
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, and characterization of lightly sulfonated multigraft acrylate-based copolymer superelastomers†‡
Konstantinos Misichronis ab,
Weiyu Wangc,
Shiwang Chengb,
Yangyang Wangc,
Umesh Shresthaa,
Mark Dadmunab,
Jimmy W. Maysab and
Tomonori Saito*b
ab,
Weiyu Wangc,
Shiwang Chengb,
Yangyang Wangc,
Umesh Shresthaa,
Mark Dadmunab,
Jimmy W. Maysab and
Tomonori Saito*b
aDepartment of Chemistry, University of Tennessee, Knoxville, TN 37996, USA
bChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: saitot@ornl.gov
cCenter for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
First published on 29th January 2018
Abstract
Multigraft copolymer superelastomers consisting of a poly(n-butyl acrylate) backbone and polystyrene side chains were synthesized and the viscoelastic properties of the non-sulfonated and sulfonated final materials were investigated using extensional rheology (SER3). The non-linear viscoelastic experiments revealed significantly increased true stresses (up to 10 times higher) after sulfonating only 2–3% of the copolymer while the materials maintained high elongation (<700%). The linear viscoelastic experiments showed that the storage and loss modulus are increased by sulfonation and that the copolymers can be readily tuned and further improved by increasing the number of branching points and the molecular weight of the backbone. In this way, we show that by tuning not only the molecular characteristics of the multigraft copolymers but also their architecture and chemical interaction, we can acquire thermoplastic superelastomer materials with desired viscoelastic properties.
Introduction
Thermoplastic elastomers (TPEs) exhibit the properties and performance of cross-linked rubber but can be processed as thermoplastics. They are low modulus, flexible materials that can be stretched repeatedly to at least twice their original length at room temperature and return approximately to their initial length and shape upon stress release. The typical design of TPEs is based upon an ABA triblock copolymer architecture with the hard phase formed by component A (which behaves as a physical crosslinker), dispersed in the soft rubbery phase of component B. Most common TPEs include polystyrene–poly(butadiene)–polystyrene (SBS) (e.g. Styroflex®) and polystyrene–poly(isoprene)–polystyrene (SIS) (e.g. Kraton® D SIS series) which are widely used in combinations with other ingredients (paraffinic oils, polyolefins, tackifying resins) in applications such as tire treads, adhesives, sealants, paving, footwear, packaging and many others.1–4Multigraft copolymers5–7 with a rubbery backbone and glassy side chains have emerged as a new class of TPEs, so-called superelastomers, with superior elongation at break and low residual strains as compared to conventional TPEs. Mays et al. synthesized a series of well-defined poly(isoprene-g-styrene) multigraft copolymers with trifunctional, tetrafunctional and hexafunctional branch points.8 These studies showed that topology of the multigraft copolymers and the number of junction points per molecule can be two major parameters for adjusting their mechanical properties.9–13
The use of diene-based TPEs is, however, limited by the poor oxidation and UV resistance of the polydiene backbone and the relatively low upper service temperature (<100 °C) in relation to the glass transition temperature of polystyrene. Efforts have been made to overcome these drawbacks by substituting the polydiene block with acrylates, such as poly(n-butyl acrylate) (PnBA) and other poly(alkyl acrylates), and the polystyrene block with methacrylates, such as poly(methyl methacrylate) (PMMA)14,15 but the ultimate tensile properties of these triblock copolymers were poor as compared to the traditional diene-based TPEs. Depending on the alkyl substituent of the ester groups in polyacrylates and polymethacrylates, glass transition temperature can be tuned within a much larger temperature range (e.g. from −89 °C for poly(n-nonyl acrylate) up to 259 °C for poly(adamantyl methacrylate)).16 Furthermore, synthesizing multigraft superelastomers via a macromonomer approach using free radical or emulsion polymerization is compatible with these acrylic monomers, reduces the polymerization time, and minimizes the complexity of the synthetic route. Recently developed procedures allowed for the synthesis of PS-g-PI and all-acrylic multigraft copolymers showing very good thermal and mechanical properties.17–20 The macromonomer approach is a very useful method to synthesize multigraft copolymers since it readily controls the molecular weight and polydispersity of the side chains as well as the desired average number of branch points.21 The combination of this method with free radical polymerization provides a scalable and facile polymerization procedure with relatively good control of the molecular characteristics of the final multigraft copolymers.
In an effort to also improve the tensile strength and the thermal stability of thermoplastic elastomers, post-polymerization modifications on hydrogenated SBS and SIS copolymers have been widely performed.22–37 Sulfonation of the polystyrene block is a very versatile chemical modification that has been utilized not only to enhance microphase separation but also to mechanically reinforce the final material by introducing ionic interactions within the polymer chain networks. Sulfonation results in a significant increase of the Flory–Huggins χ value and it has been shown27,28 that even low sulfonation levels can improve or alter microphase separation. Moreover, there is a critical value of sulfonation level (approximately 15–20% depending on the MW of the PS block),30,31 above which the polymer becomes non-thermally processable, poorly-soluble in most commonly used solvents and very brittle due to the highly ionically cross-linked networks. Thus, for a thermoplastic multigraft superelastomer, it is crucial to control the degree of sulfonation and find the fine line between increased tensile strength, high elongation, elasticity, and processability.
This study describes the synthesis and characterization of polystyrene-g-poly(n-butylacrylate) multigraft copolymers (abbreviated as PSm-PnBA) comprising of PnBA backbones and lightly sulfonated PS side chains synthesized by a macromonomer approach. The PS macromonomer (PSm) was synthesized by living anionic polymerization and high vacuum techniques,38 while conventional free radical polymerization was employed for the synthesis of the final multigraft copolymers (Scheme 1). The effects of the molecular weight, number of branch points and volume fraction on the properties of the final materials were investigated.
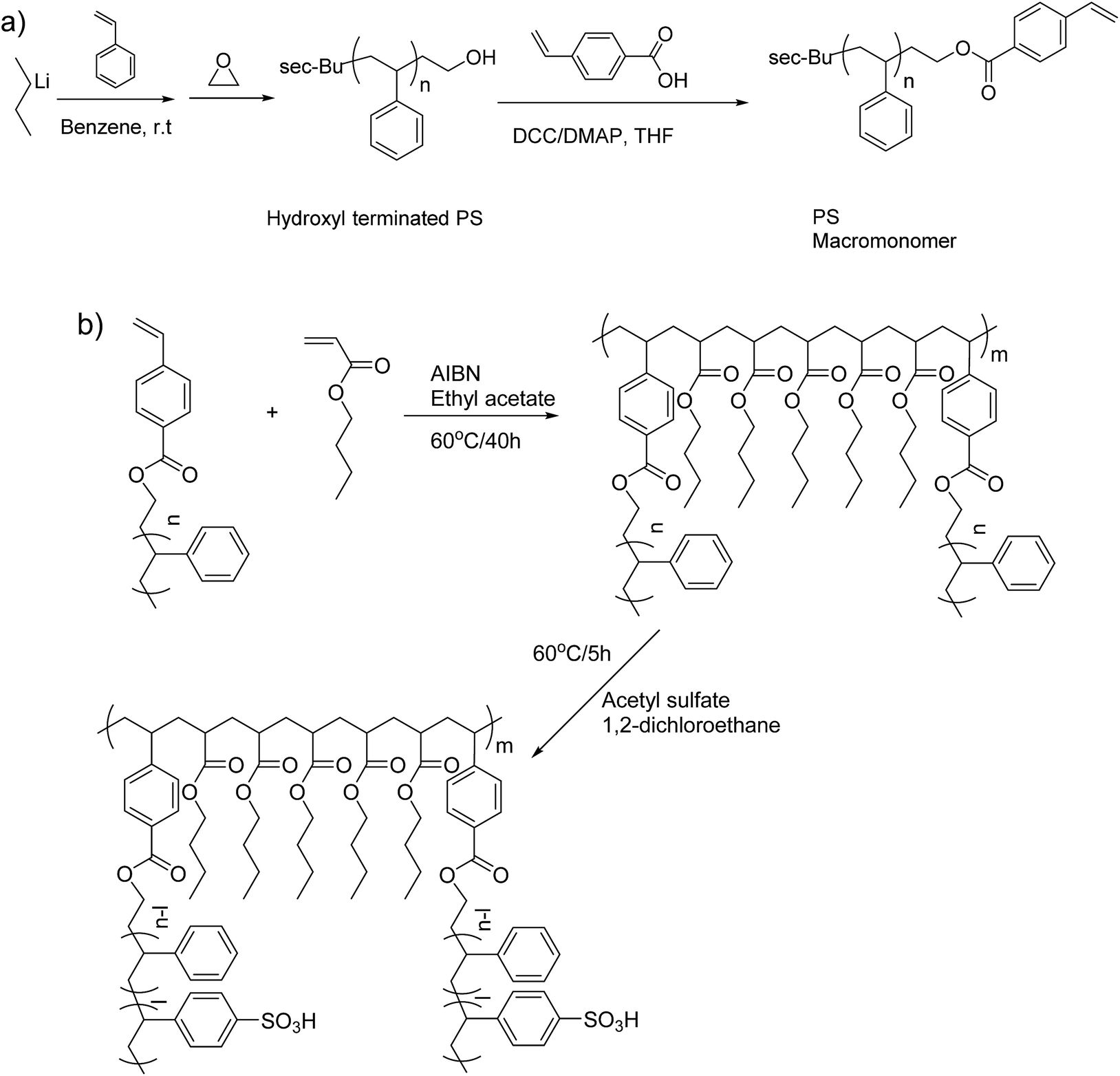 | ||
| Scheme 1 (a) Synthesis of the PS macromonomer and (b) synthetic route and post-polymerization sulfonation of the multigraft copolymers. | ||
Experimental
Materials
The PS macromonomer was synthesized in evacuated, n-butyllithium washed and benzene rinsed glass vessels. Detailed descriptions of the high-vacuum technique as well as the purification procedures for styrene (Sigma Aldrich, ≥99%), benzene (Sigma Aldrich, ≥99%) and sec-BuLi (initiator, Acros, 1.3 M in cyclohexane/hexane 92/8) required for anionic polymerization have already been reported in the literature.38 2,2′-Azobisisobutyronitrile (AIBN) (Sigma Aldrich, 98%) was purified by crystallization from methanol, while an activated alumina (Sigma Aldrich, basic) column was utilized to remove the inhibitors from toluene (solvent, Fisher Chemical) and n-butyl acrylate (n-BA) (Sigma Aldrich, ≥99%). N,N′-Dicyclohexylcarbodiimide (DCC) (Sigma Aldrich, 99%), 4-(dimethylamino) pyridine (DMAP) (Sigma Aldrich, ≥99%), 4-vinylbenzoic acid (Sigma Aldrich, 97%), dichloroethane (DCE) (Fisher Chemical), acetic anhydride (Sigma Aldrich, ≥99%) and sulfuric acid (Sigma Aldrich, 95–98%) were used as received.Macromonomer synthesis
The synthetic procedure for the PS macromonomer is shown in Scheme 1a. In an n-butyllithium/benzene rinsed glass apparatus equipped with break-seals, 40 g of styrene were introduced into 400 ml of benzene solution with 4 mmol of sec-butyllithium as initiator under high vacuum and the polymerization was left to proceed in room temperature. After 16 hours, 1.5 ml of ethylene oxide were added to the living polystyrene anion solution and the color turned from orange to transparent within 1 min. The reaction was kept at room temperature for 24 hours and was terminated with high vacuum sealed MeOH. The resulting hydroxyl terminated polystyrene (PS–OH) was precipitated three times in large excess of MeOH and dried under reduced pressure at 40 °C for 48 hours. 39.3 g of PS–OH were recovered and dissolved into 400 ml of anhydrous THF with 16 mmol of DCC and 16 mmol of DMAP. A 10 ml THF solution with 16 mmol of 4-vinylbenzoic acid were added to the solution and it was kept at room temperature. After 6 days, the polymer solution was precipitated in excess of MeOH three times in order to remove any excess of 4-vinylbenzoic acid and catalyst. The resulting PS macromonomer with polymerizable styrene end group was dried under reduced pressure at 40 °C for 48 hours and characterized with MALDI-TOFF.Multigraft copolymer synthesis and sulfonation
All copolymerizations were conducted in high vacuum sealed round bottom flasks at 60 °C in toluene using AIBN as initiator. The PS macromonomer was always the minority component while the amount of initiator was tailored depending on the desirable final molecular weight and number of branching points. More specifically, for the synthesis of PSm-PnBA-1 multigraft copolymer, 0.651 g of PS macromonomer, 3.52 g of n-BA and 22 mg of AIBN (0.134 mmol) were introduced in a round bottom flask containing 30 ml of toluene and were degassed in the high vacuum line twice. Then, the solution was put in an oil bath at 60 °C and the polymerization was left to proceed for 40 hours. After the completion of the polymerization, the flask was put in an ice bath and precipitated in 200 ml of MeOH, filtered and dried in a vacuum oven for 2 days. For the sulfonation procedure, 0.72 g of the dried PSm-PnBA-1 multigraft copolymer and 15 ml of DCE were added in a round bottom flask exhibiting a side arm sealed with a rubber septum. The flask was attached in the high vacuum line, degassed twice, removed from the high vacuum line and placed in an oil bath at 60 °C. A solution of 15 ml of DCE, 0.113 ml (1.2 × 10−3 mol) of acetic anhydride and 0.03 ml (6 × 10−4 mol) of sulfuric acid was mixed and left to react for 10 minutes at 0 °C. The mixture was then introduced with a syringe into the flask containing the PSm-PnBA-1 sample and the reaction was allowed to proceed for 5 hours under stirring at 60 °C (change of color solution to light brown). MeOH was added to terminate the reaction and the final sulfonated multigraft copolymer was precipitated in excess of MeOH, filtered, washed thoroughly with DI H2O and MeOH three times, dried in a vacuum oven for 3 days and stored at −20 °C. Both the synthesis and sulfonation procedures for the multigraft copolymers are shown in Scheme 1b.Instrumentation
Molecular characterization
Size exclusion chromatography (SEC) was carried out at 40 °C using a Polymer Laboratories GPC-120 unit equipped with a Precision Detectors PD 2040 (two-angle static light scattering (TALLS) detector), a Viscotek 220 differential viscometer and a Polymer Laboratories differential refractometer. A Polymer Laboratories PLgel; 7.5 × 300 mm; 10 μ; 500, 103, 10 × 105 and 10 × 106 Å column set was employed and the calibration range was 600–7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500000 g mol−1 using PS standards. The mobile phase was THF at a flow rate of 1.0 ml min−1. 1H-NMR was carried out on a Varian Mercury 500 instrument. All samples were dissolved in deuterated chloroform (CDCl3).
500000 g mol−1 using PS standards. The mobile phase was THF at a flow rate of 1.0 ml min−1. 1H-NMR was carried out on a Varian Mercury 500 instrument. All samples were dissolved in deuterated chloroform (CDCl3).
Thermal analysis
Differential scanning calorimetry (DSC) was used to investigate the thermal properties of the samples. A TA Instruments Q2000 was utilized in temperatures ranging from −140 °C to 160 °C at a heating rate of 10 °C min−1 with a 2 minute isothermal hold at the maximum and minimum temperatures. Reported glass transition temperatures are those measured on the second of two heating runs.Morphological characterization
Small angle X-ray scattering (SAXS) measurements were carried out in transmission geometry on a three pinhole collimated system equipped with a Rigaku rotating anode (CuKα radiation λ = 1.542 Å, operating at 4.2 kW), an Osmic multilayer mirror for higher proton flux and a MARCCD 2D detector (average pixel size 78.7 × 78.7 μm). The sample to detector distance was 1.72 m and calibration was carried out using Ag-Behenate as the standard. All patterns were background corrected and normalized with respect to thickness and subsequently were radially averaged yielding 1D SAXS patterns of I(q) vs. q. The setup of the instrument enabled a resolvable range of 0.05 nm−1 ≤ q ≤ 2 nm−1. The bulk films were prepared by solution casting the polymers from a non-selective solvent (toluene) for 4 days in a porcelain crucible and then by annealing them in a vacuum oven at 130 °C (above the Tg of PS) for 3 more days. All films were quenched into liquid nitrogen after they were removed from the annealing oven in order to “trap” their thermodynamic state.Extensional rheometry and dynamic mechanical analysis (DMA)
Uniaxial extension experiments of the samples were carried out on an HR2 rheometer (TA Instruments) using the SER3 fixture while an RSA-G2 dynamic mechanical analyzer (TA Instruments) was used for complimentary temperature ramp experiments. The specimens for the tests were prepared by solution-casting the polymers in a Teflon Petri dish. Rectangular sheet-like specimens were cut out from the film by a razor blade and removed from the Teflon surface with the aid of dry ice. All the uniaxial extension and DMA experiments were performed with constant Hencky strain rates.Results and discussion
Synthesis of lightly sulfonated multigraft acrylate-based copolymers
The molecular characteristics of the PS macromonomer (PSm) as well as the multigraft superelastomers are presented in Table 1. The results show well-defined final products with narrow polydispersity for PSm (1.03) and controlled molecular weight,![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n = 10300 g mol−1 (Fig. S1‡). The PS macromonomer serves as the PS side chain of the multigraft copolymers. The free radical copolymerization of PS macromonomer with n-butyl acrylate at high monomer concentrations with an appropriate molar ratio of AIBN (4 mg of initiator per one ml of monomer) resulted in controlled molecular weights and number of branches for the final copolymers. Higher amounts of AIBN and solvent produced much shorter polymer chains for the n-butyl acrylate backbone, thus lower number of branches and lower molecular weights for the branched copolymers. Consequently, it was crucial to regulate the amount of AIBN needed, in order to achieve highly branched copolymers and high conversions of the monomers. Although the exact targeted final molecular weights for the multigraft copolymers are difficult to achieve using free radical polymerization, we were able to achieve final molecular weights with values very close to the targeted theoretical molecular weights by adjusting the appropriate ratio of AIBN/n-butyl acrylate/solvent.
n = 10300 g mol−1 (Fig. S1‡). The PS macromonomer serves as the PS side chain of the multigraft copolymers. The free radical copolymerization of PS macromonomer with n-butyl acrylate at high monomer concentrations with an appropriate molar ratio of AIBN (4 mg of initiator per one ml of monomer) resulted in controlled molecular weights and number of branches for the final copolymers. Higher amounts of AIBN and solvent produced much shorter polymer chains for the n-butyl acrylate backbone, thus lower number of branches and lower molecular weights for the branched copolymers. Consequently, it was crucial to regulate the amount of AIBN needed, in order to achieve highly branched copolymers and high conversions of the monomers. Although the exact targeted final molecular weights for the multigraft copolymers are difficult to achieve using free radical polymerization, we were able to achieve final molecular weights with values very close to the targeted theoretical molecular weights by adjusting the appropriate ratio of AIBN/n-butyl acrylate/solvent.
| Sample | MPSmacron (kg mol−1) | Mtotalna (kg mol−1) | PDI | PS weight fraction f | Number of branch points | Sulfonation degreeb (mol%) |
|---|---|---|---|---|---|---|
| a Determined by using SEC equipped with TALLS and dn/dcmultigraft = fPS × dn/dcPS + fPnBA × dn/dcPnBA with dn/dcPS = 0.187 ml g−1 and dn/dcPnBA = 0.060 ml g−1.b Determined by using 1H-NMR spectroscopy. | ||||||
| PSm-PnBA-1 | 10.3 | 195.0 | 1.75 | 0.20 | 4 | 13.5 |
| PSm-PnBA-2 | 10.3 | 125.4 | 1.80 | 0.20 | 3 | 13.5 |
1H-NMR in combination with SEC (Fig. 1a and b respectively) was used to characterize the molecular weights of the final copolymers, their PS content, the average number of branch points and the sulfonation degree. The content of PS glassy side chains was kept at ∼20 wt% since previous studies9–13 showed that the highest modulus with the highest elongation rates were obtained with a glassy side chain composition of approximately 20 wt%. The sulfonated derivatives of the two PSm-PnBA multigraft copolymers are labelled as PSSm-PnBA followed by their respective numbers (1 and 2).
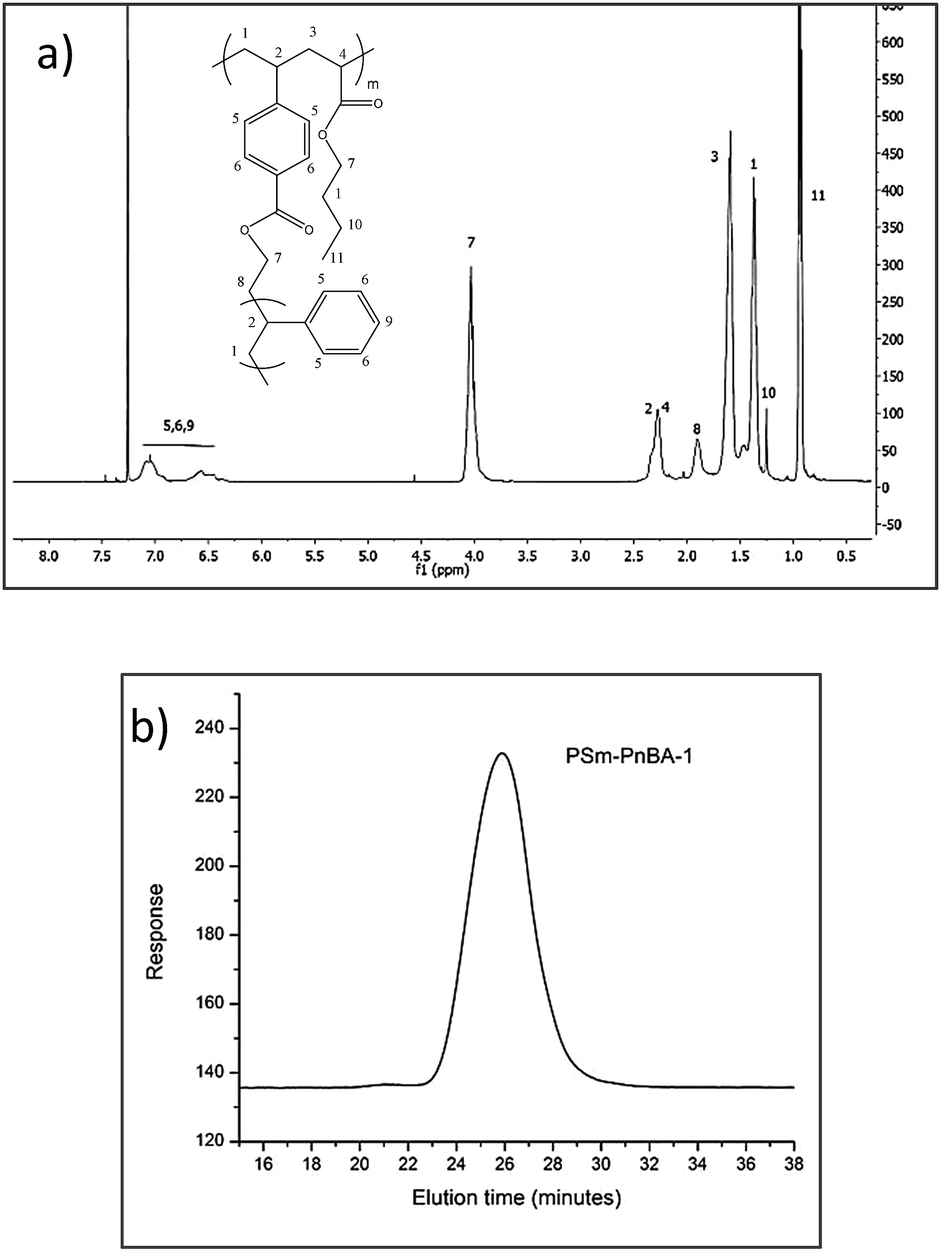 | ||
| Fig. 1 (a) 1H-NMR spectrum and (b) SEC chromatogram of the PSm-PnBA-1 multigraft copolymer. | ||
Fig. 1b shows the SEC chromatogram corresponding to PSm-PnBA-1 multigraft copolymer exhibiting a monomodal peak. The absence of any other peaks in lower molecular weights verifies that there is no unreacted PS macromonomer present in the final multigraft copolymer. The relative 1H-NMR (Fig. 1a) peaks corresponding to PS aromatic rings (6.3–7.3 ppm) and to PnBA backbone acrylate group (methylene next to ester, 4 ppm) respectively, confirm the successful random copolymerization of the monomers.
In order to calculate the branching points of the multigraft copolymers, we used the final molecular weight of each multigraft copolymer, the “fixed” molecular weight of the PS macromonomer and the final weight fraction of each component calculated by 1H-NMR spectroscopy. For both multigraft copolymers, the PS weight fraction is 20%, thus the total molecular weight of PS content is 195k × 20/100 = 39k and 125.4k × 20/100 = 25.08k for PSm-PnBA-1 and PSm-PnBA-2 respectively. By dividing the total molecular weight of PS content for each of the two multigraft copolymers with the molecular weight of the PS macromonomer, we get the average number of branching points for PSm-PnBA-1 and PSm-PnBA-2 respectively. This indicates that PSm-PnBA-1 sample exhibits mostly 4 branching points per chain while PSm-PnBA-2 exhibits 2 and 3 branching points per chain.
The sulfonation degree of PS side chains (20% of the total molecular weight) was kept at 13.5% in order to maintain processability which gives ion exchange capacity (IEC) of 0.26 mmol g−1. During sulfonation, controlling the molar ratio of acetyl sulfate/styrene is very important for achieving the desired sulfonation level, and in this study the ratio was kept at 0.5 (i.e. [acetyl sulfate]/[styrene] = 0.5). The sulfonation level of each multigraft copolymer (SL%) was calculated by using 1H-NMR and the following equation:22
 | (1) |
DSC measurements were performed for both PSm-PnBA-1 and PSm-PnBA-2 as well as for their relative sulfonated samples PSSm-PnBA-1 and PSSm-PnBA-2. For all samples, only one glass transition temperature was observed (Fig. S2a and b‡). This suggests that the rapid cooling rate in DSC did not provide sufficient time for microphase separation to occur, which is consistent with observed results in other experiments with similar graft copolymers.39,40 Since poly(n-butyl acrylate) is the majority component, the glass transition temperature is shifted towards lower temperatures and closer to the Tg of PnBA. Sulfonation of the PS block shifts the Tg towards higher temperatures, increasing it from −46 °C for PSm-PnBA-1 to −40 °C for PSSm-PnBA-1 (sulfonated) and from −42 °C for PSm-PnBA-2 to −39 °C for PSSm-PnBA-2 (sulfonated).
The increase of Tg is associated with the ion content and the ionomeric effect which is caused by regions with restricted mobility that act as physical cross-links.40,41 The Tg of both non-sulfonated samples is slightly lower than expected, which can be possibly attributed to small traces of solvent trapped in the copolymer, even though the samples were dried thoroughly. Also from Fig. S2a and b,‡ it is observed that the curve indicating the glass transition, occurs over a relatively broad range of temperatures and thus, it may introduce an error of ±2–3 °C in the measurements.
Viscoelastic properties
Extensional rheometry (SER3) at room temperature was used to determine the non-linear viscoelastic properties of the multigraft copolymers and to investigate the impact of sulfonation on these properties. The linear viscoelastic characterization was performed by small amplitude oscillatory shear.Sulfonation has a significant impact on the linear viscoelastic response for PSm-PnBA-1 and PSm-PnBA-2 (Fig. 2a and b). In the case of PSm-PnBA-1, storage and loss modulus are increased by approximately two orders of magnitude at room temperature after sulfonation. The change of slope is due to the change from liquid-like to solid-like behavior after sulfonation. The crossover for the PSm-PnBA-1 before sulfonation is associated with the relaxation behavior of the system and the sample is liquid-like at low frequencies and solid-like at high frequencies. For PSm-PnBA-2, G′ and G′′ increase by one order of magnitude with sulfonation. Before sulfonation, the loss and storage modulus of PSm-PnBA-1 are lower than those of PSm-PnBA-2 which is caused by the longer distance among the PS side chains and the less dense packing of those chains due to the higher molecular weight of the PnBA backbone of the PSm-PnBA-1 multigraft copolymer. On the other hand, after sulfonation, the loss and storage modulus of PSSm-PnBA-1 are much higher than those of PSSm-PnBA-2 since there are more sulfonated PS side chains per macromolecule in PSSm-PnBA-1. The sulfonated PS chains act as physical crosslinks forming ionic bonds between the pendant –SO3−H+ groups which dominate the dynamics of both systems. Increasing the number of branching points only by 1 (from 3 to 4) storage and loss modulus increase drastically.
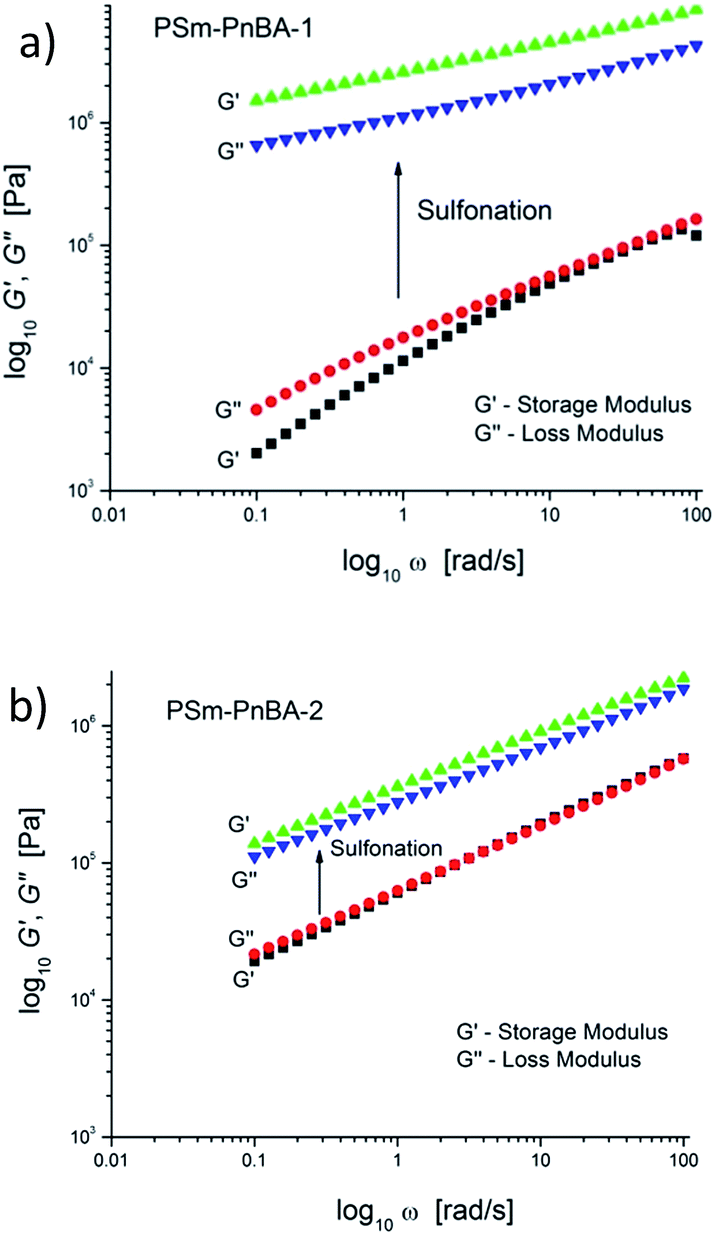 | ||
| Fig. 2 Linear viscoelastic data in room temperature for (a) PSm-PnBA-1 and (b) PSm-PnBA-2 copolymers before and after sulfonation. Storage and loss modulus increase drastically after sulfonation for both samples. | ||
In a recent study,42 Filippidi et al. investigated a similar mechanism where they introduced strong iron charges (Fe3+) into a loosely cross-linked amorphous epoxy network with high catechol content in order to form dynamic iron–catechol cross-links. They showed that this mussel inspired system enhances stiffness and toughness without trading off elasticity, due to the incorporation of ionomeric nanodomains into a cross-linked network. The non-linear viscoelastic measurements for PSm-PnBA-1 and PSSm-PnBA-1 copolymers were conducted at three different strain rates 0.1, 1 and 10 s−1 (Fig. S3a and b‡). The true stress is significantly increased after sulfonation for all strain rates without sacrificing elongation (true strain) very much. Fig. 3 shows the notable increase in true stress after sulfonation for each one of the three strain rates measured. For 0.1 s−1 (Fig. 3a), true stress for PSm-PnBA-1 is 0.365 MPa while after sulfonation true stress reaches 10.6 MPa for the same true strain value (ε = 2). Both PSm-PnBA-1 and PSSm-PnBA-1 (sulfonated) samples follow the same trend for the rest of the strain rates (Fig. 3b and c). True stress increases from 1.74 MPa to 33.9 MPa and from 5.86 MPa to 84.9 MPa for 1 s−1 and 10 s−1 respectively for ε = 2. The elongation (true strain) at break before sulfonation for PSm-PnBA-1 was at 2060% (ε = 3) for strain rates 1 s−1 and 10 s−1 and at 1380% (ε = 2.6) with strain rate equal to 0.1 s−1, while after sulfonation the elongation at break was decreased (but still remained significant) to ∼700% (ε = 2) for all three strain rates tested. Similar behavior is observed for the PSm-PnBA-2 sample and its sulfonated derivative PSSm-PnBA-2 where true stress increases as the strain rate increases for both samples (Fig. S4a and b‡). On the other hand, elongation (true strain) at break for both PSm-PnBA-2 and PSSm-PnBA-2 remains similar for all strain rates tested. Fig. 4 shows the increase in true stress after sulfonation for all three different strain rates. The low degree of sulfonation does not alternate significantly the elongation of both PSm-PnBA-2 and PSSm-PnBA-2 and the materials maintain their elasticity while true stress increases 2 to 4 times more than the non-sulfonated case. For 0.1 s−1 (Fig. 4a), true stress for PSm-PnBA-2 is 1.71 MPa while after sulfonation true stress reaches 4.1 MPa for the same true strain value (ε = 2.2). Both PSm-PnBA-2 and PSSm-PnBA-2 samples follow the same trend for the rest of the strain rates (Fig. 4b and c), where the true stress increases from 4.8 MPa to 15.1 MPa and from 16.2 MPa to 36.1 MPa for 1 s−1 and 10 s−1 respectively. Sulfonation has a greater impact on the non-linear viscoelastic properties of PSm-PnBA-1 than those of PSm-PnBA-2, which can be explained by the fact that the PS side chains of PSm-PnBA-1 form more ionic bonds per macromolecule as compared to the lower average numbers of –SO3−H+ groups in PSm-PnBA-2. Sulfonating one more PS branch in combination with the increase of the total molecular weight of the PSm-PnBA-1 multigraft copolymer, gives a much higher increase of true stress after sulfonation as compared to PSm-PnBA-2. This increase in true stress for both PSSm-PnBA-1 and PSSm-PnBA-2 multigraft copolymers indicates the significant impact on the viscoelastic properties caused by sulfonating only a very small fraction (2–3%, IEC ∼0.26 mmol g−1) of the final material (i.e. sulfonating 13.5 wt% of the PS chains which occupy 20 wt% of the whole multigraft copolymer). Slightly altering the properties of the glassy side chains, i.e. tuning the PS chemical structure by adding –SO3−H+ groups that can form ionic bonds, and changing the number of branching points of the copolymers will allow one to take advantage of the favourable branched architecture and manipulate the thermoplastic and elastic properties of the copolymer towards a desired direction.
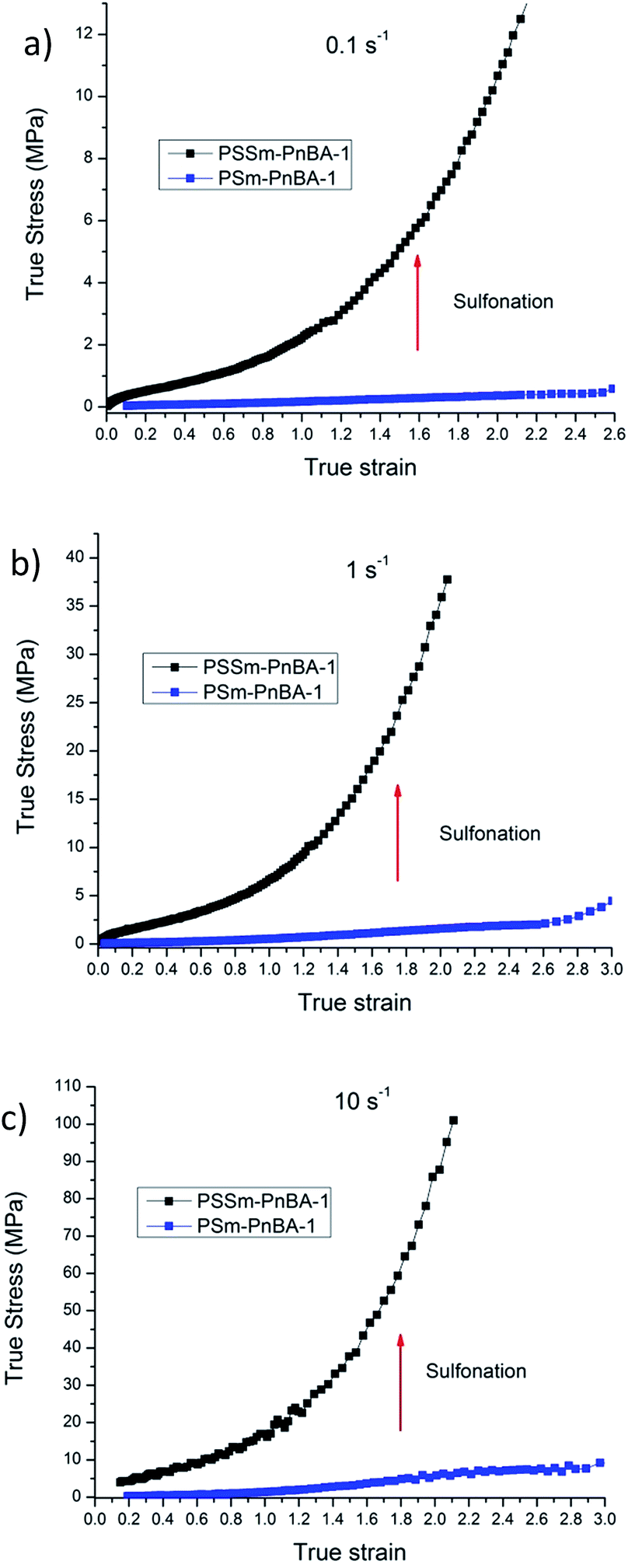 | ||
| Fig. 3 Non-linear viscoelastic data comparison between PSm-PnBA-1 and PSSm-PnBA-1 for (a) 0.1 s−1 (b) 1 s−1 (c) 10 s−1. | ||
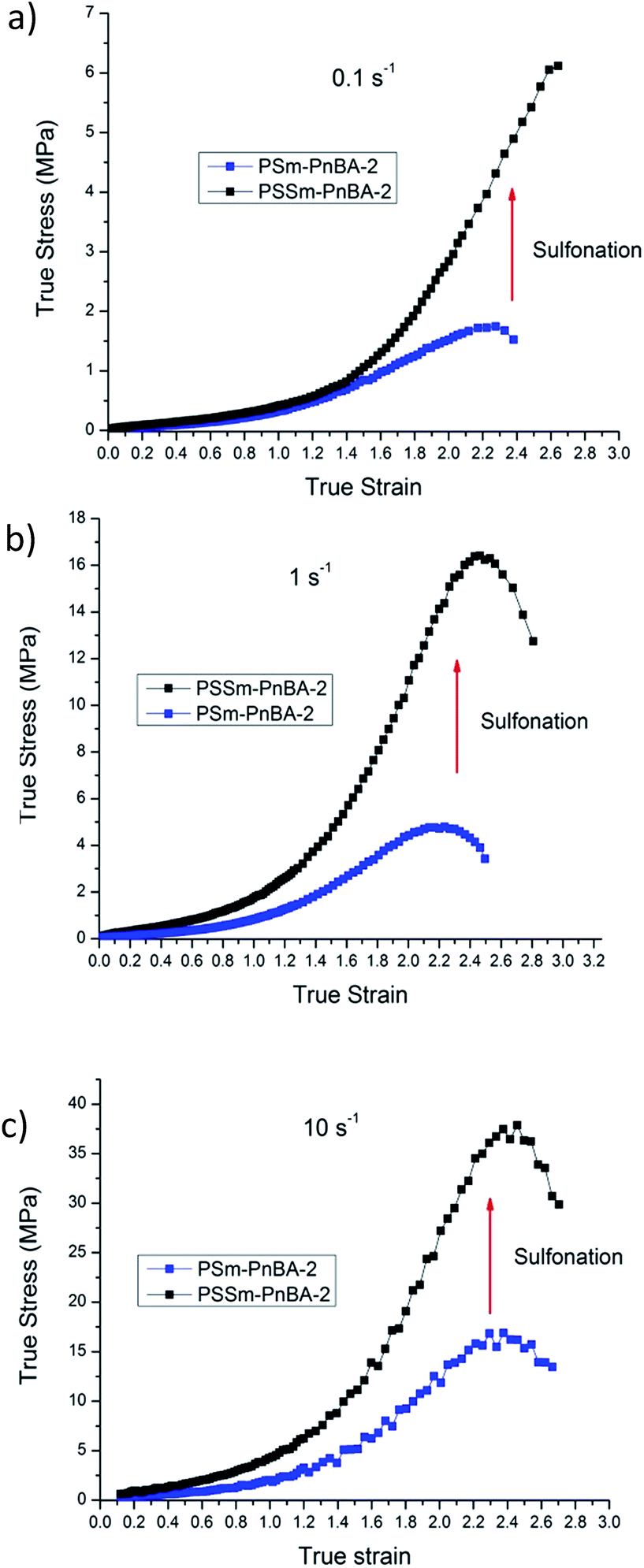 | ||
| Fig. 4 Non-linear viscoelastic data comparison between PSm-PnBA-2 and PSSm-PnBA-2 for (a) 0.1 s−1 (b) 1 s−1 (c) 10 s−1. | ||
Morphology
Fig. 5 shows the SAXS patterns of PSm-PnBA-1, PSSm-PnBA-1, PSm-PNBA-2 and PSSm-PnBA-2 (Fig. 5a–d respectively). All samples (non-sulfonated and sulfonated) were annealed at 130 °C and they appear to exhibit no long range order since no scattering peaks are observed. In all cases, the total volume fraction of PS block is ΦPS = 0.19 (calculated from the equation ΦA = fAdA/(fAdA + fBdB), where dA = dPS = 1.05 g ml−1 and dB = dPnBA = 1.087 g ml−1 and fA = 0.2 and fB = 0.8 are the weight fractions of PS and PnBA, respectively, determined by 1H-NMR spectroscopy). It is well-known43 that the product of the Florry–Huggins interaction parameter χ with the degree of polymerization N and in combination with the volume fraction Φ can provide information about the predicted morphology of a given block copolymer. The values of χAB for a block copolymer with A and B blocks can be estimated by the molar volumes and the solubility parameters of the two components via eqn (2):
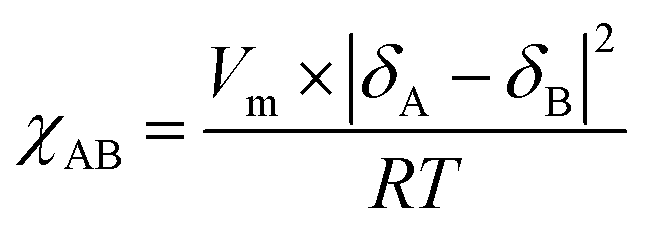 | (2) |
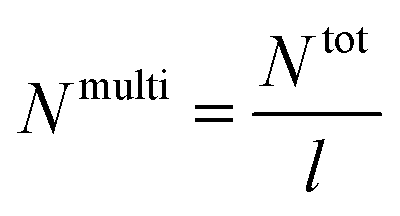 | (3) |
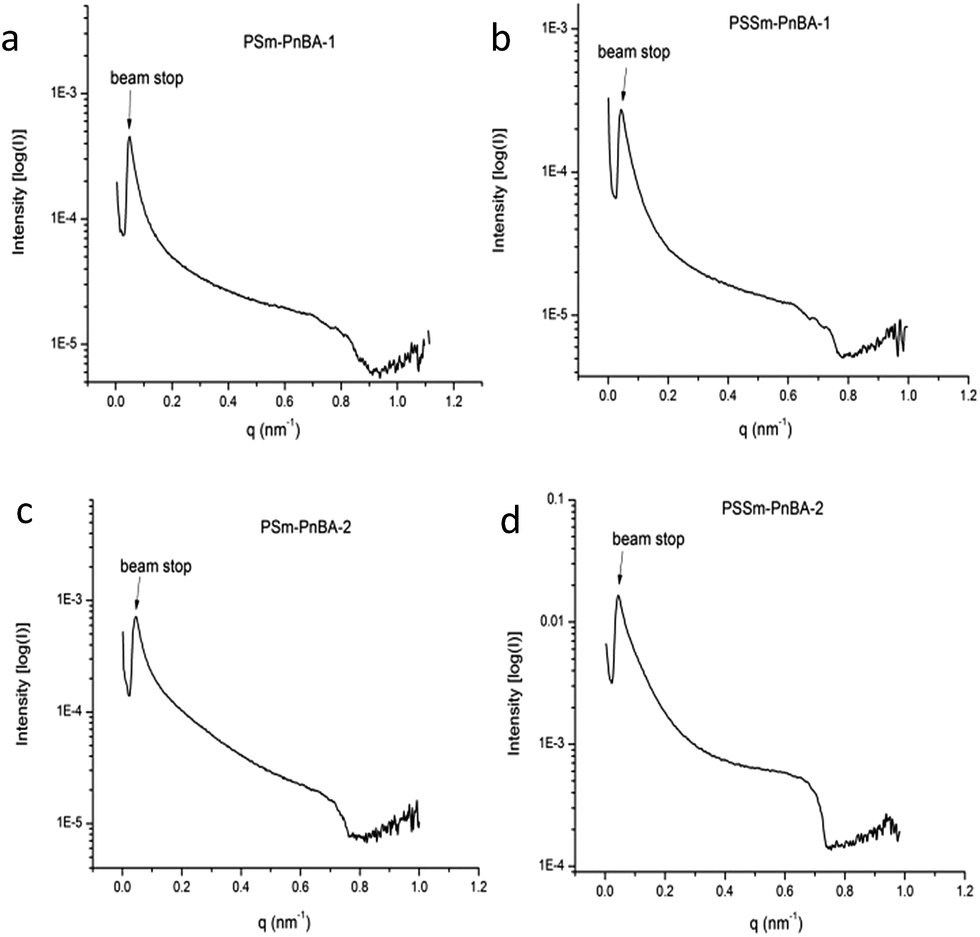 | ||
| Fig. 5 SAXS data of (a) PSm-PnBA-1 (b) PSSm-PnBA-1 (c) PSm-PnBA-2 and (d) PSSm-PnBA-2 multigraft copolymers. | ||
| Sample | ΦPS | Florry–Huggins interaction parameter χ | Degree of polymerizationa Nmulti | χN |
|---|---|---|---|---|
| a Calculated using eqn (3), where Ntot = NA + NB = (PSn/MSt0) + (PnBAn/MnBA0) and MSt0 = 104 g mol−1 and MnBA0 = 128.17 g mol−1. |
||||
| PSm-PnBA-1 | 19 | 0.0011 | 325 | 0.36 |
| PSm-PnBA-2 | 19 | 0.0011 | 279 | 8.38 |
| PSSm-PnBA-1 | 19 | 0.0258 | 325 | 0.31 |
| PSSm-PnBA-2 | 19 | 0.0258 | 279 | 7.19 |
Conclusions
In conclusion, the concept of synthesizing a PS macromonomer (side chains) through anionic polymerization and then copolymerizing it with n-butyl acrylate using free radical polymerization proves to be very efficient since the strategy does not require tedious multi-step anionic polymerization or any fractionation or separation. The overall time for the synthesis of the multigraft copolymers with controlled molecular weight and number of branches was significantly reduced and the method could be readily adoptable in industry. Slight sulfonation of PS-g-PnBA multigraft superelastomers (2–3 wt% total) shows that we can further improve their viscoelastic properties and increase true stress significantly, while maintaining high elongation, not only by tuning their molecular characteristics but also by altering their chemical structure. On the other hand sulfonation of PS side chains does not seem to have any significant impact on the morphology of the multigraft copolymers, which remain weakly microphase separated but do not exhibit any long range order.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Materials Sciences and Engineering Division. A portion of this research was conducted at the Center for Nanophase Materials Sciences, which is a DOE Office of Science User Facility.Notes and references
- R. J. Spontak and P. N. Patel, Curr. Opin. Colloid Interface Sci., 2000, 5, 334–341 CrossRef CAS.
- K. Knoll and N. Nießner, Macromol. Symp., 1998, 132, 231–243 CrossRef CAS.
- R. N. Legge, Rubber Chem. Technol., 1989, 62, 529–547 CrossRef.
- R. N. Legge, G. Holden and E. H. Schröeder, Thermoplastic Elastomers – A Comprehensive Review, Hanser Publishers, Munich, 1987 Search PubMed.
- D. Uhrig and J. W. Mays, Polym. Chem., 2011, 2, 69–76 RSC.
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747–3792 CrossRef CAS PubMed.
- R. Schlegel, et al., Macromolecules, 2011, 44, 9374–9383 CrossRef CAS.
- D. Uhrig and J. W. Mays, Macromolecules, 2002, 35, 7182–7190 CrossRef CAS.
- Y. Zhu, et al., Macromolecules, 2006, 39, 4428–4436 CrossRef CAS.
- R. Schlegel, et al., Eur. Polym. J., 2009, 45, 2902–2912 CrossRef CAS.
- D. Uhrig, R. Schlegel, R. Weidisch and J. W. Mays, Eur. Polym. J., 2011, 47, 560–568 CrossRef CAS.
- U. Staudinger, et al., Eur. Polym. J., 2008, 44, 3790–3796 CrossRef CAS.
- R. Schlegel, et al., Polymer, 2009, 50, 6297–6304 CrossRef CAS.
- D. J. Tong and R. Jerome, Polymer, 2000, 41, 2499–2510 CrossRef.
- D. J. Tong, et al., Polymer, 2001, 42, 3503–3514 CrossRef.
- C. C. Cypcar, P. Camelio, V. Lazzeri, J. L. Mathias and B. Waegell, Macromolecules, 1996, 29, 8954–8959 CrossRef CAS.
- A. Goodwin, et al., Ind. Eng. Chem. Res., 2015, 54, 9566–9576 CrossRef CAS.
- W. Wang, et al., Ind. Eng. Chem. Res., 2015, 54, 1292–1300 CrossRef CAS.
- H. Li, et al., J. Colloid Interface Sci., 2015, 453, 226–236 CrossRef CAS PubMed.
- Y. Luo, X. Wang, Y. Zhu, B.-C. Li and S. Zhu, Macromolecules, 2010, 43, 7472–7481 CrossRef CAS.
- X. Leng, et al., RSC Adv., 2016, 6(36), 30320–30329 RSC.
- J. Yeo, et al., Nanotechnology, 2012, 245703 CrossRef CAS PubMed , (11pp).
- S. Kim, et al., J. Membr. Sci., 2013, 427, 85–91 CrossRef CAS.
- Y. A. Elabd and E. Napadenski, Polymer, 2004, 45, 3037–3043 CrossRef CAS.
- J.-E. Yang and J.-S. Lee, Electrochim. Acta, 2004, 50, 617–620 CrossRef CAS.
- Y. A. Elabd, E. Napadenski, J. M. Sloan, D. M. Crawford and C. W. Walker, J. Membr. Sci., 2003, 217, 227–242 CrossRef CAS.
- Y. A. Elabd, E. Napadenski, C. W. Walker and K. I. Winey, Macromolecules, 2006, 39, 399–407 CrossRef CAS.
- X. Lu, W. P. Steckle and R. A. Weiss, Macromolecules, 1993, 26, 6525–6530 CrossRef CAS.
- A. Buonerba, V. Speranza and A. Grassi, Macromolecules, 2013, 46, 778–784 CrossRef CAS.
- R. A. Weiss, A. Sen, C. L. Willis and L. A. Pottick, Polymer, 1991, 32, 1867–1874 CrossRef CAS.
- R. A. Weiss, A. Sen, C. L. Willis and L. A. Pottick, Polymer, 1991, 32, 2785–2792 CrossRef CAS.
- J. Won, et al., J. Membr. Sci., 2003, 214, 245–257 CrossRef CAS.
- K. A. Mauritz, R. I. Blackwell and F. L. Beyer, Polymer, 2004, 45, 3001–3016 CrossRef CAS.
- B. Kim, J. Kim and B. Jung, J. Membr. Sci., 2005, 250, 175–182 CrossRef CAS.
- E. A. Elabd, C. W. Walker and F. L. Beyer, J. Membr. Sci., 2004, 231, 181–188 CrossRef.
- R. I. Blackwell and K. A. Mauritz, Polymer, 2004, 45, 3457–3463 CrossRef CAS.
- J. Kim, et al., Macromol. Rapid Commun., 2002, 23, 753–756 CrossRef CAS.
- D. Uhrig and J. W. Mays, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6179–6222 CrossRef CAS.
- J. L. De La Fuente, M. Fernandez-Garcia and E. L. Madruca, J. Appl. Polym. Sci., 2001, 80, 783–789 CrossRef CAS.
- T. Saito, et al., Macromolecules, 2008, 41, 3503–3512 CrossRef CAS.
- A. Eisenberg, B. Hird and R. B. Moore, Macromolecules, 1990, 23, 4098–4107 CrossRef CAS.
- E. Filippidi, et al., Science, 2017, 358, 502–505 CrossRef CAS PubMed.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, Wiley-Interscience Publication, New York, 4th edn, 1999 Search PubMed.
- S. P. Carvallo, E. F. Lucas, G. Gonzales and L. S. Spinelli, J. Braz. Chem. Soc., 2013, 24, 1998–2007 Search PubMed.
- L. Zhang, J. Lin and s. Lin, J. Phys. Chem. B, 2008, 112, 9720–9728 CrossRef CAS PubMed.
- W. Wang, et al., Macromolecules, 2014, 47, 7284–7295 CrossRef CAS.
- J. Mijovic, M. Sun, S. Pejanovic and J. W. Mays, Macromolecules, 2003, 36, 7640–7651 CrossRef CAS.
Footnotes |
| † This manuscript has been authored by UT-Battelle, LLC under Contract No. DE-AC05-00OR22725 with the U. S. Department of Energy. The publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan). |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08641e |
| This journal is © The Royal Society of Chemistry 2018 |