DOI:
10.1039/C7RA09090K
(Paper)
RSC Adv., 2018,
8, 4494-4502
Pitch-tunable pillar arrays for high-throughput culture and immunohistological analysis of tumor spheroids†
Received
17th August 2017
, Accepted 8th January 2018
First published on 24th January 2018
Abstract
Tumor spheroids are multicellular, three-dimensional (3D) cell culture models closely mimicking the microenvironments of human tumors in vivo, thereby providing enhanced predictability, clinical relevancy of drug efficacy and the mechanism of action. Conventional confocal microscopic imaging remains inappropriate for immunohistological analysis due to current technical limits in immunostaining using antibodies and imaging cells grown in 3D multicellular contexts. Preparation of microsections of these spheroids represents a best alternative, yet their sub-millimeter size and fragility make it less practical for high-throughput screening. To address these problems, we developed a pitch-tunable 5 × 5 mini-pillar array chip for culturing and sectioning tumor spheroids in a high throughput manner. Tumor spheroids were 3D cultured in an alginate matrix using a twenty-five mini-pillar array which aligns to a 96-well. At least a few tens of spheroids per pillar were cultured and as many as 25 different treatment conditions per chip were evaluated, which indicated the high throughput manner of the 5 × 5 pillar array chip. The twenty-five mini-pillars were then rearranged to a transferring pitch so that spheroid-containing gel caps from all pillars can be embedded into a specimen block. Tissue array sections were then prepared and stained for immunohistological examination. The utility of this pitch-tunable pillar array was demonstrated by evaluating drug distribution and expression levels of several proteins following drug treatment in 3D tumor spheroids. Overall, our mini-pillar array provides a novel platform that can be useful for culturing tumor spheroids as well as for immunohistological analysis in a multiplexed and high throughput manner.
Introduction
Three-dimensional (3D) cell cultures, such as tumor spheroids cultured in hydrogels,1–8 on polymer scaffolds,2,7,9–13 on microcarrier beads14,15 or in hanging droplets,16 have been actively adopted in biomedical and pharmaceutical research, especially in the cancer research field. Significant differences in cellular phenotypes and gene expression profiles have been established between two dimensional (2D) and 3D environments in cultures of both primary and established cell lines of cancer and stem cells.17–22 Based on the several advantages of these 3D culture models, tumor spheroids may serve as a replacement for human tissues that have limited availability and a high degree of tissue complexity. Multicellular tumor spheroids maintain three dimensionality and physiologically relevant organization. The in vivo tissue-like characteristics of tumor spheroids has been demonstrated by the presence of cell–cell and cell–extracellular matrix (ECM) interactions associated with intracellular signaling for cell proliferation and behavior.20,21 The use of 3D multicellular tumor spheroids enables rapid and reproducible assay systems for evaluating differential responsiveness to various drugs and the underlying mechanisms.5,6,23 Development of clinically relevant 3D cell cultures has been adapted towards high-throughput screening and analysis platforms.
To establish clinically relevant 3D cell-based assays, evaluation of intracellular events in tumor spheroids is required. The ability to measure molecular changes following drug exposure or under certain conditions is crucial for enhancing the predictability of 3D cell-based, drug-screening systems.5,6 Although whole spheroids have been used for 3D cell-based high-content imaging (HCI), critical limitations exist in their immunostaining and image acquisition. Unlike traditional 2D cell culture that can be easily stained with colorimetric and fluorescent dyes for HCI, cells in multicellular spheroids are difficult to stain uniformly due to poor penetration of dyes and antibodies into dense network of cells and ECM.24,25 Immunostaining to detect various proteins of interest enclosed in multicellular 3D structure of intact spheroids often require specific methods such as tissue clearing procedure to increase macromolecular diffusion or convection (Fig. S1†). Furthermore, imaging cells in 3D cell structures is difficult even with a confocal microscope due to light refraction and optical attenuation in dense 3D cell cultures, resulting in limited range of imaging depth.26–28 Specialized techniques such as phase fluorimetry and two-photon microscopy have been used to measure oxygen distribution29 and other cellular events30 in 3D cell cultures, yet they are not widely available. Preparation of histological sections and subsequent immunostaining is an alternative approach.31–33
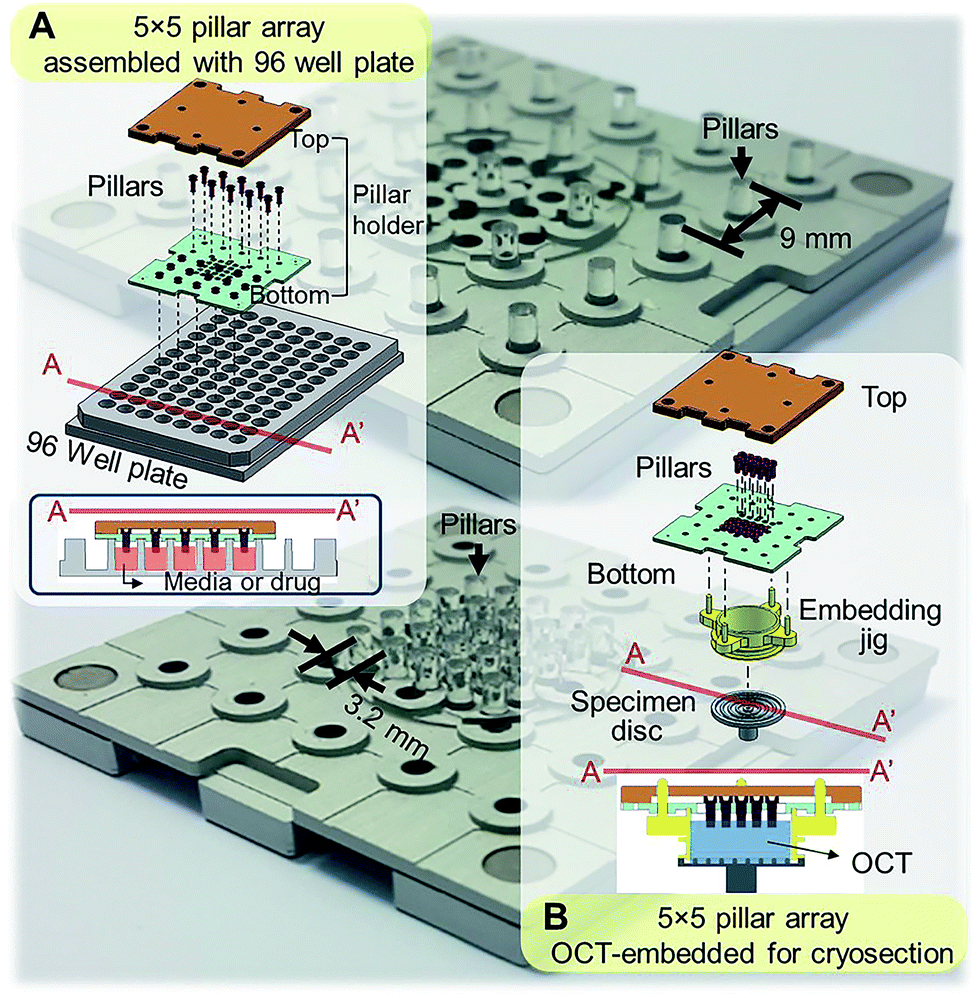
Standard tissue processing protocols for cryo- or paraffin sections may not be suitable for tumor spheroids. Due to small sizes and fragile structure, harvesting spheroids from culture plates and embedding in cryomount or paraffin is extremely difficult and time consuming. To address these limitations, our group has developed mini-pillar array platforms as reported recently.34 A 3 × 3 pillar array platform has been successfully introduced to culture tumor cells into 3D spheroids using 96-well plates and to transfer these tumor spheroids from the pillars to cryomount or paraffin specimen blocks for sectioning and immunocytochemical assays.34 The present study was to improve throughput of the pillar array platform by using a pitch-tunable chip design. The previous 3 × 3 pillar array was fabricated to have a 9 mm pillar-to-pillar distance to fit multiwell dimensions of 96-well format. The 3 × 3 array dimension could not be modified to arrange more pillars due to the disk diameter of standard specimen chuck (discs), i.e., 30 mm. In order to engineer more pillars on a chip that can be transferred to one specimen block, a pitch-tunable 5 × 5 pillar array platform has been developed. Pillar holes were positioned so that nine pillars can be arranged with either a 9 mm pillar-to-pillar distance for culturing spheroids in 96-well plate format or a 3.2 mm distance for OCT mounting on the specimen disc (Fig. 1). Herein, we present a successful 3D culture of several human tumor spheroids in alginate droplets on the 25 pillars of 5 × 5 pillar array using HT-29 and DLD-1 human colorectal adenocarcinoma cells and AsPC-1 and PANC-1 human pancreatic adenocarcinoma cells. At least a few tens of spheroids per pillar were cultured and as many as 25 different treatment conditions per chip were evaluated, which indicated the high throughput manner of the 5 × 5 pillar array chip. Feasibility of high throughput screening on the 5 × 5 pillar array was demonstrated by evaluation of drug accumulation and drug-induced changes in cell viability and intra- and extra-cellular proteins expression.
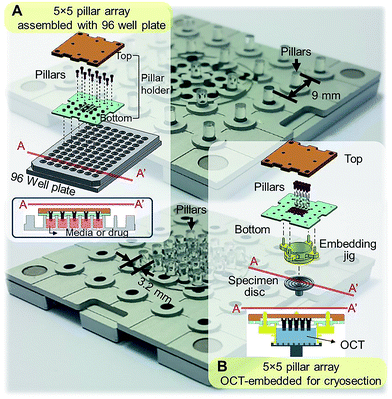 |
| | Fig. 1 Schematic illustration of the assembly of a 5 × 5 pillar array for tumor spheroid culture and cryosection. (A) Assembly of 5 × 5 pillars at 9 mm distance for 3D cell cultures in 96-well format. Cross-sectional view of the pillars placed into the 96-well plate is also shown. (B) Assembly of 5 × 5 pillars at 3.2 mm distance on the pillar holder for embedding and cryosectioning. Each pillar has a diameter of 2 mm. | |
Materials and methods
Cell culture
Human colorectal cancer cell line HT-29 was purchased from the Korean Cell line Bank (Seoul, Korea). HT-29 was cultured in RPMI 1640 (Gibco BRL, Grand Island, NY) media supplemented with 100 μg mL−1 streptomycin, 100 units per mL penicillin, 250 ng mL−1 amphotericin B and 10% fetal bovine serum (FBS, Welgene, Daegu, Korea) in a humidified atmosphere (5% CO2/95% air) at 37 °C.
Pillar preparation
The mini-pillars were custom-made by MBD Co. (Cat. No. Cellvitro™ 55Cryo, E-mail: 873087@mbdker.co.kr). Mini-pillar inserts with 2 mm diameter were made with polystyrene by injection molding. The pillars were inserted in 9 mm-pitch holes on the pillar holder for cell culture in 96-well plates (Fig. 1A) and in 3.2 mm-pitch holes for cryosection (Fig. 1B). The mini-pillars were sterilized before pre-coating and cell loading by boiling in 70% ethanol for 30 min followed by UV irradiation as described previously.34 Poly-L-lysine (PLL) pre-coating was used to stabilize the loading of hydrogel (alginate, collagen, or Matrigel) onto the tip of the mini-pillar.34 When using alginate as hydrogel, the pillar tip was subjected to barium coating for cell loading: 2 μL of 100 mM barium chloride (BaCl2, Sigma-Aldrich) was dispensed on the surface of the pillar and dried in a biosafety cabinet for 3 h. BaCl2 was used as a cross-linking agent for the alginate hydrogel since barium ion chelates with carboxylic groups on alginate.
3D spheroids culture & drug exposure
Cells in log phase growth were harvested and cell suspensions were prepared at 2 × 106 mL−1. Alginate solution (1%) was prepared by mixing 100 μL of 3% alginate (wt/vol) in distilled water with 200 μL of culture media. The cell suspension was mixed with 1% alginate solution at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio and 1 × 103 cells were loaded in 1 μL of cell–alginate mixture onto the tip of mini-pillars (Fig. 2A).34 For alginate hydrogels, the gelation time after loading of the cell–alginate suspension was kept within 1–2 min before immersion in media to prevent over-drying. Subsequent culturing of cell–alginate was performed in 96-well plates under regular cell culture conditions (Fig. 2B). Media change was done by transferring the pillar holder containing 3D spheroids to a new 96-well plate filled with fresh media, and drug exposures were done by placing culturing pillars directly in the drug-containing media (Fig. 2C). Medium changes were done every 2 days and cells were cultured for 6 days.
:1 ratio and 1 × 103 cells were loaded in 1 μL of cell–alginate mixture onto the tip of mini-pillars (Fig. 2A).34 For alginate hydrogels, the gelation time after loading of the cell–alginate suspension was kept within 1–2 min before immersion in media to prevent over-drying. Subsequent culturing of cell–alginate was performed in 96-well plates under regular cell culture conditions (Fig. 2B). Media change was done by transferring the pillar holder containing 3D spheroids to a new 96-well plate filled with fresh media, and drug exposures were done by placing culturing pillars directly in the drug-containing media (Fig. 2C). Medium changes were done every 2 days and cells were cultured for 6 days.
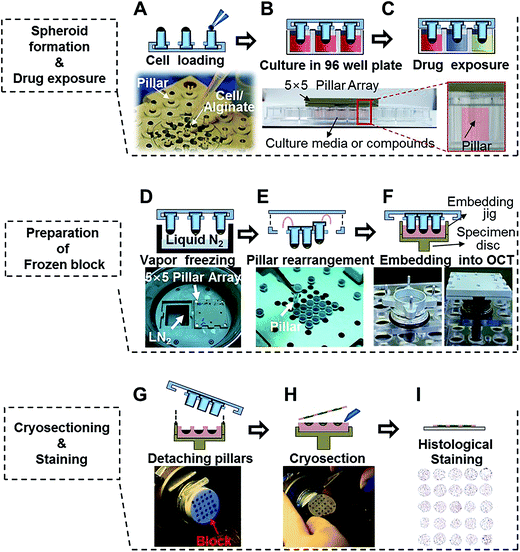 |
| | Fig. 2 Experimental procedures used for 5 × 5 pillar arrays for culturing and cryosectioning tumor spheroids. (A) Loading 1 μL human cell–alginate mixture at the tip of the pillars. (B) Culturing the cells in 3D for 4 to 6 days. (C) Exposing 3D-cultured tumor spheroids to drugs by immersing the pillars into 96-wells containing drug solutions. (D) Freezing the spheroid-loaded pillar array over vapor-phase liquid nitrogen. (E) Rearranging pillars to the center of the array for micro-section on a single microscope slide. (F) Embedding the pillar array in OCT and freezing at −20 °C to prepare the OCT/spheroids block. (G) Removing the embedding jig and the pillars. (H) Sectioning the OCT/spheroids block with a cryo-microtome. (I) Staining cryosections on a microscope slide, followed by image acquisition. | |
Preparation of frozen sections
After culturing or drug exposure, the pillar holder containing tumor spheroids was transferred to a liquid nitrogen chamber and incubated for 30 min (Fig. 2D). When frozen, pillars were rearranged by taking out 9 mm-pitch holes and placing into 3.2 mm-pitch holes (Fig. 2E). The rearranged pillars were then stamped in the OCT solution using an embedding jig (custom-made by http://www.mbdbiotech.com/. EM001) and freezing at −20 °C for 20 min in a cryotome chamber. The pillar holder and the embedding jig were made of aluminum and used repeatedly after autoclave sterilization (Fig. 2F). After 10–15 min, the pillar array chip was carefully removed, leaving the tumor spheroids alginate cap array, in the frozen OCT block (Fig. 2G). The jig was designed in such a way that culture samples were placed beneath the surface of the OCT block which helped with initial alignment of microtome angle and precise cutting of the whole array into one section. The OCT block was placed in the microtome for cutting (Fig. 2H).35 Tumor spheroid array sections were obtained in 10 μm thickness, and approximately 20 consecutive sections were obtained from one OCT block (Fig. 2I).
Drug distribution
To determine drug uptake in 3D spheroids, spheroids were exposed to doxorubicin (DOX) in solution for 30 min at 37 °C or to DOX liposome formulations for 2 h at 40 °C. Non-temperature-sensitive liposomes (NTSL) and temperature-sensitive liposomes (TSL) were prepared by the thin film hydration method.36,37 Briefly, the composition of NTSL (HPSC:CHOL:DSPE-mPEG-2000 = 12.6:4.2:4.2 by weight ratio), TSL1 (DPPC:MPPC:DSPE-mPEG-2000 = 21.6:2.6:1.0 by weight ratio) and TSL2 (DSPC:MSPC:DSPE-mPEG-2000 = 21.6:2.6:1.0 by weight ratio) was dispersed in chloroform and dried onto a round bottom flask using a rotary evaporator under vacuum (Buchi Rotavapor R-210, Flawil, Switzerland). The lipid film was hydrated at 40 °C with 300 mM citric acid buffer (pH 4). Fluorescence images were acquired from cryosections after fixation & degelation of alginate matrix, followed by slide mounting as described above.
Western blot analysis
Proteins were extracted from HT-29 spheroids using a RIPA lysis buffer containing 150 mM NaCl, 1% NP-40, 0.5% (w/v) sodium deoxycholate (Na-DCA), 0.1% SDS, a protease inhibitor, and 50 mM Tris–HCl (pH 8.0). Total protein in lysates was quantified using the Bradford assay (Quick Start™ Bradford Protein Assay, BIO-RAD, USA) 50 μg of total protein were resolved on 8% SDS gel under reducing condition and transferred onto nitrocellulose membrane (GE Healthcare Life Sciences Whatman™). The membrane was blocked with 5% skim milk in TBS with 0.05% Tween-20 (TBS-T) at room temperature, and incubated with antibodies against cleaved poly ADP ribose polymerase 1 (c-PARP-1) (1:2000, Abcam, ab2317) and β-actin (1:5000, Thermo Scientific, MA5-15739) at 4 °C overnight. After exposing to horseradish peroxidase-conjugated secondary antibodies, proteins were visualized by SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). The band intensity was analyzed using ImageJ software (version 1.49) and expression level was defined as the ratio of target protein relative to β-actin.
Hematoxylin and immunohistological staining
Hematoxylin and immunohistological staining on the cryosections was conducted following fixation and alginate removal steps. Cryosections were fixed in 95% ethanol for 15 min. Degelation of alginate matrix was done by 30 min-incubation in 75 mM EDTA solution following ethanol fixation. Hematoxylin staining was done using a common staining protocol. In brief, pre-fixed sections were stained in Harris hematoxylin solution for 1 min and subjected to cytoplasm de-staining in 0.6% (vol/vol) acid alcohol for 15 s followed by 30 s hematoxylin bluing in 0.3% ammonia water. The sections were then subjected to a series of dehydration steps using 95% absolute ethanol before xylene rinse as clearing step. Finally, micro-tissue sections were mounted using a drop of Permount (mountant) for microscopic observation (AX70, Olympus, Japan). The hematoxylin stained slides were scanned on a Panoramic MIDI digital scanner (3DHISTECH Ltd, Budapest, Hungary). Immunofluorescence staining was done for E-cadherin (1:100, Cell Signaling, 3195S), collagen type I (1:200, Abcam, ab34710), laminin (1:200, Abcam, ab14509), connective tissue growth factor (CTGF) (1:200, Abcam, ab6992), transforming growth factor beta (TGF-β) (1:400, Abcam, ab92486) and c-PARP-1 (1:100, Abcam, ab2317). Incubation with primary antibodies was done at 4 °C in a humidified chamber overnight. After blocking non-specific binding using 10% normal goat serum for 60 min, secondary antibody labeled with goat anti-rabbit IgG (H + L) secondary antibody 594 (Thermo Scientific, A27016) and 488 (Thermo Scientific, A11034) was added for 2 h at room temperature in a humidified chamber. Slides were then counter-stained with DAPI and mounted for confocal microscopy (LSM 510 Meta, Carl Zeiss, Oberkochen, Germany). Diameter of spheroids or objects was calculated using DAPI staining images and the area obtained from ImageJ software assuming a circular shape of spheroids based on an equation (area = πr2). Fluorescence intensity was calculated using ZEN software (Carl Zeiss). For quantitative comparison, data were normalized to DAPI intensity. Tissue clearing and immunostaining for collagen type I (1:300, Abcam, ab34710) and Ki-67 (1:300, Santa Cruz, sc-15402) was performed either using a manual procedure provided by K. Chung (http://www.chunglab.org), or by using X-CLARITY system (Logos Biosystems, Anyang-si, Korea).
Results & discussion
Experimental procedure for 3D culturing of tumor spheroids on the pillars
Fig. 2 demonstrates the experimental procedures of high-throughput 3D cell culture using the 5 × 5 pitch-tunable pillar and preparation of cryosections for high-content histological analysis of spheroids. Demonstration of these experimental procedures is provided in ESI Video 1.† The first step involves loading cells in a hydrogel mixture on the tip of each pillar and then culturing the cells by immersing the array tip upside-down into 96-well plates (Fig. 2A). Cells were cultured and exposed to multiple drugs for cytotoxicity assessments (Fig. 2B and C). For preparation of cryo-blocks, the spheroids on pillars were frozen in the vapor phase of liquid nitrogen, pulled off and relocated into short-pitch positions (Fig. 2D and E). This rearranged array, was then embedded into OCT solution (Fig. 2F). After freezing in a cryotome chamber (−20 °C), blocks were sectioned and stained to measure changes in biological signals under various experimental conditions (Fig. 2G–I).
3D culture of human tumor spheroids on 5 × 5 pillar arrays
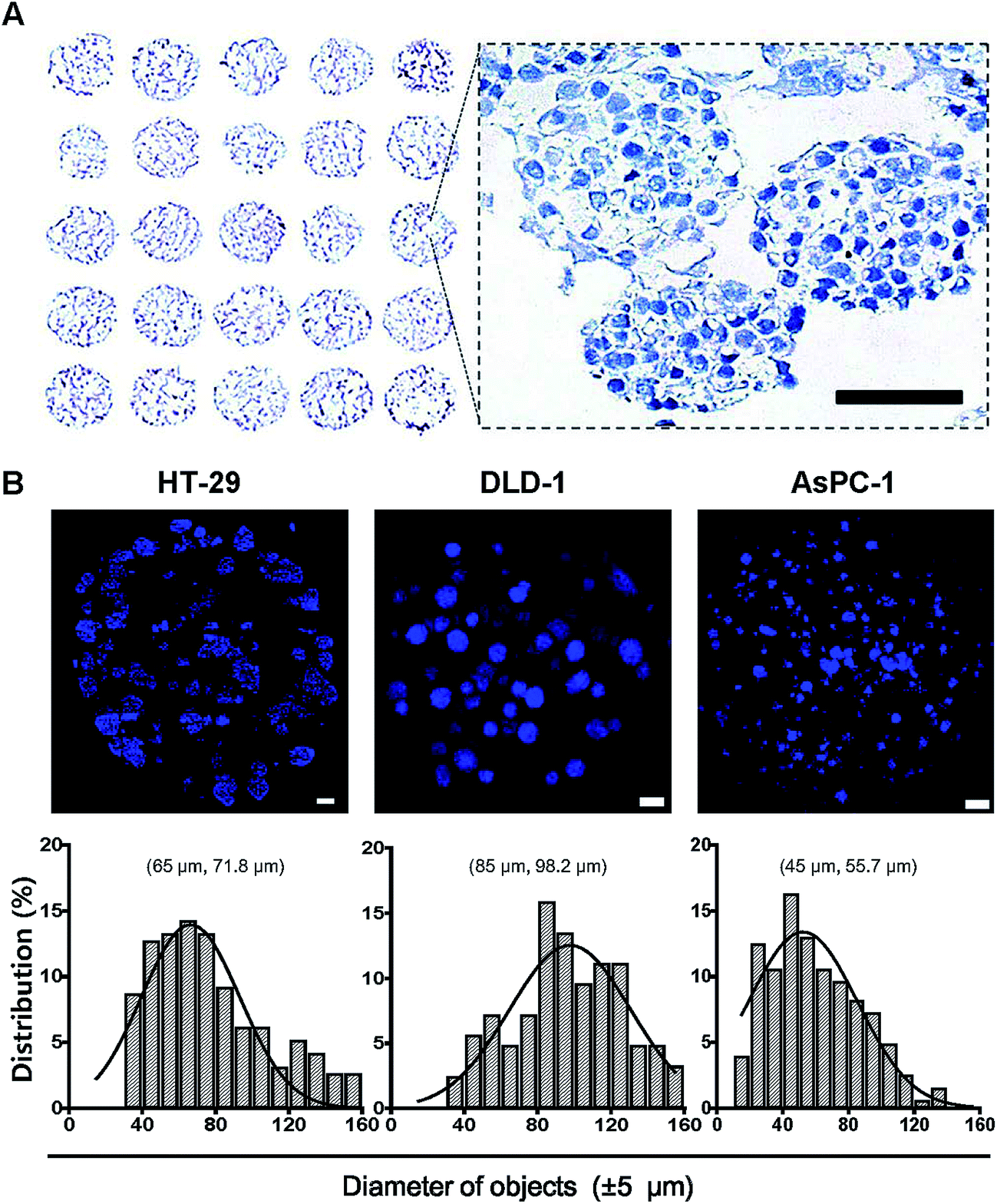
The utility and applicability of pitch-tunable pillar arrays was demonstrated by appropriate cell growth and formation of 3D spheroids using human colorectal and pancreatic adenocarcinoma cells. HT-29 spheroids were successfully cultured for 4 and 8 days before transferring to the OCT block for cryosection. Sections of the spheroids array were stained with hematoxylin for histological evaluation (Fig. 3A). Multiple spheroids developed in a single alginate droplet with the volume as small as 1 μL. There were as many as 60 spheroids in each droplet. A representative enlarged image of a single droplet stained with hematoxylin clearly demonstrates that HT-29 cells grew into 3D cellular aggregates. The spheroids exhibited an even distribution within the gel and cells in 3D spheroids were found to be compact in structure which facilitated close cell–cell interactions. Integrity of the tumor spheroids was confirmed by high cell viability as indicated by prominent nuclear staining as well as the absence of necrotic regions. The average diameter of HT-29 spheroids cultured for 4 days was about 90 μm, and varied between 70 and 150 μm (ESI Video 2†). Tumor spheroids of HT-29, DLD-1 and AsPC-1 were stained with DAPI (Fig. 3B). Nuclear DAPI staining was performed on the cryosections of HT-29 spheroids, hence the diameters of objects measured represent the cross-sectional sizes of the spheroids (median 71.8 μm). DAPI staining of whole spheroid was done on the pillars (DLD-1 and AsPC-1 in Fig. 3B). DLD-1 spheroids showed significantly larger size compared to AsPC-1; median size of 98.2 μm and 55.7 μm for DLD-1 and AsPC-1 spheroids, respectively.
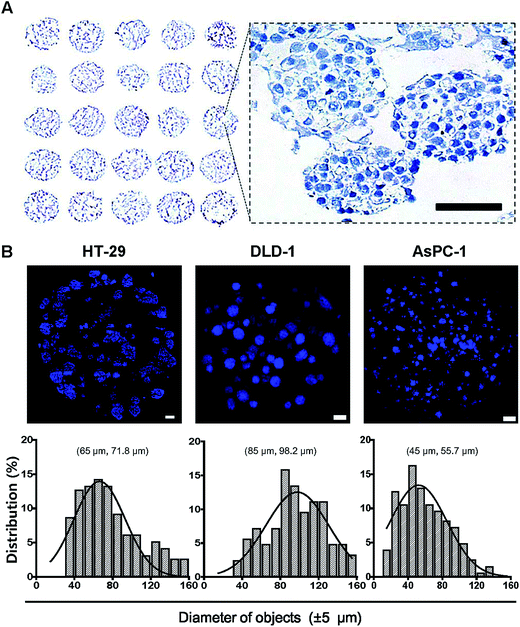 |
| | Fig. 3 Cross sectional images of tumor spheroids cultured on the 5 × 5 pillar array. HT-29, DLD-1 and AsPC-1 cells were cultured in alginate for 4 to 6 days to form spheroids on pillar tips and stained with DAPI. (A) Hematoxylin staining of HT-29 spheroids was done on a cryosections of 5 × 5 pillar array. The enlarged image shows the morphology of spheroids. Scale bar, 50 μm. (B) Sectional images and size distribution of tumor spheroids of HT-29, DLD-1, AsPC-1 cells. DAPI staining was performed either on cryosections (HT-29) or on pillar tips (DLD-1 and AsPC-1) followed by optical sectioning. Scale bar, 100 μm. Size distribution, (mode and median). | |
Drug penetration assay of 3D spheroids
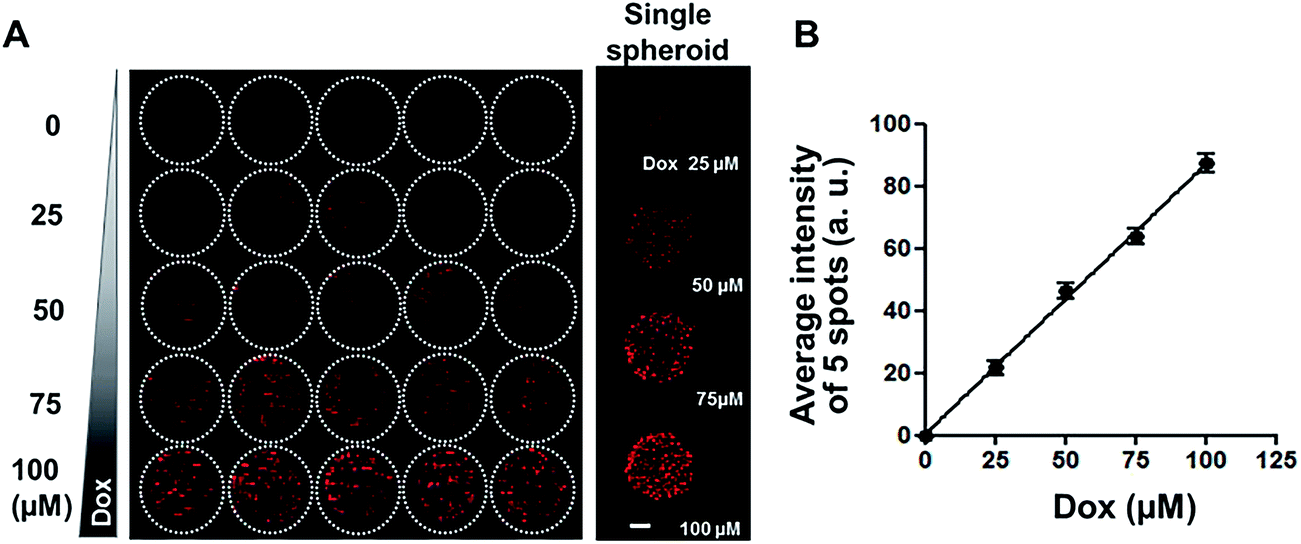
Since diffusion of drugs into spheroids is an important consideration for the evaluation of drug efficacy, fluorescent DOX was used as a model compound to test drug distribution, in our spheroid array chip. HT-29 spheroids in 1% alginate droplets on 5 × 5 pillars were cultured for 6 days and then exposed to a range of DOX concentrations (0, 25, 50, 75, and 100 μM, five replicates each) for 2 hours, and then fluorescence intensity per droplet was measured from the confocal image of OCT cryosections (Fig. 4A). Proportional relationship between fluorescence intensity and DOX concentration showed that DOX diffusion into spheroids appeared to be concentration-dependent (Fig. 4B).
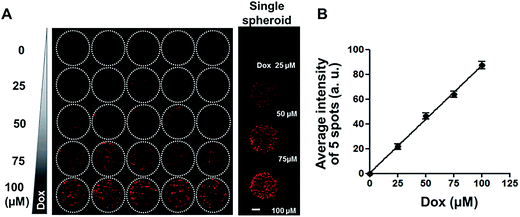 |
| | Fig. 4 Uptake of DOX in HT-29 spheroids grown on 5 × 5 pillar array. (A) Composite fluorescence images of 5 × 5 array of HT-29 cell spheroids exposed to DOX. HT-29 spheroids cultured for 5 days were exposed to DOX for 30 min (5 replicates per concentration) and cryosectioned for confocal imaging. Representative images of HT-29 spheroids exposed to 0, 25, 50, 75, or 100 μM DOX are shown in the right panel. Scale bar, 50 μm. (B) Linear relationship between average fluorescence intensity and DOX concentration, showing concentration-dependent uptake of DOX into HT-29 spheroids. | |
Drug uptake assay with temperature-sensitive liposomes containing DOX
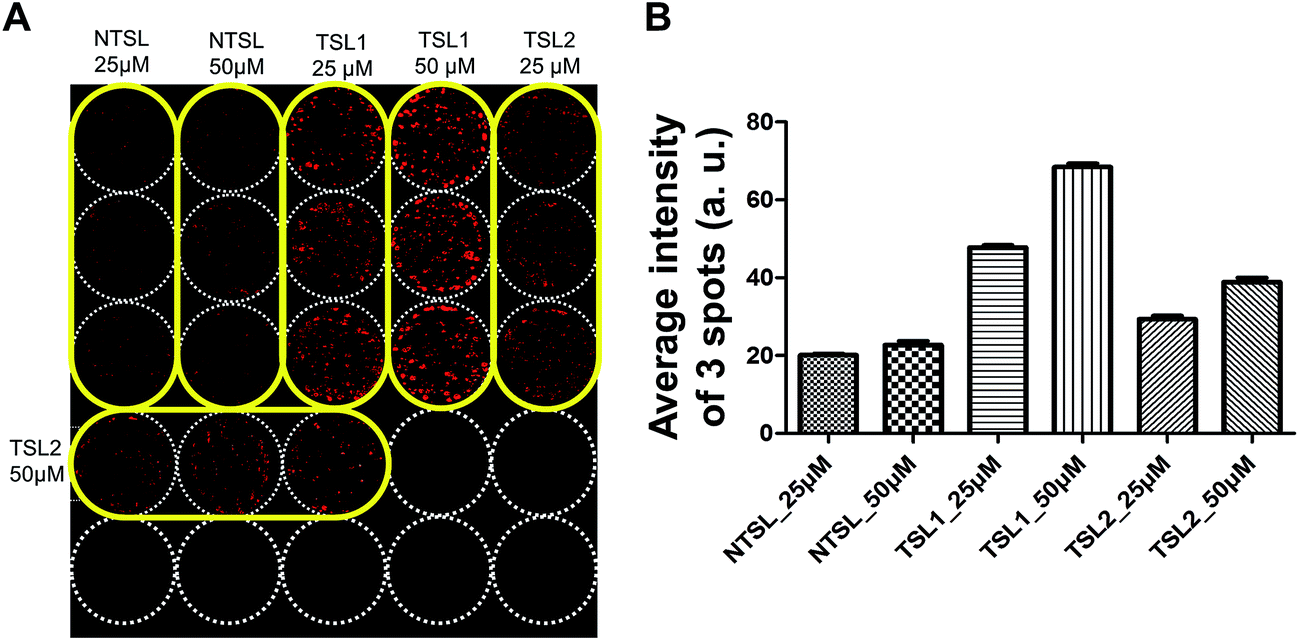
Drug delivery systems (DDS) such as liposomes are often utilized for the delivery of anticancer drugs.38,39 Drug delivery into human tumors may be hindered by the size of delivery vehicle where many factors including increased interstitial fluid pressure, high cell density and ECM may hinder the distribution of DDS through multi-cellular layers of the tumor avascular region.40 3D tumor spheroid cultures are the best models to test these formulations for their efficacy. HT-29 spheroids cultured on the pillars were used to test the distribution of drugs delivered in TSLs containing DOX, for 2 h at 40 °C (Fig. 5). TSLs are stable and retain DOX below transition temperatures. They are designed to release encapsulated DOX at a specific temperature:41 TSL1 and TSL2 formulations were designed to release DOX at 40.5 °C and 41.07 °C, respectively. NTSL is expected to release DOX at 47.5 °C. As expected, HT-29 spheroids exposed to TSL1 showed greater DOX accumulation compared to TSL2 and NTSL (Fig. 5B). Our data was in agreement with the results from a previous study:34 TSL2 was shown to release approximately 10% of incorporated DOX, while TSL1 released 70% at 40 °C. Although we could not differentiate as to whether the liposomes were actually distributed within spheroids, or whether DOX is just released at the transition temperature, while still in media, the latter case may be the more plausible explanation.
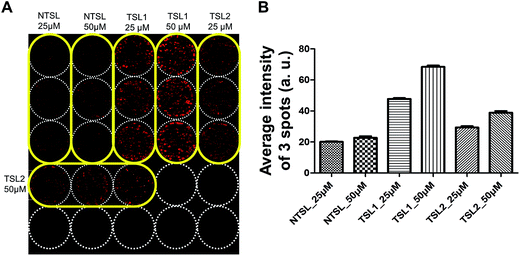 |
| | Fig. 5 Uptake of TSLs containing DOX in HT-29 spheroids grown on a 5 × 5 pillar array. HT-29 spheroids cultured for 5 days were exposed to TSLs containing DOX for 2 hours at 40 °C and cryosectioned for image acquisition. (A) Image of the 5 × 5 array of HT-29 spheroids exposed to temperature-sensitive liposomes with different transition temperatures (40.5 °C for TSL1; 41.07 °C for TSL2) or NTSL in triplicate per concentration. (B) Average fluorescence intensity determined from triplicate pillar spots loaded with HT-29 spheroids following drug exposure. | |
Anticancer drug evaluation
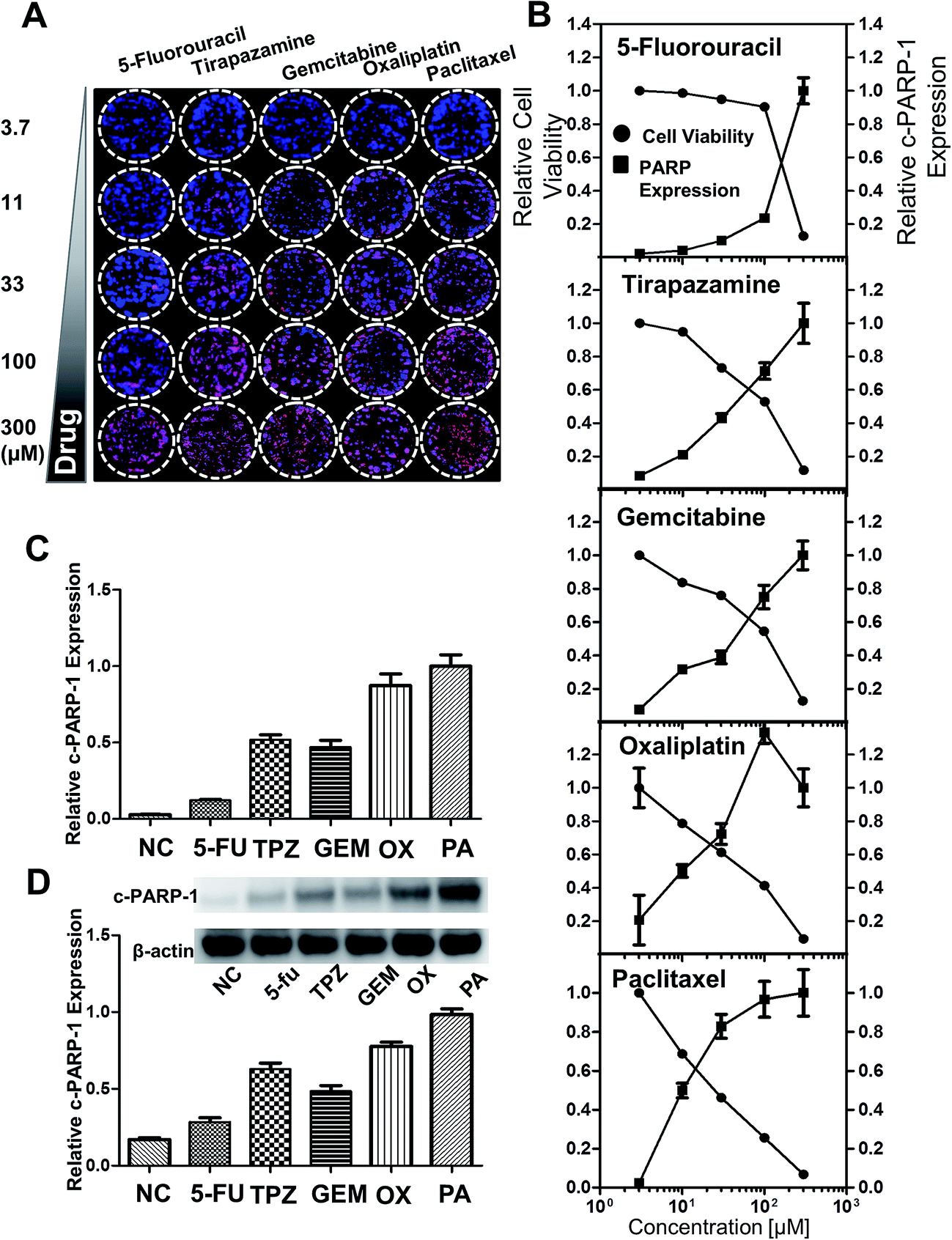
To monitor the cytotoxicity of a drug directed against the HT-29 spheroids, expression of intracellular molecules associated with apoptotic events such as the cleaved form of c-PARP-1 was measured. c-PARP-1 is a member of a family of proteins involved in DNA repair and a biomarker for apoptosis. HT-29 spheroids were exposed to 5-fluorouracil (5-FU), tirapazamine (TPZ), gemcitabine (GEM), oxaliplatin (OX), and paclitaxel (PA) for 2 days. Spheroids were then stained for c-PARP-1 and nuclei. The intensity of DAPI staining (blue) indicated the number of cells, whereas c-PARP-1 staining (red) represented the level of apoptosis induction due to drug exposure (Fig. 6A). The relative cell viability was calculated as fluorescence intensity ratio of {(DAPI − c-PARP-1)/DAPI} and plotted against drug concentration. The relative expression level of c-PARP-1 within the group was also calculated by taking the ratio of fluorescence signals at each concentration to that of the highest concentration (300 μM). c-PARP-1 expression increased with increasing drug concentrations for all five drugs, and consequently, cell viability decreased (Fig. 7B). At 33 μM, OX and PA showed a greater than 50% decrease in cell viability, whereas 5-FU did not show significant changes in viability up to 100 μM42 (Fig. 6B). The profile of drug sensitivity in HT-29 spheroids was examined at 33 μM by fluorescence intensity of c-PARP-1 and confirmed by Western blot analysis (Fig. 6C and D). Our data demonstrated that HT-29 spheroids have differential sensitivity to the 5 different drugs, with 5-FU showing minimal and OX and PA the highest activity. As we showed previously,34 hypoxia-selective TPZ induced significant levels of apoptosis, which may be attributed to the existence of hypoxic condition in 3D spheroid cultures.16
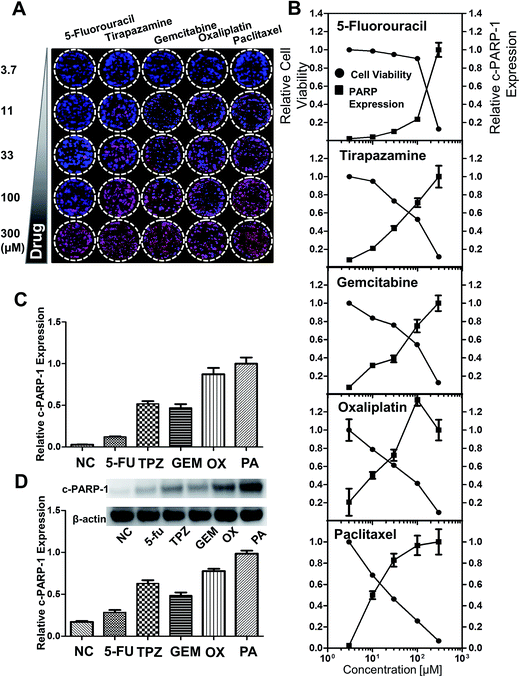 |
| | Fig. 6 Analysis of drug-induced cell death in HT-29 spheroids grown on the pillar array. Spheroids cultured for 6 days were exposed to 5-FU, TPZ, GEM, OX, and PA for 2 days, and subjected to cryosection and immunocytochemical staining. (A) Composite image of the 5 × 5 spheroid array, showing a concentration-dependent increase in c-PARP-1 expression (red) with DAPI (blue) for nucleus. (B) Relative cell viability and c-PARP-1 expression as a function of drug concentration. The fluorescent intensities were obtained from the array image in (A). (C) Relative c-PARP-1 expression obtained from the pillar array at 33 μM dosage of the five compounds. (D) Relative c-PARP-1 expression obtained from Western blots at 33 μM for the five compounds. | |
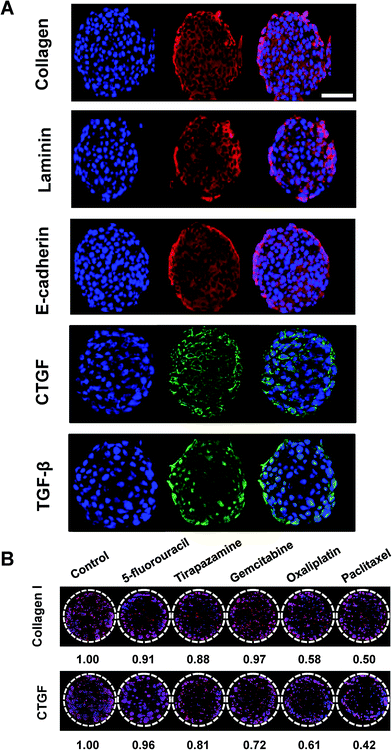 |
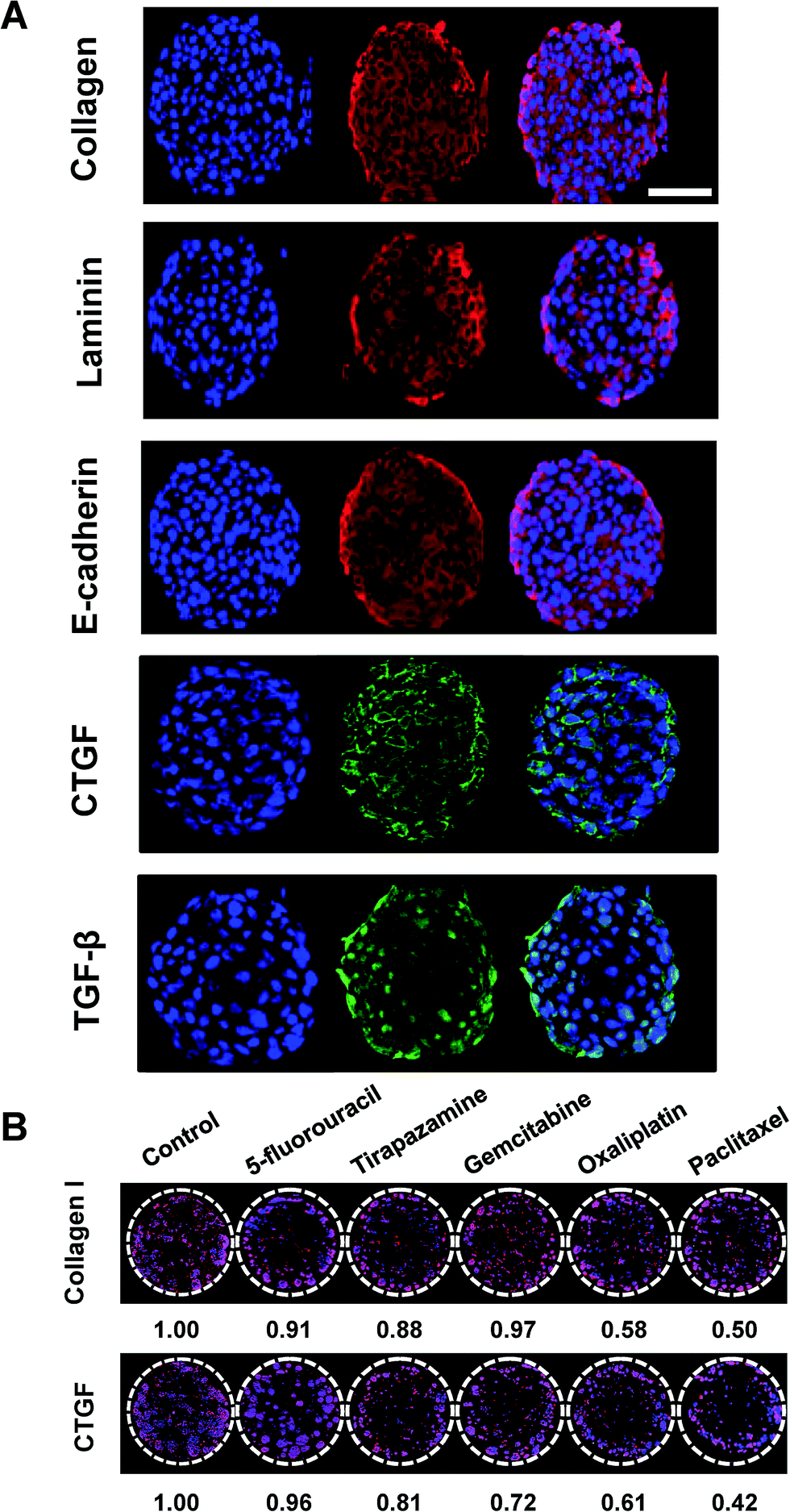
| | Fig. 7 Immunofluorescence detection of intracellular and matricellular proteins in HT-29 cells grown as tumor spheroids on pillar arrays. (A) Representative sectional images of a single HT-29 spheroid immunologically stained for several proteins including type I collagen, laminin, E-cadherin, CTGF, and TGF-β. Scale bar, 50 μm. (B) Changes in type I collagen and CTGF expression was evaluated in HT-29 spheroids following exposure to a panel of anticancer drugs. Multiple spheroids were shown on the cross-sectional images of one pillar spot in (B). | |
Immunohistological analysis of tumor spheroids
Auto- and juxtacrine growth-signaling and tumor-associated stromal-signaling mediated by ECMs are known to contribute to environment-mediated drug resistance (EMDR) in solid tumors.43 Thus, the in vivo-like tissue characteristics were evaluated, not only by morphology but also by the expression of these cellular and matricellular signaling molecules by immunofluorescence staining on sections of the spheroids array. HT-29 spheroids encapsulated in 1% alginate on the 5 × 5 pillar array were cryosectioned and stained. The most prominent features of the multicellular tumor spheroid cultures was the deposition of ECM proteins such as type I collagen and laminin (Fig. 7A). Cell–cell adhesion protein, E-cadherin and growth signaling proteins such as CTGF and TGF-β33 were also successfully detected (Fig. 7A). Nuclear staining was used to observe the relative localization of these proteins, within the spheroids. The expression of laminin appeared to be more discrete around nuclei as compared to the diffused distribution of E-cadherin and collagen type I throughout the cytoplasm. CTGF was mainly expressed in the perinuclear region, while TGF-β was found either in cytoplasm or nuclei. CTGF is well known as a secreted matricellular protein that modulates important signaling pathways leading to cell adhesion and migration, myofibroblast activation, epithelial–mesenchymal transition (EMT), and ECM deposition.44 TGF-β is one of the key signaling molecules related to cell migration and invasion.7 Within each spheroid, TGF-β was highly expressed in the periphery of the spheroid rather than in the core region, which can be understood by the fact that the cells in the periphery tend to grow faster than those in the center of the spheroid. Expression levels of collagen type I and CTGF was decreased after exposure to a panel of anticancer drugs (Fig. 7B). The degree of changes appeared to be correlated to the viability changes measured by c-PARP-1 expression: 5-FU showed the lowest efficacy in cytotoxicity as well as in reducing protein expressions whereas OA and PA showed the highest (Fig. 6). Overall, the expression of cellular and extracellular proteins indicated that the culture conditions used in the study were facilitating cell–cell and cell–ECM interactions in 3D, which supports the conclusion that human cancer cells grown as 3D spheroid on the mini-pillar array exhibit the in vivo human tumors microenvironments.
Limitations and perspectives
The present method was not intended for generating 3D reconstructed images of a whole spheroid. Instead, cross-sectional images of multiple spheroids can be analyzed to provide representative information of the group of spheroids. Actual size of spheroids may also vary, which may reflect nodule heterogeneity in vivo tumors. Further improvement of the present method can be achieved by modifying the design of pillar and array chip for an increased throughput and ease of handling, which is currently underway.
Conclusions
Quantitative measurements of molecular markers on tissue sections using immunohistochemistry are routinely performed in biomedical research. Multiplexed high content analysis (HCA) has emerged as an important platform technology based on methodology for multiplexed immunohistochemistry, imaging and quantitation.45 Multiplex analysis combined with multi-sample processing methods such as tissue microarray (TMA) provided high-throughput molecular profiling as well as proteomic analysis of cancer.46,47 As the importance of three-dimensionality in cell biology has become appreciated, in vitro 3D culture models are now being actively utilized in early drug screening.21,48 Our pitch-tunable array chip provides an efficient method for preparation of micro-tumor sections amenable to multiplexed HCA for evaluation of compound activity.49 In addition, our pillar array, analogous to TMA, may provide high-throughput advantages and improve the early drug discovery process.
Author contributions
D. W. L., J. K. and H. J. H. contributed equally to this work by conducting the majority of the experiments and gathering the data presented here. B. C. S. and M. Y. L. helped in analyzing the data. H.-J. K. developed and designed the study, analyzed the data and interpreted the results. All authors contributed to the preparation of the manuscript.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korea government (MEST) (No. 2016R1A2B2011832 and 2012R1A5A2047939).
References
- N. A. Franken, H. M. Rodermond, J. Stap, J. Haveman and C. van Bree, Nat. Protoc., 2006, 1, 2315–2319 CrossRef CAS PubMed.
- J. Friedrich, C. Seidel, R. Ebner and L. A. Kunz-Schughart, Nat. Protoc., 2009, 4, 309–324 CrossRef CAS PubMed.
- W. L. Haisler, D. M. Timm, J. A. Gage, H. Tseng, T. C. Killian and G. R. Souza, Nat. Protoc., 2013, 8, 1940–1949 CrossRef CAS PubMed.
- J. L. Horning, S. K. Sahoo, S. Vijayaraghavalu, S. Dimitrijevic, J. K. Vasir, T. K. Jain, A. K. Panda and V. Labhasetwar, Mol. Pharm., 2008, 5, 849–862 CrossRef CAS PubMed.
- D. W. Lee, S. H. Yi, S. H. Jeong, B. Ku, J. Kim and M. Y. Lee, Sens. Actuators, B, 2013, 177, 78–85 CrossRef CAS.
- M. Y. Lee, R. A. Kumar, S. M. Sukumaran, M. G. Hogg, D. S. Clark and J. S. Dordick, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 59–63 CrossRef CAS PubMed.
- G. Mehta, A. Y. Hsiao, M. Ingram, G. D. Luker and S. Takayama, J. Controlled Release, 2012, 164, 192–204 CrossRef CAS PubMed.
- H. L. Wong, M. X. Wang, P. T. Cheung, K. M. Yao and B. P. Chan, Biomaterials, 2007, 28, 5369–5380 CrossRef CAS PubMed.
- D. W. Hutmacher, Biomaterials, 2000, 21, 2529–2543 CrossRef CAS PubMed.
- L. A. Kunz-Schughart, J. P. Freyer, F. Hofstaedter and R. Ebner, J. Biomol. Screening, 2004, 9, 273–285 CrossRef CAS PubMed.
- S. J. Kwon, D. W. Lee, D. A. Shah, B. Ku, S. Y. Jeon, K. Solanki, J. D. Ryan, D. S. Clark, J. S. Dordick and M. Y. Lee, Nat. Commun., 2014, 5, 3739 CAS.
- J. Lee, G. D. Lilly, R. C. Doty, P. Podsiadlo and N. A. Kotov, Small, 2009, 5, 1213–1221 CAS.
- L. Shapiro and S. Cohen, Biomaterials, 1997, 18, 583–590 CrossRef CAS PubMed.
- A. M. Fernandes, T. G. Fernandes, M. M. Diogo, C. L. da Silva, D. Henrique and J. M. Cabral, J. Biotechnol., 2007, 132, 227–236 CrossRef CAS PubMed.
- Y. Yang, F. M. Rossi and E. E. Putnins, Biomaterials, 2007, 28, 3110–3120 CrossRef CAS PubMed.
- Y. C. Tung, A. Y. Hsiao, S. G. Allen, Y. S. Torisawa, M. Ho and S. Takayama, Analyst, 2011, 136, 473–478 RSC.
- M. J. Bissell and D. Radisky, Nat. Rev. Cancer, 2001, 1, 46–54 CrossRef CAS PubMed.
- N. Gjorevski, A. Ranga and M. P. Lutolf, Development, 2014, 141, 1794–1804 CrossRef CAS PubMed.
- N. He, X. Wang, N. Kim, J. S. Lim and S. Yoon, J. Mol. Graphics Modell., 2013, 43, 41–46 CrossRef CAS PubMed.
- F. Hirschhaeuser, H. Menne, C. Dittfeld, J. West, W. Mueller-Klieser and L. A. Kunz-Schughart, J. Biotechnol., 2010, 148, 3–15 CrossRef CAS PubMed.
- F. Pampaloni, E. G. Reynaud and E. H. Stelzer, Nat. Rev. Mol. Cell Biol., 2007, 8, 839–845 CrossRef CAS PubMed.
- R. H. Shoemaker, Nat. Rev. Cancer, 2006, 6, 813–823 CrossRef CAS PubMed.
- D. W. Lee, Y. S. Choi, Y. J. Seo, M. Y. Lee, S. Y. Jeon, B. Ku, S. Kim, S. H. Yi and D. H. Nam, Anal. Chem., 2014, 86, 535–542 CrossRef CAS PubMed.
- A. M. Al-Abd, J. H. Lee, S. Y. Kim, N. Kun and H. J. Kuh, Cancer Sci., 2008, 99, 423–431 CrossRef CAS PubMed.
- K. Huang, H. Ma, J. Liu, S. Huo, A. Kumar, T. Wei, X. Zhang, S. Jin, Y. Gan, P. C. Wang, S. He, X. Zhang and X. J. Liang, ACS Nano, 2012, 6, 4483–4493 CrossRef CAS PubMed.
- A. Bullen, Nat. Rev. Drug Discovery, 2008, 7, 54–67 CrossRef CAS PubMed.
- J. P. Celli, I. Rizvi, A. R. Blanden, I. Massodi, M. D. Glidden, B. W. Pogue and T. Hasan, Sci. Rep., 2014, 4, 3751 CrossRef PubMed.
- L. le Roux, A. Volgin, D. Maxwell, K. Ishihara, J. Gelovani and D. Schellingerhout, Mol. Imaging, 2008, 7, 214–221 Search PubMed.
- S. C. Lesher-Perez, G. A. Kim, C. H. Kuo, B. M. Leung, S. Mong, T. Kojima, C. Moraes, M. D. Thouless, G. D. Luker and S. Takayama, Biomater. Sci., 2017, 5, 2106–2113 RSC.
- P. Joshi and M. Y. Lee, Biosensors, 2015, 5, 768–790 CrossRef PubMed.
- C. Godugu, A. R. Patel, U. Desai, T. Andey, A. Sams and M. Singh, PLoS One, 2013, 8, e53708 CAS.
- M. S. Kim, S. Kwon, T. Kim, E. S. Lee and J. K. Park, Biomaterials, 2011, 32, 1396–1403 CrossRef CAS PubMed.
- S.-E. Yeon, D. Y. No, S.-H. Lee, S. W. Nam, I.-H. Oh, J. Lee and H.-J. Kuh, PLoS One, 2013, 8, e73345 CAS.
- J. Kang, D. W. Lee, H. J. Hwang, S. E. Yeon, M. Y. Lee and H. J. Kuh, Lab Chip, 2016, 16, 2265–2276 RSC.
- A. H. Fischer, K. A. Jacobson, J. Rose and R. Zeller, CSH Protoc., 2008, pdb.prot4991 Search PubMed.
- H. D. Han, Y. W. Jeon, H. J. Kwon, H. N. Jeon, Y. Byeon, C. O. Lee, S. H. Cho and B. C. Shin, Acta Biomater., 2015, 24, 279–285 CrossRef CAS PubMed.
- K. Na, S. A. Lee, S. H. Jung, J. Hyun and B. C. Shin, Colloids Surf., B, 2012, 91, 130–136 CrossRef CAS PubMed.
- S.-I. Lee, S.-K. Nagayya-Sriraman, S. Shanmugam, R. Baskaran, C.-S. Yong, S.-K. Yoon, H.-G. Choi and B.-K. Yoo, Biomol. Ther., 2011, 19, 231–236 CrossRef CAS.
- J. H. Choi, Y. J. Lee and D. Kim, J. Pharm. Invest., 2017, 47, 51–64 CrossRef CAS.
- J. H. Baker, K. E. Lindquist, L. A. Huxham, A. H. Kyle, J. T. Sy and A. I. Minchinton, Clin. Cancer Res., 2008, 14, 2171–2179 CrossRef CAS PubMed.
- Y. W. Jeon, S. H. Cho, H. D. Han and B. C. Shin, J. Korean Chem. Soc., 2014, 58, 575–579 CrossRef CAS.
- S. H. Aktas, H. Akbulut, N. Akgun and F. Icli, Cancer Biomarkers, 2012, 12, 135–140 CrossRef CAS PubMed.
- M. B. Meads, R. A. Gatenby and W. S. Dalton, Nat. Rev. Cancer, 2009, 9, 665–674 CrossRef CAS PubMed.
- A. Jacobson and J. L. Cunningham, Fibrog. Tissue Repair, 2012, 5, S8 CrossRef PubMed.
- E. C. Stack, C. Wang, K. A. Roman and C. C. Hoyt, Methods, 2014, 70, 46–58 CrossRef CAS PubMed.
- M. R. Groseclose, P. P. Massion, P. Chaurand and R. M. Caprioli, Proteomics, 2008, 8, 3715–3724 CrossRef CAS PubMed.
- O. P. Kallioniemi, U. Wagner, J. Kononen and G. Sauter, Hum. Mol. Genet., 2001, 10, 657–662 CrossRef CAS PubMed.
- J. W. Haycock, Methods Mol. Biol., 2011, 695, 1–15 CAS.
- D. L. Taylor and K. A. Giuliano, Drug Discovery Today: Technol., 2005, 2, 149–154 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09090k |
| ‡ D. W. L., J. K. and H. J. H. contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *cgh
*cgh