
Ni-Catalyzed 1,2-iminoacylation of alkenes via a reductive strategy†
Chuan
Wang  *a
*a
*a
Abstract
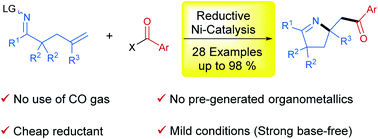
In this protocol, we developed a reductive strategy for 1,2-iminoacylation of alkenes. Under the catalysis of the Ni-biquinoline system, various oxime esters incorporating a pendant terminal olefinic unit were successfully reacted with acid chlorides or anhydrides as electrophilic acylating reagents in the presence of Zn as a reductant, furnishing a series of pyrrolines in moderate to excellent yields. This reaction is distinguished by safe and mild reaction conditions that avoid the use of CO gas as a carbonyl source, pregenerated organometallics and strong bases as reaction additives.
- This article is part of the themed collection: Organic Chemistry Frontiers HOT articles for 2018
 Please wait while we load your content...
Please wait while we load your content...

