
ReI-Catalyzed highly regio- and stereoselective C–H addition to terminal and internal alkynes†

Abstract
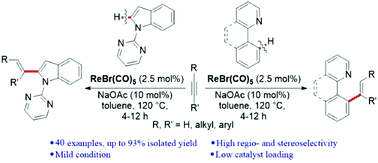
We have developed an effective ortho C–H functionalization of arylpyridines and detachable N-pyrimidyl indoles by terminal and internal alkynes using a Re(I) catalyst providing an efficient access to various E-selective alkenylation products. The catalytic reaction is compatible with various aliphatic alkynes, aromatic terminal alkynes and internal alkynes, and structurally different nitrogen heterocycles. Deuterium-labeling experiments indicate that significant deuterium scrambling occurs with the directing groups and acetylenic sp C–H bonds before the migratory insertion.
 Please wait while we load your content...
Please wait while we load your content...

