
Visible-light-mediated allylation of alkyl radicals with allylic sulfones via a deaminative strategy†
Miao-Miao
Zhang
a and
Feng
Liu
 *ab
*ab
aJiangsu Key Laboratory of Neuropsychiatric Diseases and Department of Medicinal Chemistry, College of Pharmaceutical Sciences, Soochow University, 199 Ren-Ai Road, Suzhou, Jiangsu 215123, People's Republic of China. E-mail: fliu2@suda.edu.cn
bKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, People's Republic of China
First published on 19th October 2018
Abstract
A visible-light-mediated radical allylation of primary amine-derived Katritzky salts with allylic sulfones via C(sp3)–N bond activation is reported, furnishing the corresponding alkenes in satisfactory yields. This mild protocol provides an efficient strategy for site-specific C(sp3)–C(sp3) bond formation using abundant feedstock primary amines, particularly α-amino acids as alkyl radical precursors. This photocatalytic reaction demonstrates a broad substrate scope and good functional group tolerance.
Introduction
The development of synthetic methods for the generation of alkyl radicals from naturally abundant and cheap feedstock-derived radical precursors is of considerable interest for chemists.1 Notably, a decarboxylative strategy using readily available carboxylic acids for radical alkylation,2 especially with redox-active esters,3,4 has received significant attention, as well as the use of amino acid-derived redox-active esters as precursors of α-aminoalkyl radicals.5 Like carboxylic groups prevalent in molecules, the presence of amino (NH2) groups in a variety of molecules, from simple building blocks to natural products and medicinally relevant compounds,6 makes them a perfect synthetic handle for further transformations or late-stage modifications. However, the generation of alkyl radicals via C(sp3)–N bond activation still remains under-explored though the conversion of C(sp2)–NH2 into radical precursors has been well established via the formation of diazonium salts.7 Fortunately, bench-stable Katritzky pyridinium salts which are readily obtained from the condensation of primary amines with 2,4,6-triphenylpyrylium in one step8 can act as alkyl radical precursors.9 Of these few reports, Watson and co-workers described a nickel-catalyzed cross-coupling between redox-activated Katritzky salts and boronic acids via a C–N bond activation process.9a,b As a photoredox catalysis example, the Glorius group developed a visible-light-mediated protocol for the deaminative generation of alkyl radicals via single-electron-transfer (SET) reduction of redox-active Katritzky salts (E1/2 = −0.93 V vs. SCE in DMF10), followed by a Minisci-type reaction to give alkylated heterocycles (Scheme 1).9c Despite these reports, the use of a deaminative strategy, especially the C(sp3)–N bond activation in the design and development of new radical reactions is still desirable.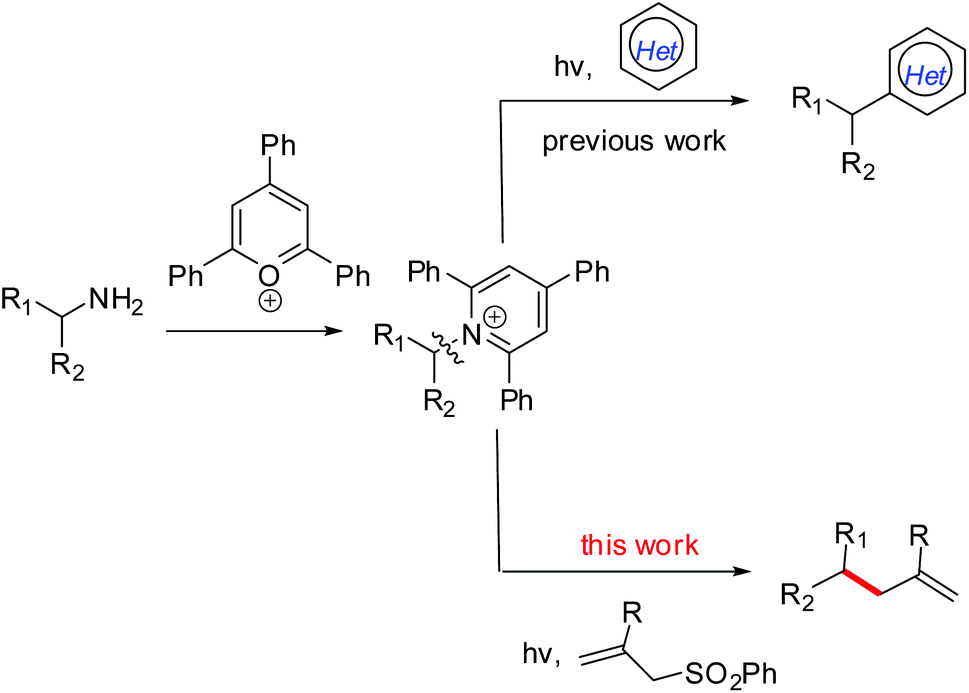 | ||
| Scheme 1 Visible-light-mediated C–C bond formation via the deaminative strategy. | ||
As one of the C–C bond formation methods, radical allylation usually involves an addition of a carbon-centered radical to a suitable allylic reagent and then a fragmentation to rebuild a double bond.11 So far, a series of alkyl radical precursors, such as alkyl halides,12 boronates,13 carboxylic acids,14 redox-active esters,15 and N-alkoxy-phthalimides,16 have been used in the radical allylation reactions, in which allyl sulfones often served as efficient radical acceptors and allylation partners. To expand the utility of primary amine-derived Katritzky salts in radical reactions, we herein report a visible-light-mediated protocol for the allylation of alkyl radicals generated by a reductive deamination process, providing a general and efficient strategy for site-specific C–C bond formation (Scheme 1).
Results and discussion
Initially, an N-Boc-aminopiperidine-derived Katritzky salt (1a) and alkyl sulfone (2a) were chosen as the model substrates for the optimization of the reaction parameters (Table 1). The tertiary amine Hünig's base (i-Pr2NEt, DIPEA) was used as the superstoichiometric reductant and Ir[(ppy)2(dtbbpy)]PF6 was employed as the photocatalyst. Upon irradiation with household blue LEDs (22 W) at ambient temperature for 3 h, we observed the formation of the desired alkene 3a in 74% yield (entry 1). We next examined the influence of the amount of DIPEA and the results showed that 5 or 10 equiv. of the reductant turned out to be inferior (entries 2 and 3). Meanwhile, other reductant triethylamine (entry 4) and other commonly used photocatalysts Ir(ppy)3 and Ru(bpy)2Cl2 (entries 5 and 6) were also tested, but they delivered product 3a in lower yields. A brief screening of organic solvents (entries 7–16) showed that a co-solvent system DCE/DMA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was the best one in terms of chemical yield (entry 13). Control experiments demonstrated that the photocatalyst and visible light were both essential for the success of the reaction (entries 17 and 18).
:1) was the best one in terms of chemical yield (entry 13). Control experiments demonstrated that the photocatalyst and visible light were both essential for the success of the reaction (entries 17 and 18).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Photocatalyst | t (h) | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.4 mmol), photocatalyst (2 mol%), DIPEA (1.6 mmol), solvent (2 mL), room temperature, irradiation with household blue LEDs (22 W). b Isolated yield. c DIPEA (10 equiv.). d DIPEA (5 equiv.). e Et3N (8 equiv.). f In the dark. | ||||
| 1 | MeCN | Ir[(ppy)2(dtbbpy)]PF6 | 3 | 74 |
| 2c | MeCN | Ir[(ppy)2(dtbbpy)]PF6 | 3 | 69 |
| 3d | MeCN | Ir[(ppy)2(dtbbpy)]PF6 | 5 | 66 |
| 4e | MeCN | Ir[(ppy)2(dtbbpy)]PF6 | 16 | 60 |
| 5 | MeCN | Ir(ppy)3 | 12 | 16 |
| 6 | MeCN | Ru(bpy)2Cl2 | 3 | 69 |
| 7 | DMA | Ir[(ppy)2(dtbbpy)]PF6 | 3 | 67 |
| 8 | DMSO | Ir[(ppy)2(dtbbpy)]PF6 | 6 | 25 |
| 9 | DCM | Ir[(ppy)2(dtbbpy)]PF6 | 3 | 71 |
| 10 | DCE | Ir[(ppy)2(dtbbpy)]PF6 | 3 | 84 |
| 11 | PhCl | Ir[(ppy)2(dtbbpy)]PF6 | 12 | 73 |
| 12 | DCM/DMA (1:1) |
Ir[(ppy)2(dtbbpy)]PF6 | 3 | 66 |
| 13 |
DCE/DMA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) :1)
|
Ir[(ppy) 2 (dtbbpy)]PF 6 | 3 | 93 |
| 14 | PhCl/DMA (1:1) |
Ir[(ppy)2(dtbbpy)]PF6 | 3 | 81 |
| 15 | DCE/DMA (2:1) |
Ir[(ppy)2(dtbbpy)]PF6 | 3 | 87 |
| 16 | DCE/DMA (1:2) |
Ir[(ppy)2(dtbbpy)]PF6 | 3 | 89 |
| 17 | DCE/DMA (1:1) |
— | 12 | 0 |
| 18f | DCE/DMA (1:1) |
Ir[(ppy)2(dtbbpy)]PF6 | 12 | 0 |
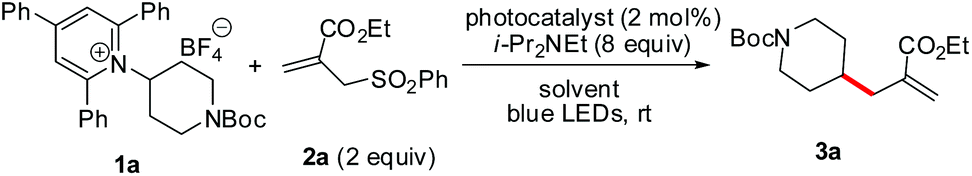
With the optimized conditions in hand, we next evaluated the scope of primary amine-derived Katritzky salts in this radical allylation reaction. As illustrated in Scheme 2, a variety of cyclic and acyclic alkyl radicals generated from Katritzky pyridinium salts were allylated in good to excellent yields (3a–n). Considering the fact that amino acids are naturally abundant feedstocks, the use of amino acids as radical precursors would be practical.5 Although these conditions were optimized for common alkyl primary amine-derived Katritzky salts, they can also be applied to the amino acid-derived Katritzky salts (3e–n). The accommodated substrates derived from natural amino acids include phenylalanine (3e), tyrosine (3g), alanine (3i), leucine (3j), isoleucine (3k), methionine (3l), aspartic acid (3m), and glutamic acid (3n). The mild nature of this protocol allows it to tolerate a wide range of functional groups, including carbamate, ester, ether, and sulfide moieties. It should be mentioned that the reaction of a benzylic pyridinium salt could also proceed smoothly to give the desired product (3d). As most of the radical reactions, the diastereoselectivity of this reaction is also poor (3c and 3k).
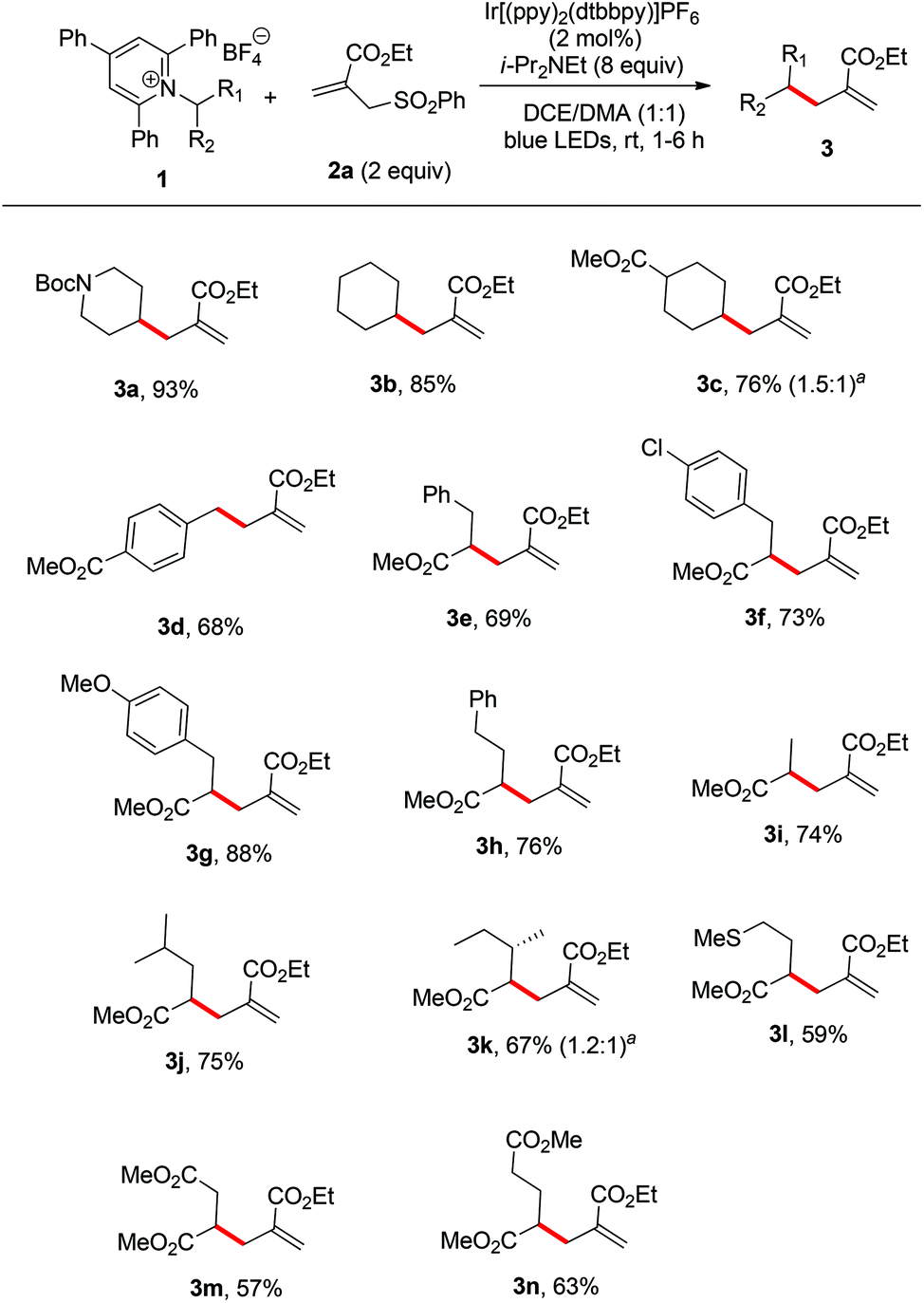 | ||
| Scheme 2 Substrate scope of Katritzky salts. Reaction conditions: 1 (0.2 mmol), 2a (0.4 mmol), Ir[(ppy)2(dtbbpy)]PF6 (2 mol%), DIPEA (1.6 mmol), DCE/DMA (1:1, 2 mL), room temperature, irradiation with blue LEDs (22 W), 1–6 h. aThe dr value was determined by 1H NMR. | ||
Furthermore, we turned our attention to defining the scope of allyl sulfones that are suitable for this radical reaction. As shown in Scheme 3, when using a glutamic acid-derived Katritzky salt (1n) as the radical precursor, a series of desired allylated products were furnished with satisfactory efficiency (4a–j). The allyl sulfones bearing methyl, ester, cyano, or aryl groups were all tolerated well under the reaction conditions though the electron-rich 2-methyl allylsulfone afforded product 4a in a lower yield compared to others. Notably, in the examples of 4f and 4g, the intact chloro and bromo atoms reserved the options for further cross-coupling. These results could substantially extend the scope of application of this radical allylation.
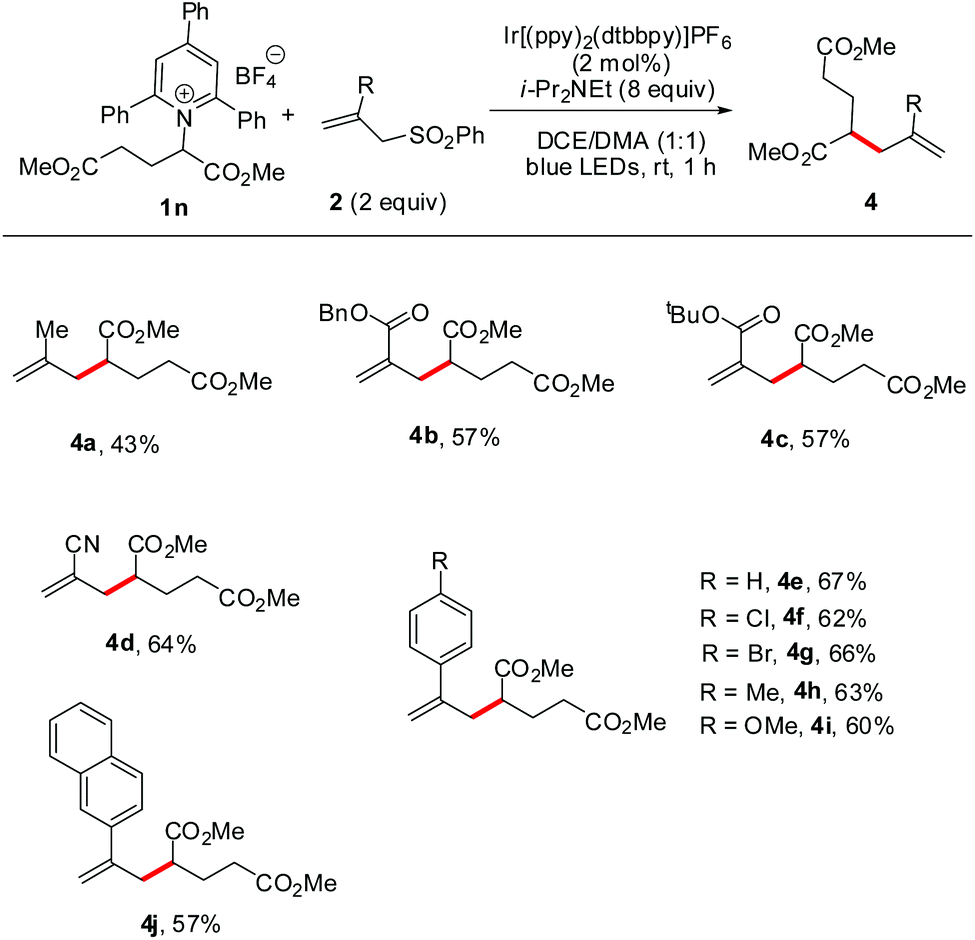 | ||
| Scheme 3 Substrate scope of allyl sulfones. Reaction conditions: 1n (0.2 mmol), 2 (0.4 mmol), Ir[(ppy)2(dtbbpy)]PF6 (2 mol%), DIPEA (1.6 mmol), DCE/DMA (1:1, 2 mL), room temperature, irradiation with blue LEDs (22 W), 1 h. | ||
To gain insights into the mechanism of this visible-light-mediated reaction, fluorescence quenching experiments were performed (Fig. S2†).17 As might be expected, the results showed that both primary amine-derived Katritzky salt 1a and i-Pr2NEt quenched the photoexcited Ir[(ppy)2(dtbbpy)]PF6 (*[Ir]3+)18 effectively but allyl sulfone 2a did not. Moreover, the addition of a radical scavenger TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl) led to the formation of 3a only in 21% yield under the standard reaction conditions, with a TEMPO-trapped product being detected by MS analysis (Fig. S1†).17 These results could support the generation of an alkyl radical intermediate via a single electron transfer (SET) process.
On the basis of the observations and previous reports,9c,12–16 a possible mechanism is depicted in Scheme 4. Firstly, redox-active Katritzky salt 1 accepts an electron from *[Ir]3+, yielding a dihydropyridine radical A and then a fragmentation driven by the reformation of an aromatic pyridine ring to give an alkyl radical B. Subsequently, the radical B adds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of allyl sulfone 2, followed by a desulfonation to give the desired product 3 or 4. Meanwhile, single-electron reduction of the [Ir]4+ intermediate by i-Pr2NEt regenerates the ground-state [Ir]3+ complex to close the photoredox cycle. On the other hand, i-Pr2NEt that serves as a stoichiometric reductant can also transfer an electron to *[Ir]3+ to afford [Ir]2+ species. This [Ir]2+ intermediate is highly reducing, enabling transfer of an electron to Katritzky salt 1. This event produces the radical B, as well as completing the photocatalytic cycle.
C bond of allyl sulfone 2, followed by a desulfonation to give the desired product 3 or 4. Meanwhile, single-electron reduction of the [Ir]4+ intermediate by i-Pr2NEt regenerates the ground-state [Ir]3+ complex to close the photoredox cycle. On the other hand, i-Pr2NEt that serves as a stoichiometric reductant can also transfer an electron to *[Ir]3+ to afford [Ir]2+ species. This [Ir]2+ intermediate is highly reducing, enabling transfer of an electron to Katritzky salt 1. This event produces the radical B, as well as completing the photocatalytic cycle.
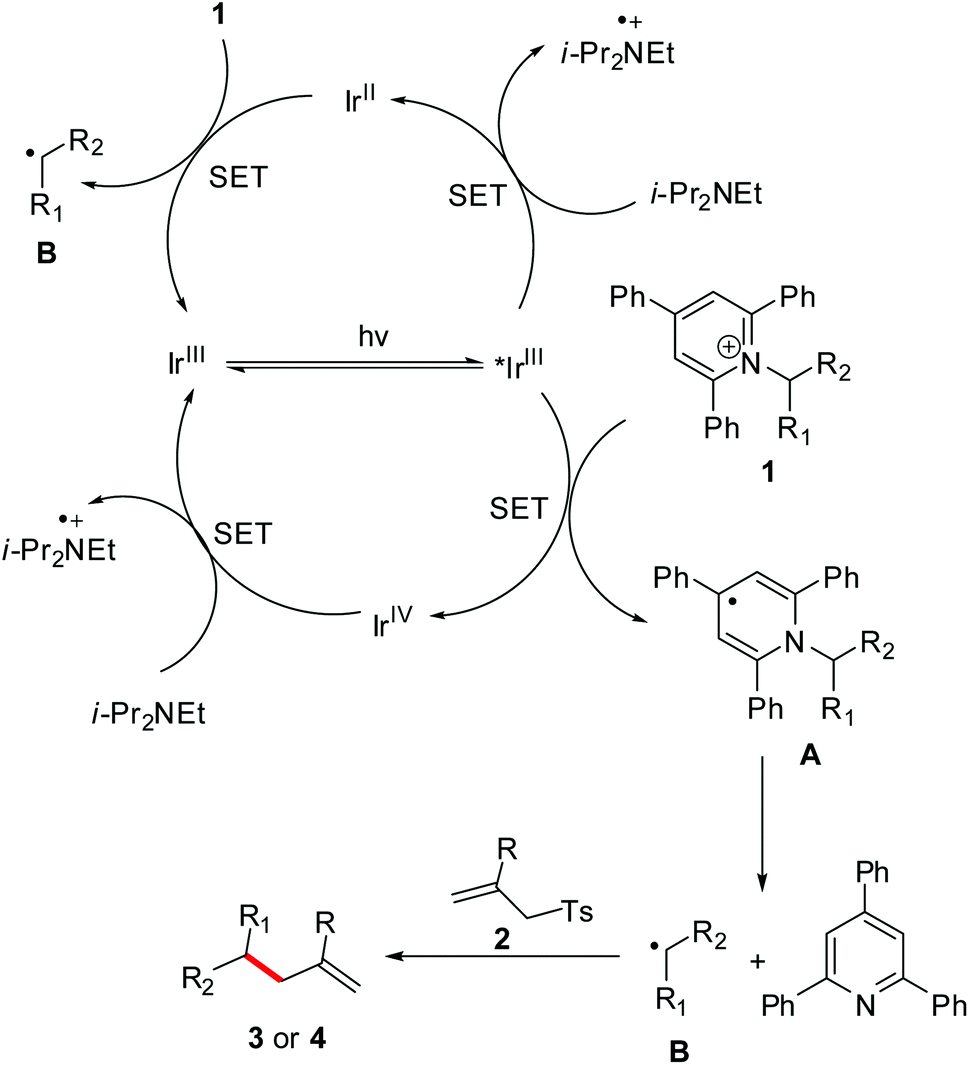 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In conclusion, we have developed a visible-light-mediated allylation reaction of Katritzky pyridinium salts with allyl sulfones via a deaminative strategy. With Ir[(ppy)2(dtbbpy)]PF6 as the photocatalyst and i-Pr2NEt as the reductant, this mild approach provides ready access to site-specific C(sp3)–C(sp3) bond formation using abundant feedstock primary amines, particularly α-amino acids as alkyl radical precursors. The generality of this protocol has been illustrated and a series of functional groups are well tolerated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (Grant No. 21302134) is acknowledged.References
- For selected reviews, see: (a) Y. Jin and H. Fu, Asian J. Org. Chem., 2017, 6, 368 CrossRef CAS; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef CAS PubMed; (c) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632 CrossRef CAS PubMed.
- For selected recent examples, see: (a) J. A. Kautzky, T. Wang, R. W. Evans and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 6522 CrossRef CAS PubMed; (b) X. Tan, Z. Liu, H. Shen, P. Zhang, Z. Zhang and C. Li, J. Am. Chem. Soc., 2017, 139, 12430 CrossRef CAS PubMed; (c) C. Liu, X. Wang, Z. Li, L. Cui and C. Li, J. Am. Chem. Soc., 2015, 137, 9820 CrossRef CAS PubMed; (d) F. Hu, X. Shao, D. Zhu, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2014, 53, 6105 CrossRef CAS PubMed; (e) F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401 CrossRef CAS PubMed.
- For selected recent examples, see: (a) C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson, A. K. Chatterjee, M. Yan and P. S. Baran, Science, 2017, 356, eaam7355 CrossRef PubMed; (b) J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, R. L. Malins, B. Vokits, S. A. Shaw, D.-H. Bao, F.-L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213 CrossRef CAS PubMed; (c) N. Suzuki, J. L. Hofstra, K. E. Poremba and S. E. Reisman, Org. Lett., 2017, 19, 2150 CrossRef CAS PubMed; (d) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801 CrossRef CAS PubMed; (e) F. Toriyama, J. Cornella, L. Wimmer, T.-G. Chen, D. D. Dixon, G. Creech and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 11132 CrossRef CAS PubMed.
- Photoredox catalysis reactions, see: (a) R. Mao, A. Frey, J. Balon and X. Hu, Nat. Catal., 2018, 1, 120 CrossRef; (b) W.-M. Cheng, R. Shang, M.-C. Fu and Y. Fu, Chem. – Eur. J., 2017, 23, 2537 CrossRef CAS PubMed; (c) D. Wang, N. Zhu, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 15632 CrossRef CAS PubMed.
- (a) R. S. Proctor, H. J. Davis and R. J. Phipps, Science, 2018, 360, 419 CrossRef CAS PubMed; (b) W.-M. Cheng, R. Shang and Y. Fu, ACS Catal., 2017, 7, 907 CrossRef CAS; (c) H. Zhang, P. Zhang, M. Jiang, H. Yang and H. Fu, Org. Lett., 2017, 19, 1016 CrossRef CAS PubMed; (d) Y. Jin, M. Jiang, H. Wang and H. Fu, Sci. Rep., 2016, 6, 20068 CrossRef PubMed.
- (a) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564 CrossRef CAS PubMed; (b) T. P. T. Cushnie, B. Cushnie and A. J. Lamb, Int. J. Antimicrob. Agents, 2014, 44, 377 CrossRef CAS PubMed; (c) N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348 CrossRef CAS.
- The generation of aryl radicals can be achieved via the reductive cleavage of C(sp2)–N bonds of the aryl diazonium salts. For a review, see: I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566 CrossRef CAS PubMed.
- A. R. Katritzky, U. Gruntz, D. H. Kenny, M. C. Rezende and H. Sheikh, J. Chem. Soc., Perkin Trans. 1, 1979, 430 RSC.
- (a) J. Liao, W. Guan, B. P. Boscoe, J. W. Tucker, J. W. Tomlin, M. R. Garnsey and M. P. Watson, Org. Lett., 2018, 20, 3030 CrossRef CAS PubMed; (b) C. H. Basch, J. Liao, J. Xu, J. J. Piane and M. P. Watson, J. Am. Chem. Soc., 2017, 139, 5313 CrossRef CAS PubMed; (c) F. J. R. Klauck, M. J. James and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 12336 CrossRef CAS PubMed.
- J. Grimshaw, S. Moore and J. T. Grimshaw, Acta Chem. Scand., Ser. B, 1983, 37, 485 CrossRef.
- For selected reviews on allylation, see: (a) J. D. Weaver, A. Recio III, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846 CrossRef CAS PubMed; (b) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed.
- B. A. Roe, C. G. Boojamra, J. L. Griggs and C. R. Bertozzi, J. Org. Chem., 1996, 61, 6442 CrossRef CAS PubMed.
- A.-P. Schaffner and P. Renaud, Angew. Chem., Int. Ed., 2003, 42, 2658 CrossRef CAS PubMed.
- L. Cui, H. Chen, C. Liu and C. Li, Org. Lett., 2016, 18, 2188 CrossRef CAS PubMed.
- C. Hu and Y. Chen, Org. Chem. Front., 2015, 2, 1352 RSC.
- (a) J. Zhang, Y. Li, R. Xu and Y. Chen, Angew. Chem., Int. Ed., 2017, 56, 12619 CrossRef CAS PubMed; (b) J. Zhang, Y. Li, F. Zhang, C. Hu and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 1872 CrossRef CAS PubMed.
- See the ESI.†.
- Regarding the redox potential of catalyst Ir[(ppy)2(dtbbpy)]PF6 (
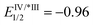 , EIV/III1/2 = +1.21,
, EIV/III1/2 = +1.21, 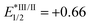 , EIII/II1/2 = −1.51), see: C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
, EIII/II1/2 = −1.51), see: C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
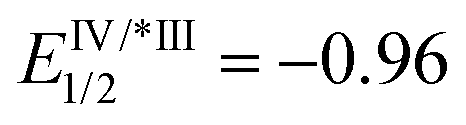
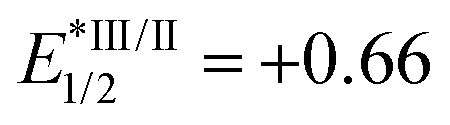
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, and copies of 1H, 13C and 19F NMR spectra for all compounds. See DOI: 10.1039/c8qo01046c |
| This journal is © the Partner Organisations 2018 |