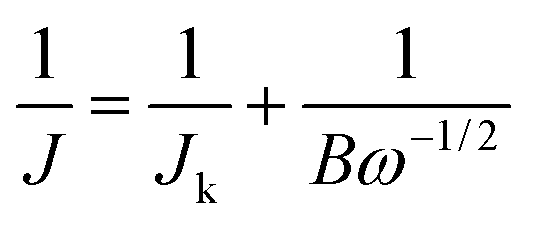 | (1) |
| B = 0.2nFD2/3v−1/6CO2 | (2) |
485 C mol−1), D the diffusion coefficient of oxygen (1.9 × 10−5 cm−2 s−1), v the kinetic viscosity of the electrolyte (0.01 cm−2 s−1), CO2 the bulk concentration of oxygen in electrolyte (1.2 × 10−6 mol cm−3), and n the number of electron exchanged during ORR process. The constant is adopted to be 0.2 when ω is expressed in rpm.46
2.5. Density functional theory(DFT) calculations
CASTEP47,48 based on the Density Functional Theory (DFT) was used to investigate the O adsorption of α-MnO2 and Zr4+ doped α-MnO2 by solving the Kohn–Sham equation of a many-body system with an iterative approach. The system was under the periodic boundary condition and computation was done in a reciprocal space. Plane-wave basis sets with a cutoff energy of 340 eV. The General Gradient Approximation (GGA)49 of Perdew, Burke and Ernzerhof (PBE)50–52 was used as the exchange–correlational functional to describe the interactions among electrons. The Brillouin zone integration was done on a grid of 3 × 2 × 1 with a Gaussian smearing parameter δ of 0.1 eV and a Self-Consistence-Field (SCF) convergence criterion of 1 × 10−5 eV.53 Initial structures were optimized by allowing the atoms positions to be relaxed while the iterative algorithm was updated by the conjugate gradient approach with convergence criteria of 0.05 eV Å−1. The crystal plane (211) of α-MnO2 surface was modeled using three-layer periodic slabs repeating in a (2 × 2) surface structure. Periodic boundary conditions were applied in all three dimensions with a vacuum region of 12 Å along the z-axis. The atoms in the top two layers of the slab were relaxed, whereas the atoms in the third layer were frozen at their bulk positions. Oxygen was placed on one side of the slab. Excluding these species and the top layer of the slab that were allowed to relax in their positions to reach the most stable configuration, all other atoms were frozen at their ideal bulk positions to simulate the bulk and reduce the computational cost.The adsorption energies (ΔEads) were defined relative to the isolated substrate and the molecules in the gas phase, as shown in eqn (3). A negative ΔEads value indicates favorable (exothermic) adsorption.
| ΔEads = E(adsorbate/catalyst) − Ecatalyst − Eadsorbate | (3) |
3. Results and discussions
3.1. Characterization
Fig. 1 shows the XRD patterns of the synthesized α-MnO2 and Zr4+ doped α-MnO2 catalysts with different Zr/Mn molar ratios. It can be seen from Fig. 1a that all the patterns can be indexed to body-centered tetragonal α-MnO2 (JCPDS NO. 44-0141, space group I4/m, a = b = 9.784 Å, c = 2.863 Å), indicating that the crystal structures of the synthesized catalysts are the same with α-MnO2. It can also be observed from Fig. 1b that the positions of the two characteristic peaks at 2θ = 28.9 and 37.6 corresponding to crystal planes (310) and (211) remain the same as that of α-MnO2 when the Zr/Mn molar ratios in the catalysts are 1:140 and 1:150. With increasing Zr/Mn molar ratio, a shift in the positions of the two characteristic peaks in Fig. 1b appears at Zr/Mn molar ratio 1:110 and the shift gets bigger at Zr/Mn molar ratio 1:90 and 1:70. The shift in the peak positions means that a lattice distortion exists. The lattice parameters were calculated by the data in Fig. 1 and shown in Table 1. The lattice parameters a, b and c of Zr4+ doped α-MnO2 become smaller compared with that of α-MnO2. The changes in the lattice parameters also show a distortion in the lattice cell. The results above indicate that α-MnO2 is successfully synthesized and element zirconium is doped into the lattice structure of α-MnO2.
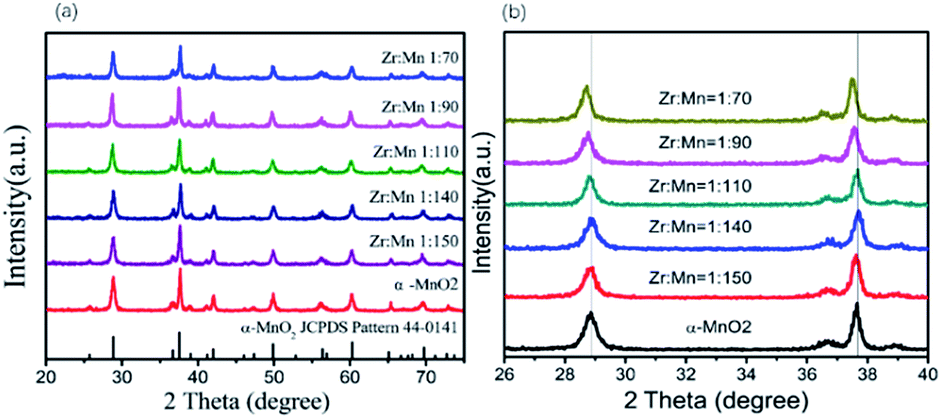 | ||
| Fig. 1 XRD patterns of Zr4+ doped α-MnO2 with different Zr/Mn molar ratios. | ||
| Sample | Lattice parameter | ||
|---|---|---|---|
| a (Å) | b (Å) | c (Å) | |
| α-MnO2 | 9.815 | 9.815 | 2.847 |
| Zr/Mn1:150 |
9.7998 | 9.7998 | 2.845 |
| Zr/Mn1:140 |
9.7976 | 9.7976 | 2.842 |
| Zr/Mn1:110 |
9.7969 | 9.7969 | 2.845 |
| Zr/Mn1:90 |
9.7960 | 9.7960 | 2.844 |
| Zr/Mn1:70 |
9.7952 | 9.7952 | 2.845 |
Fig. 2 shows the FE-SEM morphologies of α-MnO2 and Zr4+ doped α-MnO2 synthesized in this work. The two kinds of synthesized products all possess a wire shape with diameter less than 100 nm. It can also be found that the surface of α-MnO2 nanowires (Fig. 2a) is smoother than that of Zr4+ doped α-MnO2 nanowires (Fig. 2b).
 | ||
| Fig. 2 FE-SEM images of (a) α-MnO2 nanowires and (b) Zr4+ doped α-MnO2 nanowires with Zr/Mn molar ratio of 1:110. | ||
The high resolution TEM images of synthesized α-MnO2 and Zr4+ doped α-MnO2 nanowires (Zr/Mn molar ratio of 1:110) are shown in Fig. 3. The nanowires are straight in Fig. 3a and c, indicating that the crystal growth is along a preferred orientation. The diameter of synthesized α-MnO2 nanowires is in the range of 10 to 50 nm and that of synthesized Zr4+ doped α-MnO2 nanowires is in the range of 30 to 80 nm. The surfaces of α-MnO2 nanowires are clean and smooth (Fig. 3a and b). As for Zr4+ doped α-MnO2 nanowires, some attachments exist on the nanowires (Fig. 3c and d). The attachments possess an irregular morphology and its crystal structure is different from the nanowires. The different crystal structure between the attachments and nanowires illustrates that the attachments and nanowires are different materials. A semi-coherent interfacial relationship can also been found between the nanowires and the attachments as shown in Fig. 3d by the arrow, demonstrating that the attachments grow from the surface of the nanowires. The results above reveal that Zr4+ doped α-MnO2 nanowires have been successfully synthesized and few attachments differing from α-MnO2 in structure are formed at high Zr/Mn molar ratios.
 | ||
| Fig. 3 TEM images of α-MnO2 nanowires (a and b) and Zr4+ doped α-MnO2 nanowires (c and d) with Zr/Mn molar ratio of 1:110. | ||
XPS was used to probe the composition and chemical state of the synthesized nanowires, and the results are shown in Fig. 4. Compared with α-MnO2 nanowires, a signal related to element zirconium is detected in Zr4+ doped α-MnO2 nanowires as shown in Fig. 4a. Fig. 4b is the XPS spectrums of O 1s for α-MnO2 nanowires and Zr4+ doped α-MnO2 nanowires. Curve simulations were conducted and the simulated curves are also shown in Fig. 4b. The fitted curve is in accord with the measured data, demonstrating the simulated results are reasonable. The binding energy values at the peak positions 529.4 eV and 531.7 eV in the O 1s spectrum of α-MnO2 nanowires correspond to the lattice oxygen and the adsorbed oxygen, respectively.54,55 It can also be seen from Fig. 4b that the peak position corresponding to lattice oxygen shifts to 529.2 eV while the peak position corresponding to adsorbed oxygen keeps the same after Zr4+ doping, which means that the chemical state of lattice oxygen in Zr4+ doped α-MnO2 nanowires is altered compared with that in α-MnO2 nanowires.
 | ||
| Fig. 4 XPS spectrums of α-MnO2 and Zr4+ doped α-MnO2 with Zr/Mn molar ratio of 1:110. (a) Full spectrums; (b) O 1s; (c) Mn 2p; (d) Zr 3d. | ||
In order to obtain the detail information about the presence of element manganese, the XPS spectra of Mn 2p for α-MnO2 nanowires were simulated with three groups of curves colored by pink, dark yellow and dark blue, respectively (Fig. 4c). The fitted curve is in accord with the measured data, demonstrating the simulated results are reasonable. The three groups of curves correspond well to the XPS spectra of Mn3+ (pink), Mn4+ (dark yellow) and Mn4+δ (dark blue) in α-MnO2,56,57 indicating that there are three kinds of element manganese with different valences in α-MnO2 nanowires. Comparing the XPS spectra of α-MnO2 nanowires and Zr4+ doped α-MnO2 nanowires in Fig. 4c, it can be found that the peak position corresponding to Mn3+ shifts positively with 0.5 eV while the peak position corresponding to Mn4+ and Mn4+δ remains the same after Zr4+ doping. The positive shift of Mn3+ binding energies with a value less than 1 eV means that the chemical state of Mn3+ ions in Zr4+ doped α-MnO2 has been changed and Mn3+ ions tend to lose their electrons by Zr4+ doping.
Fig. 4d is Zr3d XPS spectrum of Zr4+ doped α-MnO2 nanowires. The measured data were simulated with two group curves colored with pink and green. The fitted curve is consistent with the measured data, demonstrating the simulated results are reasonable. The binding energy values of green peaks in Fig. 4d are separately 182.0 eV and 184.2 eV, which is in consistent with that of ZrO2.58 The result manifests the existence of ZrO2. The binding energy values of pink peaks in Fig. 4d are separately 181.2 eV (Zr3d5/2) and 183.4 eV(Zr3d3/2), which are 0.8 eV negative shifts compared with that of green peaks in Fig. 4d. The negative shift in Zr3d binding energies with a value less than 1 eV means that some zirconium ions in Zr4+ doped α-MnO2 tend to get electrons. Comprehensively considering the XPS results in Fig. 4, it can be concluded that Mn3+ ions, Mn4+ ions, Mn4+δ ions and Zr4+ ions co-existed in the crystal structure of synthesized Zr4+ doped α-MnO2 nanowires. Zr4+ ions occupied the positions originally belonging to element manganese in the crystal structure and resulted in a mutual action between Zr4+ ions and Mn3+ ions. The mutual action made Mn3+ ions tend to lose their electrons and Zr4+ ions tend to get electrons. The existence of Zr4+ ions in the crystal structure also changed the chemical state of the lattice oxygen. XPS analyses in Fig. 4d also indicate that there are some ZrO2 in Zr4+ doped α-MnO2, which can be considered to be the attachments shown in Fig. 3d.
3.2. Electrochemical activity of synthesized catalysts of ORR
The cathodic polarization curves of the air electrodes prepared by α-MnO2 nanowires and Zr4+ doped α-MnO2 nanowires with different Zr/Mn molar ratios were measured and the results are shown in Fig. 5a. As the potential moves negatively, the current density of the electrodes increases slowly in the potential range from 0.3 V to 0.1 V. Then the current density begins to rise rapidly with the potential shifting more negative than 0.1 V. According to the linear sweep voltammetry curves in Fig. 5a, the onset potentials of the electrodes were plotted in Fig. 5b. The onset potential of the electrodes prepared by pure α-MnO2 is 0.02 V (vs. Hg/HgO) while that of the electrodes prepared by Zr4+ doped α-MnO2 catalyst with molar ratio Zr:Mn = 1:90 shows the highest onset potential (0.082 V). It can be observed that the onset potential increases with the rise of Zr:Mn ratio and drops after reaching the peak value. To clearly show the effect of Zr/Mn molar ratios in the catalysts on the current density, the current densities at potential −0.2 V in Fig. 5a were plotted against Zr/Mn molar ratios and the curve is shown in Fig. 5c. The current density in Fig. 5c alters in a volcano shape with Zr/Mn molar ratio increasing. The largest current density (60 mA cm−2) emerges at Zr/Mn molar ratio of 1:110, which is 23% larger than that of the electrode prepared with α-MnO2 nanowires (49 mA cm−2). The current during the cathodic polarization process is caused by ORR on the surface of the electrodes. The larger the current density during the cathodic polarization process is, the more efficiently the catalyst promotes ORR. The results indicate that the electrocatalytic activity of α-MnO2 on ORR can be remarkably improved by Zr4+ doping. After Zr/Mn molar ratios exceeding 1:110, excessive zirconium forms ZrO2 crystals attached on the surfaces of Zr4+ doped α-MnO2 nanowires as shown in Fig. 3d (the attachments). The existence of ZrO2 attachments reduces the catalytical area of Zr4+ doped α-MnO2 nanowires, resulting in the decrease of the current density as shown in Fig. 5b.
 | ||
| Fig. 5 (a) Cathodic polarization curves of the air electrodes prepared with α-MnO2 nanowires and Zr4+ doped α-MnO2 nanowires with different Zr/Mn molar ratios as catalysts. (b) The onset potentials of the electrodes prepared by Zr4+ doped α-MnO2 catalyst with different Zr/Mn ratios (c) the relations between Zr/Mn molar ratios and the current densities at potential −0.2 V. | ||
The electrochemical impedance spectroscopies (EIS) of the air electrodes prepared by α-MnO2 nanowires and Zr4+ doped α-MnO2 nanowires with different Zr/Mn molar ratios as catalysts were measured and shown in Fig. 6. All the EIS plots in Fig. 6a exhibit the same shape, which are composed of a large arc in the high frequency range in the fourth quadrant linked with a very small semi-circle in the middle frequency range in the first quadrant followed by a long straight line in the low frequency range in the first quadrant.
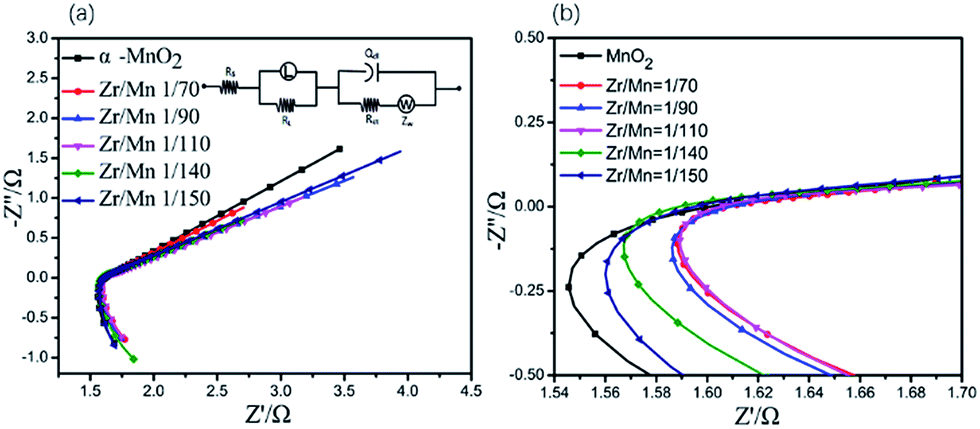 | ||
| Fig. 6 (a) EIS of the air electrodes prepared by α-MnO2 nanowires and Zr4+ doped α-MnO2 nanowires with different Zr/Mn molar ratios as catalysts. Upper inset is the equivalent circuit. (b) The amplified part of the EIS. Dot: measured data; solid line: simulated results. | ||
The reduction of oxygen molecules on the surface of the air electrodes can be considered to proceed by following two steps.59 In the first step, oxygen molecules from the air diffuse through the air electrode and absorb on the surface of the air electrode near the solution side, which can be described in eqn (4). In the second step, absorbed oxygen molecules are electrochemically reduced and forms OH−, which can be described in eqn (5).
| O2 → O2 adsorbed | (4) |
| O2 adsorbed + 4e− + 2H2O → 4OH− | (5) |
The very small semi-circle in the EIS (Fig. 6) means that the electrochemical reduction of absorbed oxygen molecules described in eqn (5) is easy to proceed. The large arc in the EIS (Fig. 6) indicates that the adsorption of oxygen molecules described in eqn (4) is difficult to proceed. This suggests that the adsorption of oxygen molecules on the surface of the air electrodes is the rate-controlling step for ORR.
The EIS data were simulated using an equivalent circuit shown in Fig. 6b, and the simulated results are also shown in Fig. 6a in solid lines. Therein, Rs represents the solution resistance, Rct the charge transfer resistance incurred during the electrochemical reactions taking place on the electrodes, Q the constant phase element modeling the non-faradaic processes, ZW the Warburg element representing the diffusion control process, L the inductance element representing the adsorption of oxygen molecules on the electrode, RL the adsorption resistance. The solid lines in Fig. 6a fit well with the measured data, demonstrating the proposed equivalent circuit are reasonable. Some of the parameters in the equivalent circuit are listed in Table 2. It is well known that the reaction rate of the electrochemical reaction step is inversely proportional to Rct.60 All the values of Rct in Table 2 are small, illustrating that the reaction described in eqn (5) is easy to proceed. The electrode prepared with Zr4+ doped α-MnO2 nanowires with Zr/Mn molar ratio of 1:110 shows the smallest value of RL. The smaller RL value is, the more easily the adsorption process described in eqn (4) proceeds. Considering the adsorption of oxygen molecules is the rate-controlling step of ORR on the air electrode, the lowest RL value reasonably explains why the catalyst with Zr/Mn molar ratio 1:110 exhibits the highest electrocatalytical activity as shown in Fig. 6.
| Sample | L (H) | RL (Ω) | Rs (Ω) | ZW (Ω) | Rct (Ω) |
|---|---|---|---|---|---|
| α-MnO2 | 1.422 | 4.685 | 1.804 | 1.57 | 29.81 |
| 1:70 |
1.367 | 3.221 | 1.565 | 1.84 | 27.34 |
| 1:90 |
1.336 | 3.195 | 1.524 | 2.68 | 24.42 |
| 1:110 |
1.391 | 3.141 | 1.308 | 2.46 | 20.64 |
| 1:140 |
1.886 | 3.721 | 1.679 | 1.67 | 29.34 |
| 1:150 |
1.53 | 4.849 | 1.9745 | 1.34 | 37.88 |
To further elucidate the pathway and kinetics of ORR, LSV curve of the electrode prepared by Zr4+ doped α-MnO2 on RDE were recorded at different rotation rates (400–1600 rpm) in O2-saturated 0.1 M KOH solution and the result is shown in Fig. 7a. The derived K–L plot of J−1 vs. ω−1/2 at a potential of −0.4 V is shown in Fig. 7b. It can be found that a good linear relationship between J−1 vs. ω−1/2. The electron transfer number n was calculated by eqn (2) based on the slope (1/B) of the K–L plot. The electron transfer number for Zr4+doped α-MnO2 is 3.8, which clearly indicate that the electrode prepared by Zr4+ doped α-MnO2 follows a desirable four-electron pathway for ORR.
 | ||
| Fig. 7 (a) LSV curves of the electrodes prepared by Zr4+ doped α-MnO2 catalyst at different rotation speed (400–1600 rpm) in oxygen-saturated 0.1 M KOH solution. (b) Koutecky–Levich (K–L) plots. (c) Chronoamperometric curves of the electrode prepared by Zr4+ doped α-MnO2 and Pt/C measured in oxygen-saturated 0.1 M KOH solution at a rotating rate 1600 rpm. | ||
The stability of catalysts is very important issue for their practical application in various energy technologies. Stability was examined during the ORR process. The durability of the electrode prepared by Zr4+ doped α-MnO2 and Pt/C as the catalyst for ORR was evaluated by chronoamperometric technique at potential −0.4 V for 7200 s. It can be seen from Fig. 7c that the ORR current density of the Zr4+ doped α-MnO2 electrode decreased by only 16.3%, but the Pt/C electrode exhibited a 30.1% decrease in current density under the same conditions. The result reveals that the performance of Zr4+ doped α-MnO2 catalyst is more stable than that of the commercial Pt/C catalyst.
3.3. Optimized configuration and O2 adsorption
To understand the influence of Zr4+ doping on the adsorption of oxygen molecules, the DFT calculations were carried out aiming at the surface of crystal plane (211) of α-MnO2 (Fig. 8a and d). Different adsorption configurations of O2 molecules were optimized by the computation methods mentioned in 2.4. The optimized adsorption structures on the surfaces are shown in Fig. 8b, c, e and f. The direct adsorption of O2 molecules on the surfaces includes the following two modes: top adsorption (Fig. 7b and e) and bridge adsorption (Fig. 7c and f). The adsorption energy of O2 molecules was calculated by eqn (3) and the results were listed in Table 3.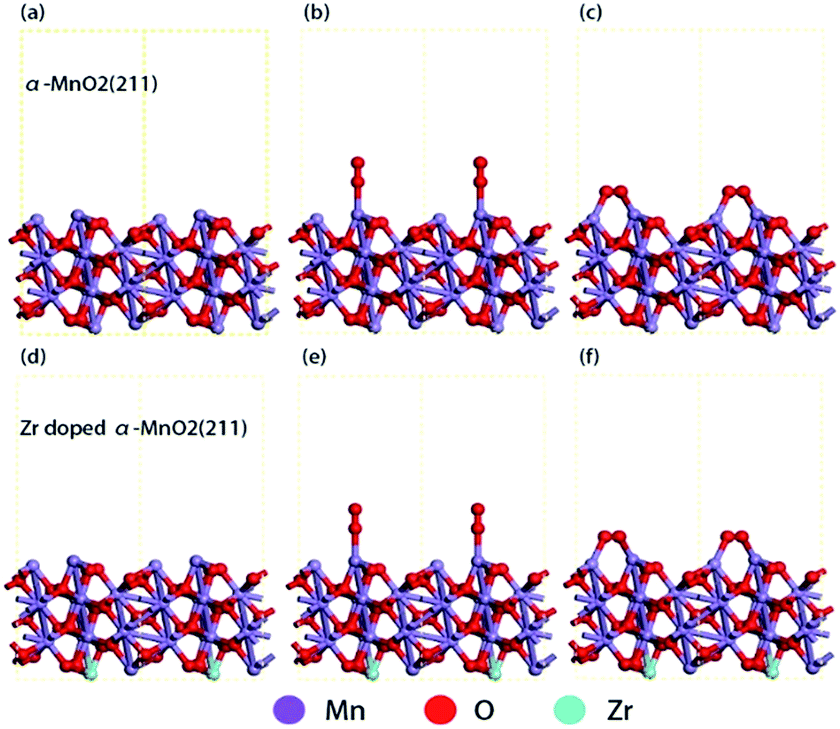 | ||
| Fig. 8 Optimized adsorption structures. (a) Side view of the optimized α-MnO2 (211) surface; (b) O2 top adsorption on α-MnO2 (211) surface; (c) O2 bridge adsorption on α-MnO2 (211) surface; (d) side view of the optimized Zr4+ doped α-MnO2 (211) surface; (e) O2 top adsorption on Zr4+ doped α-MnO2 (211); (f) O2 bridge adsorption on Zr4+ doped α-MnO2. | ||
| Surface | Top adsorption/eV | Bridge adsorption/eV |
|---|---|---|
| Crystal plane (211) of α-MnO2 | −1.2242 | −2.8777 |
| Crystal plane (211) of Zr doped α-MnO2 | −1.3313 | −3.1317 |
As shown in Table 3, the bridge adsorption energy is more negative than top adsorption energy, indicating that bridge adsorption is more favorable for O2 molecules. Comparing with α-MnO2, the bridge absorption energy of O2 molecules on Zr4+ doped α-MnO2 decreases by 0.26 eV. The more negative adsorption energy indicates an easier adsorption process and a more stable adsorption structure.61,62 The results in Table 3 reveal that the mutual action between Zr4+ and Mn3+ in Zr4+ doped α-MnO2 nanowires enhances the adsorption process of O2 molecules. The bridge adsorption configuration is supposed to be the preferential adsorbing manner for O2 molecules on the surface of Zr4+ doped α-MnO2. According to EIS analyses, the adsorption process of O2 molecules is the rate-controlling step for ORR. The results in Table 3 provide a reasonable explanation about the improvement in the performance of ORR by Zr4+ doping.
4. Conclusions
Zr4+ doped α-MnO2 nanowires were successfully synthesized by a hydrothermal method. XRD, SEM, TEM and XPS analyses indicated that Mn3+ ions, Mn4+ ions, Mn4+δ ions and Zr4+ ions co-existed in the crystal structure of synthesized Zr4+ doped α-MnO2 nanowires. Zr4+ ions occupied the positions originally belonging to element manganese in the crystal structure and resulted in a mutual action between Zr4+ ions and Mn3+ ions. The mutual action made Mn3+ ions tend to lose their electrons and Zr4+ ions tend to get electrons. The existence of Zr4+ ions in the crystal structure also changed the chemical state of the lattice oxygen. Cathodic polarization analyses showed that Zr/Mn molar ratio notably affects the ORR performance of the air electrodes prepared by Zr4+ doped α-MnO2 nanowires. The highest ORR current density of the air electrodes prepared by Zr4+ doped α-MnO2 nanowires in alkaline solution appeared at Zr/Mn molar ratio of 1:110, which was 23% higher than that prepared by α-MnO2 nanowires. The results indicated that the electrocatalytic activity of α-MnO2 for ORR remarkably improved by Zr4+ doping. EIS analyses indicated that the adsorption process of O2 molecules on the surface of the air electrodes prepared by Zr4+ doped α-MnO2 nanowires was the rate-controlling step for ORR. The calculation results based on density functional theory (DFT) revealed that the mutual action between Zr4+ and Mn3+ in Zr4+ doped α-MnO2 nanowires enhanced the adsorption process of O2 molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by the Chinese Natural Science Foundation (No. 21276181). We are grateful for the support.Notes and references
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl, Y. M. Wang, X. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, M. N. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. F. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- V. Komanicky, H. Iddir, K. C. Chang, A. Menzel, G. Karapetrov, D. Hennessy, P. Zapol and H. You, J. Am. Chem. Soc., 2009, 131, 5732–5733 CrossRef CAS PubMed.
- C. L. Lee, C. M. Syu, H. P. Chiou, C. H. Chen and H. L. Yang, Int. J. Hydrogen Energy, 2011, 36, 10502–10512 CrossRef CAS.
- L. Tammeveski, H. Erikon, A. Sarapuu, J. Kozlova, P. Ritslaid, V. Sammelselg and K. Tammeveski, Electrochem. Commun., 2012, 20, 15–18 CrossRef.
- B. P. Vinayan, K. Sethupathi and S. Ramapraabhu, Int. J. Hydrogen Energy, 2013, 38, 2240–2250 CrossRef CAS.
- G. F. Alvarez, M. Mamlouk and S. M. Senthil Kumar, J. Appl. Electrochem., 2011, 41, 925–937 CrossRef CAS.
- P. Fu, L. H. Zhou, L. H. Sun, B. H. Huang and Y. Yuan, RSC Adv., 2017, 7, 13383–13389 RSC.
- Y. Z. Xu, C. L. Chen, M. Zhou, G. Y. Fu, Y. Y. Zhao and Y. H. Chen, RSC Adv., 2017, 7, 26722–26728 RSC.
- W. X. Wang, Y. Shi, M. C. Li, Z. Y. Wang, S. F. Wu, F. C. Lyu, C. Q. Shang and Z. G. Lu, RSC Adv., 2016, 6, 110758–110764 RSC.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS PubMed.
- X. Y. Xu, C. X. Shi, Q. Li, R. Chen and T. H. Chen, RSC Adv., 2017, 7, 14382–14388 RSC.
- G. Panomsuwan, N. Saito and T. Ishizaki, RSC Adv., 2016, 6, 114553–114559 RSC.
- X. W. Bai, E. J. Zhao, W. C. Wang, Y. Wang, K. Li, L. Lin, J. C. Yang, H. Sun and Z. J. Wu, RSC Adv., 2017, 7, 23812–23819 RSC.
- S. Jin, A. G. Hubert, Y. Naoaki, N. Haruyuki, B. G. John and S. H. Yang, Nat. Chem., 2011, 3, 546–550 CrossRef PubMed.
- Y. Y. Liang, H. L. Wang, J. G. Zhou, Y. G. Li, J. Wang, T. Regier and H. J. Dai, J. Am. Chem. Soc., 2012, 134, 3517–3523 CrossRef CAS PubMed.
- J. Xu, P. Gao and T. S. Zhao, Energy Environ. Sci., 2012, 5, 5333–5339 CAS.
- Z. S. Wu, S. Yang, Y. Sun, K. Parvea, X. L. Feng and K. Mullen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., Early Ed., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- I. Roche, E. Chainet and M. Chatenet, J. Phys. Chem. C, 2007, 111, 1434–1443 CAS.
- M. L. Calegaro, F. H. B. Lima and E. A. Ticianelli, J. Power Sources, 2006, 158, 735–739 CrossRef CAS.
- Y. L. Cao, H. X. Yang, X. P. Ai and L. F. Xiao, J. Electroanal. Chem., 2003, 557, 127–134 CrossRef CAS.
- M. Lanqun, T. Sotomura, K. Nakatsu, N. Koshiba, Z. Dun and T. Ohsaka, J. Electrochem. Soc., 2002, 149, A504–A507 CrossRef.
- F. H. B. Lima, M. L. Calegaro and E. A. Ticianelli, J. Electroanal. Chem., 2006, 590, 152–160 CrossRef CAS.
- S. Devaraj and N. Munichandraiah, J. Phys. Chem. C, 2008, 112, 4406–4417 CAS.
- F. Cheng, Y. Su, J. Liang, Z. Tao and J. Chen, Chem. Mater., 2009, 22, 898–905 CrossRef.
- Y. J. Zhang, C. S. Sun, P. Lu, K. Y. Li, S. Y. Song and D. F. Xue, CrystEngComm, 2012, 14, 5892–5897 RSC.
- K. Selvakumar, S. M. S. Kumar, R. Thangamuthu, G. Kruthika and P. Murugan, Int. J. Hydrogen Energy, 2014, 39, 21024–21036 CrossRef CAS.
- F. Cheng, J. Shen, W. Ji, Z. Tao and J. Chen, ACS Appl. Mater. Interfaces, 2009, 1, 460–466 CAS.
- W. Xiao, D. Wang and X. W. Lou, J. Phys. Chem. C, 2010, 114, 1694–1700 CAS.
- L. Mao, D. Zhang, T. Sotomura, K. Nakatsu, N. Koshiba and T. Ohsaka, Electrochim. Acta, 2003, 48, 1015–1023 CrossRef CAS.
- F. Cheng, T. Zhang, Y. Zhang, J. Du, X. Han and J. Chen, Angew. Chem., Int. Ed., 2013, 52, 2474–2477 CrossRef CAS PubMed.
- D. A. Slanac, A. Lie, J. A. Paulson, K. J. Stevenson and K. P. Johnston, J. Phys. Chem. C, 2012, 116, 11032–11039 CAS.
- Q. Tang, L. Jiang, J. Qi, O. Jiang, S. Wang and G. Sun, Appl. Catal., B, 2011, 104, 337–345 CrossRef CAS.
- S. W. Liu and X. Qin, RSC Adv., 2015, 5, 15627–15633 RSC.
- W. Sun, A. Hsu and R. Chen, J. Power Sources, 2011, 196, 4491–4498 CrossRef CAS.
- I. Roche, E. Chainet and M. Vondrak, J. Phys. Chem. C, 2007, 111, 1434–1443 CAS.
- A. C. Garcia, A. D. Herrera, E. A. Ticianelli, M. Chatenet and C. Poinsignon, J. Electrochem. Soc., 2011, 158, B290–B296 CrossRef CAS.
- K. A. Walz, C. S. Johnson, J. Genthe, L. C. Stoiber, W. A. Zeltner, M. A. Anderson and M. M. Thackeray, J. Power Sources, 2010, 195, 4943–4951 CrossRef CAS.
- J. Xu, G. Chen, H. J. Zhang, W. Zheng and Y. Li, J. Appl. Electrochem., 2015, 45, 123–130 CrossRef CAS.
- V. Subramanian, H. W. Zhu and B. Q. Wei, J. Power Sources, 2006, 159, 361–364 CrossRef CAS.
- F. Hashemzadeh, M. M. K. Motlagh and A. Maghsoudipour, J. Sol-Gel Sci. Technol., 2009, 51, 169–174 CrossRef CAS.
- R. E. Davis, G. L. Horvath and C. W. Tobias, Electrochim. Acta, 1967, 12, 287 CrossRef CAS.
- K. Chen, Y. J. Hao and M. R. Zhang, RSC Adv., 2017, 7, 5782 RSC.
- M. C. Payne, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045 CrossRef CAS.
- V. Milman, B. Winkler, J. A. White, C. J. Pickard, M. C. Payne, E. V. Akhmataskaya and R. H. Nobes, Int. J. Quantum Chem., 2000, 77, 895 CrossRef CAS.
- J. A. White and D. M. Bird, Phys. Rev. B, 1976, 13, 5188 CrossRef.
- J. P. Perdrew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef PubMed.
- J. P. Perdrew, K. Burk and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef.
- Y. H. Li, G. Y. Zhong and F. Peng, Phys. Chem. Chem. Phys., 2015, 17, 21950 RSC.
- H. J. Monkhorst and J. Pack, Phys. Rev. B, 1976, 13, 5188 CrossRef.
- F. Wang, H. X. Dai, J. G. Deng, G. M. Bai, K. M. Ji and Y. X. Liu, Environ. Sci. Technol., 2012, 46, 4034–4041 CrossRef CAS PubMed.
- J. B. Jia, P. Y. Zhang and L. Chen, Appl. Catal., B, 2016, 189, 210–218 CrossRef CAS.
- D. A. Pena, B. S. Uphade and P. G. Smirniotis, J. Catal., 2004, 221, 421–431 CrossRef CAS.
- E. Pargoletti, G. Cappelletti, A. Minguzzi, S. Rondinini, M. Leoni, M. Marelli and A. Vertova, J. Power Sources, 2016, 325, 116–128 CrossRef CAS.
- C. D. Wagner, W. M. Riggs, L. E. Davis and J. F. Moulder, Handbook Of X-Ray Photoelectron Spectroscopy, ed. G. E. Mullenberg, Perkin-Elmer Corporation Press, Minnesota, 1979, vol. 2, p. 100 Search PubMed.
- Z. Shi, J. J. Zhang, Z. S. Liu, H. J. Huang and D. P. Wikinson, Electrochim. Acta, 2006, 51, 1905–1916 CrossRef CAS.
- Z. He and F. Mansfeld, Energy Environ. Sci., 2009, 2, 215–219 CAS.
- L. Li, Z. D. Wei, S. G. Chen, X. Q. Qi, W. Ding, M. R. Xia, R. Li, K. Xiong, Z. H. Deng and Y. Y. Gao, Chem. Phys. Lett., 2012, 539, 88–93 Search PubMed.
- M. H. Yu, Z. K. Wang and C. Hou, Adv. Mater., 2017, 29, 1602868 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |