
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric phase-transfer catalysed β-addition of isoxazolidin-5-ones to MBH carbonates†
Vito
Capaccio
ab,
Katharina
Zielke
a,
Andreas
Eitzinger
a,
Antonio
Massa
b,
Laura
Palombi
 b,
Kirill
Faust
c and
Mario
Waser
*a
b,
Kirill
Faust
c and
Mario
Waser
*a
aInstitute of Organic Chemistry, Johannes Kepler University Linz, Altenbergerstr. 69, 4040 Linz, Austria. E-mail: mario.waser@jku.at
bDipartimento di Chimica e Biologia, Università di Salerno, Via Giovanni Paolo II, 132, 84084 Fisciano, SA, Italy
cInstitute of Catalysis, Johannes Kepler University Linz, Altenbergerstr. 69, 4040 Linz, Austria
First published on 12th October 2018
Abstract
A novel high yielding, enantio- and diastereoselective protocol for the synthesis of α-allylated highly functionalised β-amino acid derivatives by adding isoxazolidin-5-ones to MBH carbonates under asymmetric phase-transfer catalysis has been developed.
Introduction
Chiral phase-transfer catalysis is one of the most powerful strategies to achieve the catalytic non-covalent asymmetric control of prochiral nucleophiles in stereoselective transformations (i.e. C–C bond forming reactions via alkylations or conjugate additions and heterofunctionalization reactions).1 Among the many enantioselective applications that rely on the use of chiral phase-transfer catalysts (PTCs), asymmetric syntheses of chiral natural and non-natural amino acids have attracted maybe the most interest.2 Here asymmetric α-functionalization reactions of glycine Schiff bases 1 to access chiral α-amino acids have been a very thoroughly investigated topic.2–4 On the other hand, the synthesis of chiral β-amino acids under chiral phase-transfer catalysis has so far mainly been associated with Mannich type approaches,1 while the use of simple isoxazolidin-5-ones 2 as starting materials for the asymmetric syntheses of α,α-difunctionalized β-amino acids emerged only recently as a powerful and complementary strategy.5–7 While Briere and co-workers have used compounds 2 as starting materials in asymmetric PT-catalysed transformations,5 the groups of Shibasaki and Cossy independently developed asymmetric transition metal-catalysed allylation approaches by using these interesting pronucleophiles.6In general, also the use of easily available Morita–Baylis–Hillman adducts 3 under asymmetric organocatalysis allows for highly enantioselective allylation reactions,8 thus resulting in a complementary strategy to transition metal-catalysed allylation approaches. Very interestingly, more than 10 years ago O'Donnell's group already demonstrated that the use of chiral Cinchona alkaloid-based PTCs allows for the highly enantioselective β-addition of glycine Schiff bases 1 to allylic acetates 3′ (Scheme 1A).9,10 Besides this inspiring initial report,9 it was also impressively shown that, based on the nature of the employed catalyst, the nucleophilic attack on allylic substrates 3 can either occur in the β- or in the γ-position.11,12
 | ||
| Scheme 1 Known reaction of glycine Schiff bases 1 with MBH acetates 3′ and targeted reaction of β-amino acid-based compounds 2 with MBH carbonates 3. | ||
Our groups have over the last years focused on the syntheses of chiral amino acids by using asymmetric phase-transfer catalysis,4 and we were now interested in the achievement of the stereoselective α-allylation of β-amino acid precursors 2 by reacting them with the simple MBH-carbonates 3 under asymmetric phase-transfer conditions (Scheme 1B).
Inspired by the work of O'Donnell,9 we rationalized that the PT-catalysed addition of 2 should also predominantly occur to the β-position of acceptor 3, which would thus result in a complementary protocol (compared to the previous reports by Cossy and Shibasaki6) for the allylation of the masked β-amino acids 2.
As chiral catalysts we focused on the use of easily available Cinchona derivatives A,1 our own bifunctional system B,13 our tartaric acid-based ammonium salt C,4 and on the commercially available Maruoka catalysts D14 (Fig. 1).
 | ||
| Fig. 1 Chiral PTCs used in this study. | ||
Results and discussion
We started our screening by investigating the reaction between the benzyl-substituted isoxazolidin-5-one 2a and the parent MBH carbonate 3a (Table 1 gives an overview of the most significant results obtained using the chiral PTCs A–D under a variety of different conditions). The first reactions were carried out with tetrabutylammonium bromide (TBAB) as an achiral PTC. As anticipated the addition of 2a to the conjugated acceptor 3a occurs exclusively to the β-position, giving the (E)-configurated product 5a as the main product (entry 1). By focusing on chiral Cinchona alkaloid-based PTCs A next, we unfortunately realized that, independent of the solvent and/or the nature/amount of the base, only racemic product can be obtained (entries 2–6). Interestingly also, the amount of base has a crucial effect on the conversion rate (compare entries 2 and 5). Based on our recent progress in the design and use of bifunctional ammonium salts like compound B,13 we next tested this catalyst, but again no enantioenrichment could be observed (entry 7). Intriguingly, the reaction proceeded significantly faster even with only 1.1 equiv. of base compared to the reactions with catalysts A.|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entrya | Cat. | Solvent | Base | T [°C] | t [h] | Yieldb [%] | E/Zc | e.r.d |
| a Using 0.1 mmol 2a and 0.15 mmol 3a (c = 0.05 M). b Isolated yield. c Determined by 1H NMR. d Determined by HPLC using a chiral stationary phase. | ||||||||
| 1 | TBAB (10%) | THF | Cs2CO3 (20 eq.) | 25 | 16 | 94 | 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
— |
| 2 | A2 (5%) | THF | Cs2CO3 (20 eq.) | 25 | 16 | 84 | 6:1 |
48:52 |
| 3 | A2 (5%) | CH2Cl2 | Cs2CO3 (20 eq.) | 25 | 16 | 39 | 5:1 |
49:51 |
| 4 | A2 (5%) | CH2Cl2 | K3PO4 (20 eq.) | 25 | 16 | 43 | 3:1 |
46:54 |
| 5 | A2 (5%) | THF | Cs2CO3 (1.1 eq.) | 25 | 16 | <5 | — | — |
| 6 | A1 (5%) | THF | Cs2CO3 (1.1 eq.) | 25 | 72 | 54 | 4:1 |
51:49 |
| 7 | B (5%) | THF | Cs2CO3 (1.1 eq.) | 25 | 48 | 75 | 5:1 |
50:50 |
| 8 | C (5%) | THF | Cs2CO3 (3 eq.) | 25 | 24 | 60 | 6:1 |
49:51 |
| 9 | D1 (5%) | THF | Cs2CO3 (3 eq.) | 25 | 24 | 86 | 6:1 |
74:26 |
| 10 | D1 (2%) | THF | Cs2CO3 (3 eq.) | 25 | 72 | 73 | 6:1 |
71:29 |
| 11 | D1 (10%) | THF | Cs2CO3 (3 eq.) | 25 | 24 | 95 | 7:1 |
75:25 |
| 12 | D2 (5%) | THF | Cs2CO3 (3 eq.) | 25 | 24 | 82 | 5:1 |
85:15 |
| 13 | D3 (5%) | THF | Cs2CO3 (3 eq.) | 25 | 24 | 26 | 5:1 |
50:50 |
| 14 | D2 (5%) | THF | Cs2CO3 (1.1 eq.) | 25 | 24 | <5 | — | — |
| 15 | D2 (5%) | CH2Cl2 | Cs2CO3 (1.1 eq.) | 25 | 96 | 60 | 3:1 |
78:22 |
| 16 | D2 (5%) | THF | K2CO3 (3 eq.) | 25 | 24 | <5 | — | — |
| 17 | D2 (5%) | MTBE | Cs2CO3 (3 eq.) | 25 | 72 | 86 | 7:1 |
83:17 |
| 18 | D2 (5%) | iPr2O | Cs2CO3 (3 eq.) | 25 | 48 | 90 | 7:1 |
87:13 |
| 19 | D2 (5%) | iPr2O | Cs2CO3 (3 eq.) | −20 | 96 | 90 | 10:1 |
94:6 |
| 20 | D2 (5%) | iPr2O | Cs2CO3 (3 eq.) | −30 | 96 | 50 | 12:1 |
94:6 |
| 21 | D2 (5%) | THF | Cs2CO3 (3 eq.) | −40 | 96 | 23 | 7:1 |
96:4 |
| 22 | D1 (5%) | THF | Cs2CO3 (3 eq.) | −40 | 96 | 39 | 7:1 |
85:15 |

Briere and co-workers recently described several asymmetric phase-transfer catalysed transformations of compounds 2 and in these reports5 they also found that catalyst classes A and B are not suited to achieve any face-differentiation with this unique pronucleophile. In contrast, the more rigid Maruoka catalysts D were found to be much better suited in their case studies, and we therefore tested the commercially available derivatives D1–3 as well as our own tartaric acid-based spiroammonium salt C4 next.
While ammonium salt C was found to be not selective (entry 8), the first experiment carried out with catalyst D1 gave product 5a in high yield and with a promising initial e.r. of 74:26 (entry 9). Carrying out the reaction with different catalyst loadings showed no pronounced effect (entries 9–11) and we thus used 5 mol% of catalyst for the further optimization. Also, different concentrations and stoichiometric ratios of the reagents were tested at that point (not given in the table), but no notable change in performance was observed. Testing catalysts D2 and D3 next, we found that the spiro-bis-binaphthyl-based derivative D2 allows for a clear increase in the selectivity (entry 12), while the bifunctional D3 did not give any asymmetric induction at all (entry 13). It was also found that 3 equiv. of Cs2CO3 in an ether solvent is the combination of choice to achieve reasonable yields and selectivities with catalyst D2 (entries 14–16) and we therefore screened different ethers next. While MTBE did not result in any improvement, diisopropylether (iPr2O) allowed for a slightly higher e.r. at room temperature (entry 18). The selectivity could be further improved by lowering the temperature from −20 to −40 °C (entries 19–22), although the reaction slowed down significantly at temperatures below −20 °C (no product formation in iPr2O at −40 °C and only a very slow reaction in THF at −40 °C).
Based on that extensive screening, we thus chose the conditions given in entry 19 (5 mol% of D2 in iPr2O at −20 °C) to carry out the investigation of the application scope next (Scheme 2).
 | ||
| Scheme 2 Application scope for the asymmetric reaction of β-amino acid-based compounds 2 with MBH carbonates 3 (all reactions were carried out under the conditions shown in entry 19, Table 1; the E:Z ratios were determined by NMR of the crude product mixture; the enantiomeric ratios are given for the major diastereomere and were measured by HPLC using a chiral stationary phase). | ||
It turned out that the reaction performs best with ethyl ester-based MBH-carbonates 3 (compare products 5a–c). Structural changes on the nucleophile 2 were relatively well tolerated (see products 5d–m), although some reduced enantioselectivities were observed when accessing the halide- and CF3-substituted products 5h, 5i, 5j, 5l. On the other hand, variations on the acceptor 3 turned out to have a rather minor effect only (products 5n–w), thus resulting in an overall satisfyingly robust protocol to obtain these novel highly functionalized β-amino acid derivatives shown in Scheme 2 with reasonable yields and high diastereo- and enantioselectivities. The only real limitation that we observed so far was when we carried out the reaction with an α-i-propyl containing isoxazolidin-5-one 2, which did not result in any product formation at all (under the racemic as well as the asymmetric reaction conditions).
Concerning the absolute configuration of products 5 it must be admitted that we have not yet been able to obtain crystals of satisfying quality of the enantioenriched products. We only obtained good crystals of racemic 5f which also proved the (E)-double bond configuration (which is in accordance to NMR investigations).15
Finally, we also investigated the atmospheric pressure hydrogenation of compound 5a under heterogeneous conditions (using Pd/C, Scheme 3). Here we found that double bond hydrogenation to obtain compound 6 occurs easily, albeit with some erosion of d.r., followed by N–O cleavage to 7 when using a slightly larger amount of hydrogenation catalyst.
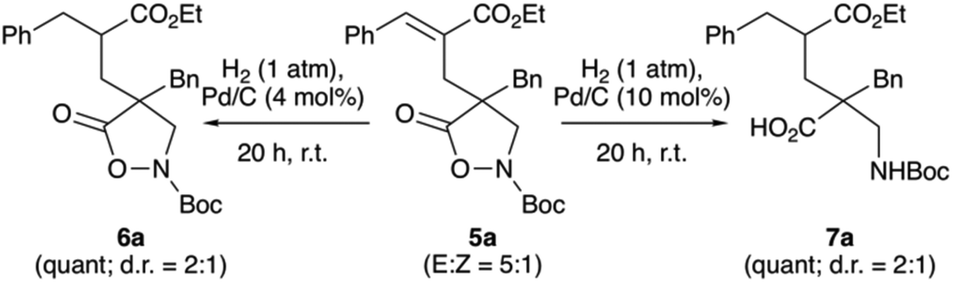 | ||
| Scheme 3 Heterogenous atmospheric pressure hydrogenation of compound 5a. | ||
Conclusions
We developed a novel metal-free α-allylation protocol to access highly functionalized β-amino acid derivatives 5 by carrying out the asymmetric phase-transfer catalysed addition of isoxazolidin-5-ones 2 to MBH carbonates 3. Very interestingly, a variety of commonly employed asymmetric PTCs did not give any noteworthy levels of enantioselectivities for this new transformation, while the reaction proceeds with satisfying enantio- and diastereoselectivities only when using the commercially available Maruoka type catalyst D2.Experimental details16
General procedure
The Baylis–Hillman carbonates 3a–m (1.5 eq., 0.15 mmol), benzyl-substituted isoxazolidin-5-ones 2a–k (1.0 eq., 0.10 mmol) and catalyst D2 (0.05 eq., 0.005 mmol, 5.4 mg) were charged to a Schlenk tube (under argon atmosphere), dissolved in 2.0 ml of isopropyl ether, and the mixture cooled to −20 °C. After 20 minutes, Cs2CO3 (3.0 eq., 0.30 mmol, 98 mg) was quickly added and the mixture was stirred at −20 °C for 96 h (1000 rpm). Then, the mixture was filtered over a pad of Na2SO4, washed with DCM, the solvent evaporated and the residue dried in vacuo. The resulting crude product was purified by chromatography (silica gel, heptane–ethyl acetate, 15/1 to 10/1) to afford enantioenriched products 5a–w.Analytical data for 5a
[α]25D = 43.9° (c = 0.41, CHCl3). 1H NMR (300 MHz, CDCl3, 298 K) δ 7.83 (s, 1H), 7.30–7.06 (m, 8H), 7.04–6.94 (m, 2H), 4.21 (q, J = 7.1 Hz, 2H), 3.82–3.70 (m, 2H), 3.06 (s, 2H), 2.96 (d, J = 13.9 Hz, 1H), 2.58 (d, J = 13.9 Hz, 1H), 1.40 (s, 9H), 1.28 (t, J = 7.1 Hz, 3H) ppm; 13C NMR (75 MHz, CDCl3, 298 K) δ 175.5, 167.7, 154.9, 143.5, 134.9, 134.9, 130.4, 128.9, 128.8, 128.7, 128.7, 128.0, 127.4, 83.6, 61.4, 54.4, 49.8, 40.8, 31.7, 28.1, 14.1 ppm; IR (film): ν (cm−1) 2977, 2938, 1794, 1712, 1454, 1369, 1254, 1202, 1146, 1095, 1022, 848, 754, 701; HRMS (MALDI-FT ICR): m/z calcd for C27H31NNaO6 [M + Na]+ = 488.20456, found: 488.20359; The enantioselectivity was determined by HPLC (Chiralpak AD-H column, n-hexane:i-PrOH = 95:5, 0.5 mL min−1, 10 °C, tmajor = 46.8 min, tminor = 61.4 min).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by the Austrian Science Funds (FWF): Project No. P30237 (financed by the Austrian National Foundation for Research, Technology and Development and Research Department of the State of Upper Austria). The NMR spectrometers used in Linz were acquired in collaboration with the University of South Bohemia (CZ) with financial support from the European Union through the EFRE INTERREG IV ETC-AT-CZ program (project M00146, “RERI-uasb”). This work was also supported by the Ministero dell'Università e della Ricerca (MIUR) and the University of Salerno (Italy). V. Capaccio gratefully acknowledges financial support from Erasmus+ for traineeships program.Notes and references
- For selected reviews: (a) T. Hashimoto and K. Maruoka, Chem. Rev., 2007, 107, 5656 CrossRef CAS PubMed; (b) T. Ooi and K. Maruoka, Angew. Chem., Int. Ed., 2007, 46, 4222 CrossRef CAS PubMed; (c) S.-s. Jew and H.-g. Park, Chem. Commun., 2009, 7090 RSC; (d) S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312 CrossRef CAS PubMed; (e) J. Novacek and M. Waser, Eur. J. Org. Chem., 2013, 637 CrossRef CAS; (f) R. Herchl and M. Waser, Tetrahedron, 2014, 70, 1935 CrossRef CAS; (g) S. Kaneko, Y. Kumatabara and S. Shirakawa, Org. Biomol. Chem., 2016, 14, 5367 RSC; (h) J. Tan and N. Yasuda, Org. Process Res. Dev., 2016, 20, 129 CrossRef; (i) L. Zong and C.-H. Tan, Acc. Chem. Res., 2017, 50, 842 CrossRef CAS PubMed; (j) J. Schörgenhumer, M. Tiffner and M. Waser, Beilstein J. Org. Chem., 2017, 13, 1753 CrossRef PubMed.
- For reviews on asymmetric phase-transfer catalysis in the synthesis of chiral amino acids: (a) K. Maruoka and T. Ooi, Chem. Rev., 2003, 103, 3013 CrossRef CAS PubMed; (b) M. J. O'Donnell, Acc. Chem. Res., 2004, 37, 506 CrossRef PubMed; (c) B. Lygo and B. I. Andrews, Acc. Chem. Res., 2004, 37, 518 CrossRef CAS PubMed.
- For selected very recent examples: (a) X. Huo, J. Fu, X. He, J. Chen, F. Xie and W. Zhang, Chem. Commun., 2018, 54, 599 RSC; (b) N. A. De Simone, R. Schettini, C. Talotta, C. Gaeta, I. Izzo, G. Della Sala and P. Neri, Eur. J. Org. Chem., 2017, 5649 CrossRef CAS; (c) J. Miguelez, H. Miyamura and S. Kobayashi, Adv. Synth. Catal., 2017, 359, 2897 CrossRef CAS; (d) U. Filp, A. L. Pees, C. Taddei, A. Pekosak, A. D. Gee, A. D. Windhorst and A. J. Poot, Eur. J. Org. Chem., 2017, 5154 CrossRef CAS; (e) S. Wen, X. Li and Y. Lu, Asian J. Org. Chem., 2016, 5, 1457 CrossRef CAS.
- For contributions by our group: (a) M. Waser, K. Gratzer, R. Herchl and N. Müller, Org. Biomol. Chem., 2012, 10, 251 RSC; (b) K. Gratzer and M. Waser, Synthesis, 2012, 44, 3661 CrossRef CAS PubMed.
- (a) T. Cadart, C. Berthonneau, V. Levacher, S. Perrio and J.-F. Briere, Chem. – Eur. J., 2016, 22, 15261 CrossRef CAS PubMed; (b) T. Cadart, V. Levacher, S. Perrio and J.-F. Briere, Adv. Synth. Catal., 2018, 360, 1499 CrossRef CAS.
- (a) J.-S. Yu, H. Noda and M. Shibasaki, Angew. Chem., Int. Ed., 2018, 57, 818 CrossRef CAS PubMed; (b) M. N. de Oliveira, S. Arseniyadis and J. Cossy, Chem. – Eur. J., 2018, 24, 4810 CrossRef PubMed.
- During the preparation of this manuscript highly selective Mannich and Michael addition reactions of compounds 2 were reported: J.-S. Yu, H. Noda and M. Shibasaki, Chem. – Eur. J., 2018 DOI:10.1002/chem.201804346.
- (a) R. Rios, Catal. Sci. Technol., 2012, 2, 267 RSC; (b) T.-Y. Liu, M. Xie and Y.-C. Chen, Chem. Soc. Rev., 2012, 41, 4101 RSC; (c) H. Pellissier, Tetrahedron, 2017, 73, 2831 CrossRef CAS; (d) T. N. Reddy and V. J. Rao, Tetrahedron Lett., 2018, 59, 2859 CrossRef.
- P. V. Ramachandran, S. Madhi, L. Bland-Berry, M. V. R. Reddy and M. J. O'Donnell, J. Am. Chem. Soc., 2005, 127, 13450 CrossRef CAS PubMed.
- For metal-catalysed additions of 1 to 3: (a) C.-G. Chen, X.-L. Hou and L. Pu, Org. Lett., 2009, 11, 2073 CrossRef CAS PubMed; (b) Y. Yuan, B. Yu, X.-F. Bai, Z. Xu, Z.-J. Zheng, Y.-M. Cui, J. Cao and L.-W. Xu, Org. Lett., 2017, 19, 4896 CrossRef CAS PubMed.
- (a) F. Zhong, J. Luo, G.-Y. Chen, X. Dou and Y. Lu, J. Am. Chem. Soc., 2012, 134, 10222 CrossRef CAS PubMed; (b) G. Zhu, J. Yang, G. Bao, M. Zhang, J. Li, Y. Li, W. Sun, L. Hong and R. Wang, Chem. Commun., 2016, 52, 7882 RSC.
- For a very recent asymmetric PT-catalysed β-addition protocol of β-ketoesters to compounds 3 see: R. Takagi, E. Fujii and H. Kondo, J. Org. Chem., 2018, 83, 11191 CrossRef CAS PubMed.
- (a) J. Novacek and M. Waser, Eur. J. Org. Chem., 2014, 802 CrossRef CAS PubMed; (b) M. Tiffner, J. Novacek, A. Busillo, K. Gratzer, A. Massa and M. Waser, RSC Adv., 2015, 5, 78941 RSC; (c) J. Novacek, J. A. Izzo, M. J. Vetticatt and M. Waser, Chem. – Eur. J., 2016, 22, 17339 CrossRef CAS PubMed.
- (a) T. Ooi, M. Kameda and K. Maruoka, J. Am. Chem. Soc., 2003, 125, 5139 CrossRef CAS PubMed; (b) R. He, S. Shirakawa and K. Maruoka, J. Am. Chem. Soc., 2009, 131, 16620 CrossRef CAS PubMed.
- CCDC 1870182† contains the supplementary crystallographic data for 5f.
- Full experimental and analytical details can be found in the online ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures, analytical details of all compounds and copies of NMR spectra and HPLC chromatograms. CCDC 1870182. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qo01057a |
| This journal is © the Partner Organisations 2018 |