
Combining JBCA and Marfey's methodology to determine the absolute configuration of threonines: the case of gunungamide A, a new cyclic depsipeptide containing chloropyrrole from the sponge Discodermia sp.†
Guillermo
Tarazona
 a,
Rogelio
Fernández
*a,
Patricia G.
Cruz
a,
Marta
Pérez
a,
Jaime
Rodríguez
*b,
Carlos
Jiménez
b and
Carmen
Cuevas
a
a,
Rogelio
Fernández
*a,
Patricia G.
Cruz
a,
Marta
Pérez
a,
Jaime
Rodríguez
*b,
Carlos
Jiménez
b and
Carmen
Cuevas
a
aNatural Products Department, PharmaMar S.A., Pol. Ind. La Mina Norte, Avda. de los Reyes 1, 28770 Colmenar Viejo (Madrid), Spain. E-mail: rfernandez@pharmamar.com
bDepartamento de Química. Facultade de Ciencias e Centro de Investigacións Científicas Avanzadas (CICA) Universidade da Coruña. A Coruña E-15071, Spain. E-mail: jaime.rodriguez@udc.es
First published on 29th October 2018
Abstract
A combination of configurational analysis based on coupling constants (JBCA) and Marfey's aminoacid derivatization allowed us to distinguish between the different possible diastereoisomers of threonines in cyclopeptides, when potentially some of the amino acids could be racemized/epimerized during the acid hydrolysis process. The strategy was applied to a new cyclic depsipeptide having an unusual chloropyrrol ring, gunungamide A (2), isolated from a marine sponge belonging to the genus Discodermia. Further validation of this approach was demonstrated with the peptide stellatolide (1), where epimerization of a threonine residue during hydrolysis in the Marfey's analysis resulted in a mistaken configuration. This approach can be extended to elucidate the absolute stereochemistry of peptides bearing amino acids with two or more contiguous chiral centers.
Introduction
Structural elucidation of new molecular scaffolds isolated from natural sources can become a very challenging exercise. Often the combination of NMR spectroscopy and mass spectrometry analysis is enough to solve the structure of such new important entities.1 However, this task can become more difficult in frameworks bearing contiguous non-protonated carbons or when the new metabolites contain several chiral centers.2 In the case of new natural peptides, which often contain a large number of stereocenters in their structures, the absolute configuration of the amino acids is solved, in most cases, by performing an acid hydrolysis followed by comparison with standards using Marfey's method3,4 and to a lesser extent by using chiral-phase chromatography.5 Although these methods are highly effective, there are exceptions when additional methodologies need to be used: (a) The presence of non-commercially available amino acids, requiring time and synthetic efforts to prepare them.6 (b) The existence of identical amino acids in the peptide skeleton, making the elucidation of the right location a difficult task.7 (c) The occurrence of amino acids with more than one chiral center which complicates the chromatographic separation of the different diastereoisomers and often requires specific and expensive columns.8 (d) Thiazole containing peptides that require an initial ozonolysis treatment before the hydrolysis.9 However, the main drawback of Marfey's methodology is the risk of partial10 or complete11 α-racemization/epimerization of the amino acids that occurs in some cases due to the strongly acidic hydrolysis conditions used, which results in irresolvable mixtures and mistaken configurations.

Although J-based configurational analysis (JBCA) has been used to establish the relative configuration of amino acids bearing two or more contiguous stereocenters,13 a combination of Marfey's method and JBCA would avoid some of the previously discussed problems. Discarding some of the possible diastereoisomers of the challenging amino acids by studying the homo and heteronuclear coupling constants prior to hydrolysis, may provide an effective and non-destructive strategy to simplify the comparison of retention times of the HPLC-MS chromatograms using Marfey's method thereby minimizing such problems.
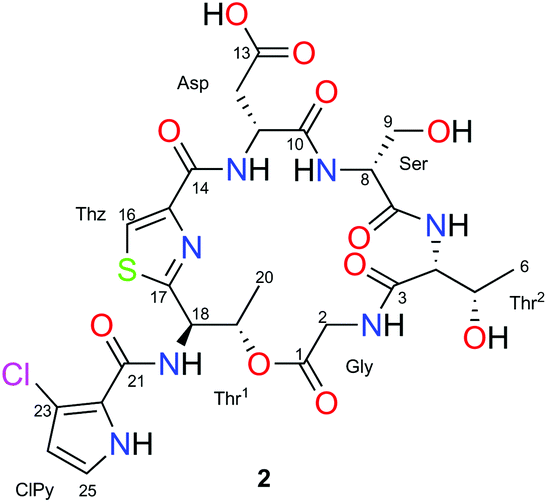
As part of our on-going search for new antitumor natural compounds,14 the bioassay-guided fractionation of an organic extract with cytotoxic activity from the marine sponge Discodermia sp., led to isolation of a new halogenated cyclic depsipeptide, named as gunungamide A (2), along with the known compound aurantoside E.15
Results and discussion
Sponges of this genus have been described as a rich source of peptide derivatives such as calyxamides16 and cyclolithistides.17,18 The most relevant is the phosphatase inhibitor calyculin A,19,20 which displays higher inhibitory activity than okadaic acid21 and discodermolide.22A sample of Discodermia sp. was collected at a depth of 100 m at Pulau Gunung in Indonesia. The methanol extract showed strong cytotoxic activity in a panel of four human cancer cell lines. Bioassay-guided fractionation of this extract by reverse-phase HPLC afforded 14.0 mg of compound 2 and 23.9 mg of aurantoside E.
The (+)-LRESI mass spectrum of 2 showed two protonated ion [M + H]+ peaks at m/z 672 and 674 in the ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 respectively, indicating the presence of a chlorine atom in the structure. The (+)-HRESI-TOFMS analysis of 2 provided the [M + H]+ ion peak at m/z 672.1518, which was in agreement with the molecular formula of C25H3035ClN7O11S (calcd for C25H3135ClN7O11S m/z 672.1485).
:1 respectively, indicating the presence of a chlorine atom in the structure. The (+)-HRESI-TOFMS analysis of 2 provided the [M + H]+ ion peak at m/z 672.1518, which was in agreement with the molecular formula of C25H3035ClN7O11S (calcd for C25H3135ClN7O11S m/z 672.1485).
The 1H and 13C NMR spectra along with the edited HSQC NMR experiment of 2 in CD3OD (Table 1) detected eight carbonyl carbon resonances (δC 175.0–161.7) and several α-aminoacid proton signals (δH 3.63–4.83) that indicated the peptide nature of compound 2. This was confirmed by the presence of five amide NH protons (δH 7.43–8.77) observed in its 1H NMR spectrum in DMSO-d6. Analysis of 2D NMR experiments of 2, including TOCSY, COSY and HMBC, allowed us to detect the presence of seven spin systems. Five of them were easily assigned to standard amino acids: aspartic acid (Asp: δH11 4.83 and δH12 3.05), glycine (Gly: δH2 4.25/3.63), serine (Ser: δH8 4.07 and δH9 3.91) and two threonines (Thr1: δH18 5.77, δH19 6.02 and δH20 1.42; Thr2: δH4 4.25, δH5 4.44 and δH6 1.20). The proton singlet (H16) at δH 8.19/δC 126.3, showing a characteristic 1JH–C value of 190 Hz (measured in a HSQC-HECADE experiment), along with its HMBC correlations to the non-protonated carbons at δC 162.7, 150.0 and 171.1, which allowed us to detect the presence of a cyclized Cys residue in the form of a thiazole moiety.
| Pos | δ C, multa | δ H, mult (J in Hz)a | δ C, multb | δ H, mult (J in Hz)b | |
|---|---|---|---|---|---|
| a In CD3OD. b In DMSO-d6. | |||||
| Gly | 1 | 170.5 C | 168.7, C | ||
| 2 | 41.9 CH2 | 4.25 d (18,0) | 40.4, CH2 | 4.11 dd (17.9, 7.6) | |
| 3.63 d (18.0) | 3.49 dd (17.9, 4.3) | ||||
| NH | 7.60 dd (7.6, 4.3) | ||||
| Thr2 | 3 | 172.9 C | 170.2, C | ||
| 4 | 60.6 CH | 4.25 d (2.7) | 58.5, CH | 4.03 dd (8.3, 2.9) | |
| 5 | 67.3 CH | 4.44 dd (6.3, 2.5) | 65.0, CH | 4.21 m | |
| 6 | 20.4 CH3 | 1.20 d (6.3) | 20.3, CH3 | 1.04 d (6.4) | |
| NH | 7.43 d (8.2) | ||||
| Ser | 7 | 173.4 C | 170.3, C | ||
| 8 | 59.5 CH | 4.07 dd (4.7, 4.7) | 58.1, CH | 3.92 m | |
| 9 | 61.8 CH2 | 3.91 dd (7.2, 4.7) | 60.4, CH2 | 3.73 dd (11.0, 4.7) | |
| 3.66 dd (11.0, 5.9) | |||||
| NH | 8.77 d (5.0) | ||||
| Asp | 10 | 175.0 C | 172.0, C | ||
| 11 | 51.1 CH | 4.83 m | 49.4, CH | 4.59 ddd (9.0, 7.8, 2.6) | |
| 12 | 36.6 CH2 | 3.05 d (5.5) | 35.3, CH2 | 2.96 dd (16.1, 2.6) | |
| 2.83 dd (16.1, 9.0) | |||||
| 13 | 173.8 C | 172.4, C | |||
| NH | 8.21 d (7.8) | ||||
| Thz | 14 | 162.7 C | 159.9, C | ||
| 15 | 150.0 C | 148.5, C | |||
| 16 | 126.3 CH | 8.19 s | 125.0, CH | 8.23 s | |
| 17 | 171.1 C | 168.5, C | |||
| Thr1 | 18 | 55.0 CH | 5.77 d (1.8) | 53.0, CH | 5.75 m |
| 19 | 72.6 CH | 6.02 dd (6.6, 1.8) | 70.4, CH | 5.78 m | |
| 20 | 17.4 CH3 | 1.42 d (6.6) | 16.8, CH3 | 1.31 d (6.4) | |
| NH | 7.90 d (9.1) | ||||
| ClPy | 21 | 161.7 C | 159.3, C | ||
| 22 | 121.2 C | 120.0, C | |||
| 23 | 115.5 C | 113.3, C | |||
| 24 | 111.6 CH | 6.26 d (2.8) | 110.2, CH | 6.28 dd (2.8, 2.9) | |
| 25 | 123.2 CH | 6.98 d (2,8) | 122.0, CH | 7.02 dd (2.9, 3.0) | |
| NH | 12.05 dd (3.0, 2.8) | ||||
The remaining spin system observed in CD3OD corresponds to two doublets at δH 6.26 (H24) and δH 6.98 (H25) which display a small coupling constant value of 2.8 Hz. Analysis of COSY and H2BC23 experiments of 2 allowed us to connect them to their corresponding carbons at δC24 111.6 and δC25 123.2, respectively and to assign them as two adjacent methine carbons. When the NMR spectra of 2 was recorded in DMSO-d6, this spin system was expanded to include an additional proton signal at δH 12.05 which was assigned to an NH group. All these data along with the HMBC correlations among the methine protons at δH 6.26 (H24) and 6.98 (H25) and the non-protonated carbons at δC 121.2 (C22) and 115.5 (C23) suggested the presence of a disubstituted pyrrol moiety. The cluster ion at m/z 127.9899/129.9869 in a 3:1 ratio (Fig. 1), observed in its (+)-HRESI-TOFMS, confirmed the presence of the chloropyrrol unit in 2. The interconnection between the chloropyrrol moiety to the amino group of the Thr1 residue through the carbonyl amide C21 was established by long range 2D NMR experiments. The HMBC correlation between the α-aminoacid proton signal δH18 5.77 and the amide carbon at δC21 161.7 allowed us to connect the Thr1 residue to the amide carbon C21. However, the presence of three consecutive non protonated carbons in the chloropyrrol moiety prevented us from detecting any key long range H–C correlations in the NMR experiments. Nevertheless, this connection was possible by a LR-HSQMBC24 experiment of 2 which displayed a four bond H–C long range correlation between methine protons H24 and H25 and the amide carbon C21 (Fig. 2, dashed arrows).
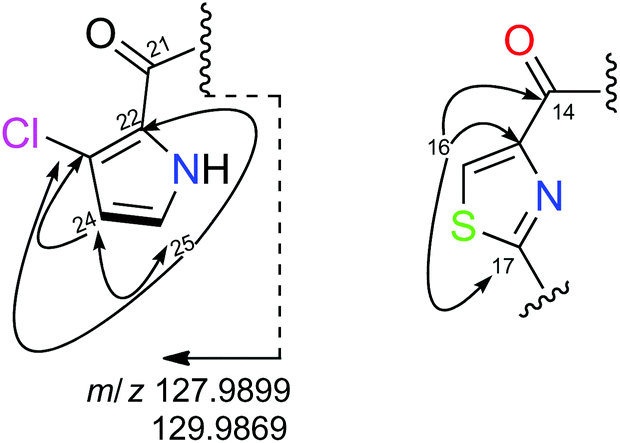 | ||
| Fig. 1 Selected H2BC (bold), HMBC (plain) correlation and MS ion fragment found for 2. | ||
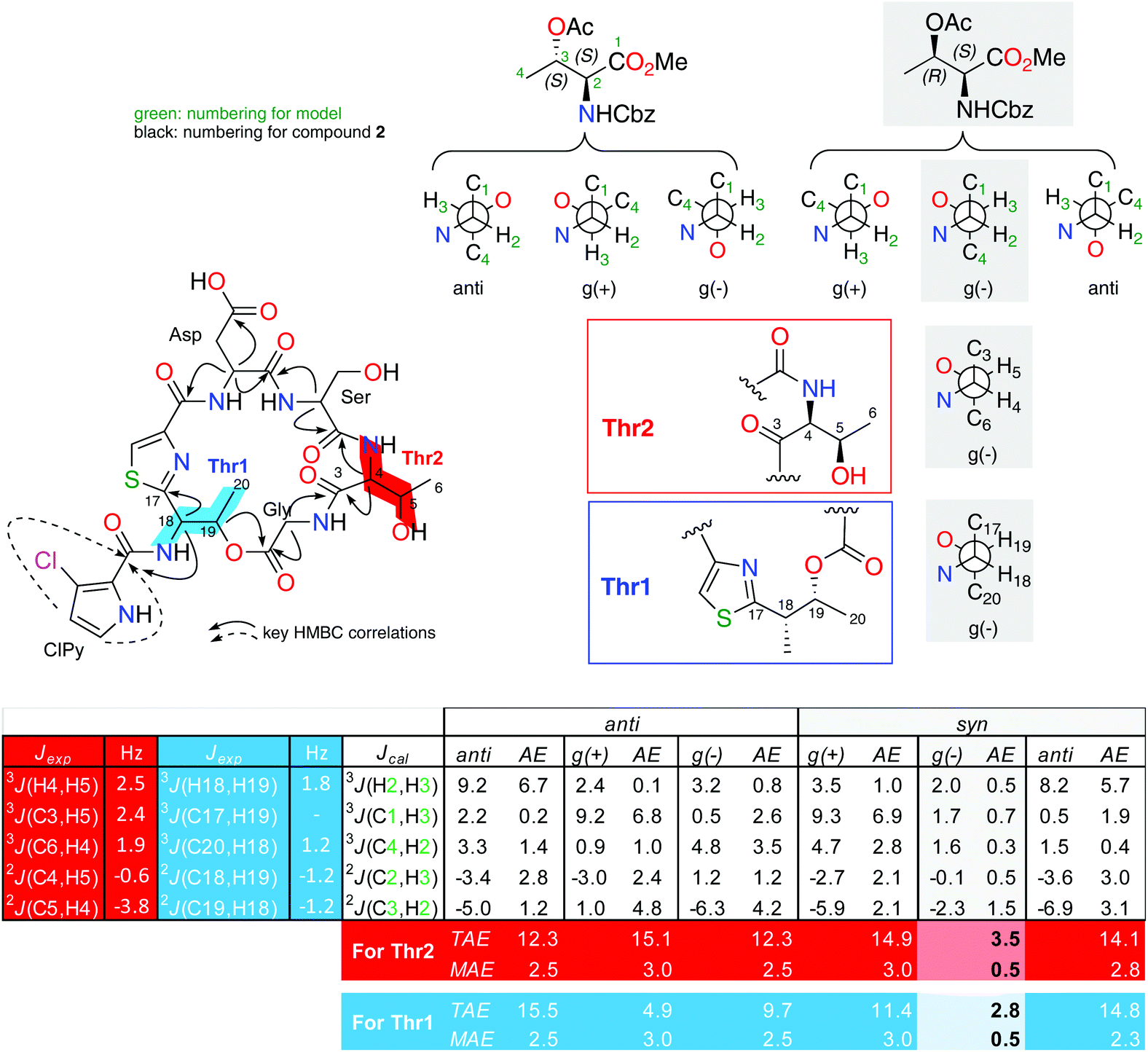 | ||
| Fig. 2 Extended J-based configuration analysis of threonines in 2. B3LYP/6-311G (d) DFT calculations of the coupling constants for the possible staggered conformers of both anti and syn configurations of the L-allo-Thr-NCBz and L-Thr-NCBz methyl esters. AE: absolute error (|Jcal − Jexp|), TAE: total absolute error (∑|Jcal − Jexp|), MAE: mean absolute error (∑|Jcal − Jexp|/n; n = number of coupling constants compared). The AE values showed in this table are for the comparison for the fragment C3–C6 (in red). For fragment C17–C20 (in blue) see ESI.† | ||
The position of the chlorine atom in the chloropyrrol moiety in 2 was proposed by 13C NMR data, which remarkably showed a broad signal at 115.5 ppm, due to the 35Cl/37Cl isotopic effect.25 The location of this atom was confirmed by comparison of its carbon and proton chemical shifts to those of similar synthetic compounds reported in the literature.26,27
Final assemble of the amino acids was achieved from standard HMBC correlations between the α-amino protons and the adjacent carbonyl groups and by HMBC correlations between the NH and α-amino protons in DMSO. All these correlations (see page S-17 in ESI†), allowed us to establish the planar structure of 2. The fragments Thz-Asp-Ser-Thr2-Gly-Thr1 (m/z 527.1547), Thz-ClPy-Thr1-Gly-Thr2 (m/z 468.0731, 470.0715), Thr1-ClPy-Thz-Asp (m/z 427.0485, 429.0449), Thr1-ClPy-Thz (m/z 311.0365, 313.0342), Thz-Thr1 (m/z 184.0529) and ClPy (m/z 127.9899, 129.9869) detected in the (+)-HRESI-TOFMS at 300 eV of 2 were in agreement with the proposed amino acid sequence (see ESI†).
At first glance, Marfey's analysis with the 1-fluoro-2-4-dinitrophenyl-5-L-alanine amide (FDAA) was applied to determine the absolute configuration of 2. An initial ozonolysis step was required to destroy the aromaticity of the thiazole in order to facilitate hydrolysis. HPLC-MS comparison of the retention times with standards allowed us to easily deduce the presence of D-Ser with a tR 6.96 min for the D-Ser-FDAA (D: 6.97 and L: 6.75 min) and D-Asp-FDAA with a tR 12.45 min (D: 12.46 and L: 11.84 min).
Taking into account that four diastereoisomers are possible for each of the two threonine residues present in 2, a more detailed analysis of the standards was performed. Although stereoisomers D and D-allo-Thr are easily distinguishable (retention times (tR) of 21.53 min for D-Thr-FDAA and 18.89 min for D-allo-Thr-FDAA) by HPLC-MS analysis, in contrast L and L-allo-Thr show almost identical retention times (tR of 16.78 min for L-Thr-FDAA and tR of 16.97 min for L-allo-Thr-FDAA).
When the analysis was repeated using UPLC conditions, the tR of L and L-allo-Thr-FDAA at 12.39 min and 13.52 min, respectively, provided better differentiation between them.
Due to the difficulties found with separation of L-Thr and L-allo-Thr in 2, along with the possible epimerization processes already observed by Martín et al. in peptides,11 we proposed that an initial J-based configurational analysis (JBCA) could be very useful in discarding some of the four possible stereoisomers. Thus, we determined the relative configuration of both Thrs present in 2 by measuring the homonuclear 1H–1H coupling constant values and the heteronuclear coupling constants using HSQC-HECADE, J-HMBC and NMR experiments.
The small values between adjacent methine protons at the two chiral centers in Thr1 and Thr2, 3JH18,H19 (1.8 Hz) and 3JH4,H5(2.5 Hz), indicated a synperiplanar disposition between pairs H18/H19 and H4/H5, respectively. In the case of Thr1, the small values of 2JC19,H18 (−1.2 Hz), 2JC18,H19 (−1.2 Hz) and 3JC20,H18 (1.2 Hz) secure the relative stereochemistry as 18S*/19R*. On the other hand, the small values of 2JC5,H4 (−3.8 Hz), 2JC4,H5 (−0.6 Hz), 3JC6,H4 (1.9 Hz) and 3JC3,H5 (2.4 Hz) measured in Thr2, suggested that the relative configuration was also 4S*/5R*(Fig. 2). These data discarded the presence of L-allo-Thr and D-allo-Thr in compound 2.
In order to quantify the coupling constants of these two C3–C6 and C17–C20 fragments present in 2, we also applied the extended JBCA proposed by Gomez-Paloma and coworkers,28 where DFT calculations of homo- and heteronuclear coupling constant data for each of the six main staggered rotamers (three for each relative stereochemical arrangement represented as syn or anti) are compared with a small model such as methyl O-acetyl-N-((benzyloxy)carbonyl)-L-threoninate (see Fig. 2), via TAE and MAE statistic parameters. B3LYP/6-311 g(d) NMR data obtained values for this simple model fits quite well with the gauche (−) rotamer for the syn-D or L-threonine.
This final analysis confirmed the full absolute stereochemistry for this natural compound (D-Ser, D-Asp, L-Thr1 and L-Thr2), and makes gunungamide (2) the first cyclic depsipeptide bearing a chloropyrrol moiety to be isolated from a marine sponge belonging to the Discodermia genus (Fig. 3).
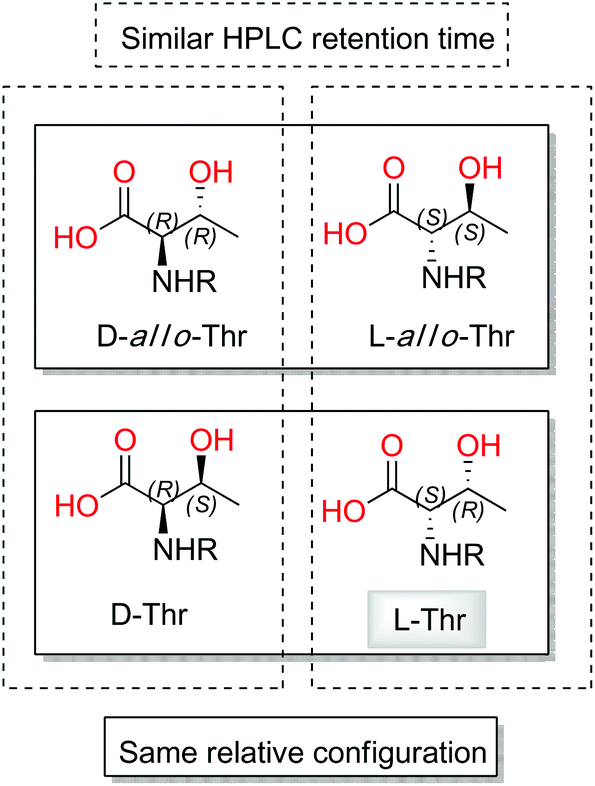 | ||
| Fig. 3 Similarities between stereoisomers of threonine. | ||
In order to validate this strategy, we applied the combination of the JBCA methodology and Marfey's analysis to the threonine residues present in the depsipeptide stellatolide A (1) which was isolated from the sponge Ecionemia acervus and synthesized by us a few years ago.11 Firstly, the application of Marfey's analysis to this cyclopeptide suggested that both Thrs corresponded to the same diastereoisomer.
However, subsequent total synthesis of 1 indicated that the two threonines have different configurations which allowed us to correct the initial proposal. Epimerization of Thr during its acid hydrolysis was proposed to explain the incorrect (mistaken) proposal (see Fig. 5).29
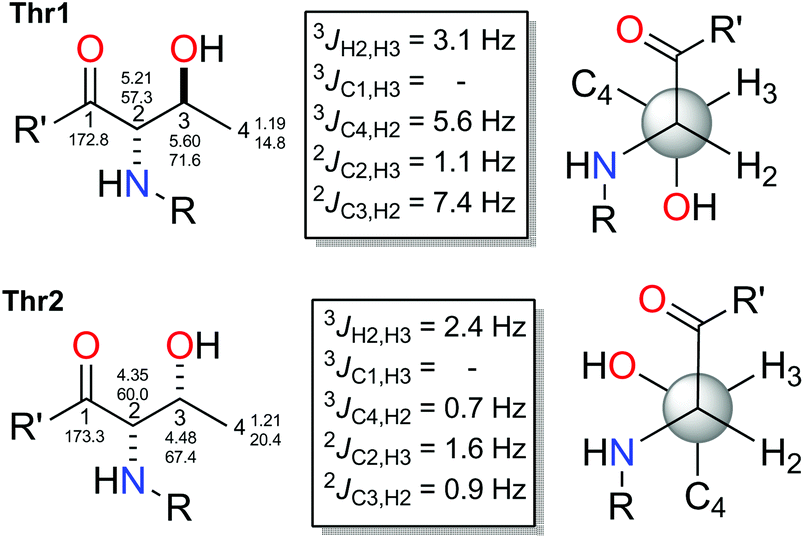 | ||
| Fig. 4 J-Based configuration analysis of threonines in stellatolide A (1). | ||
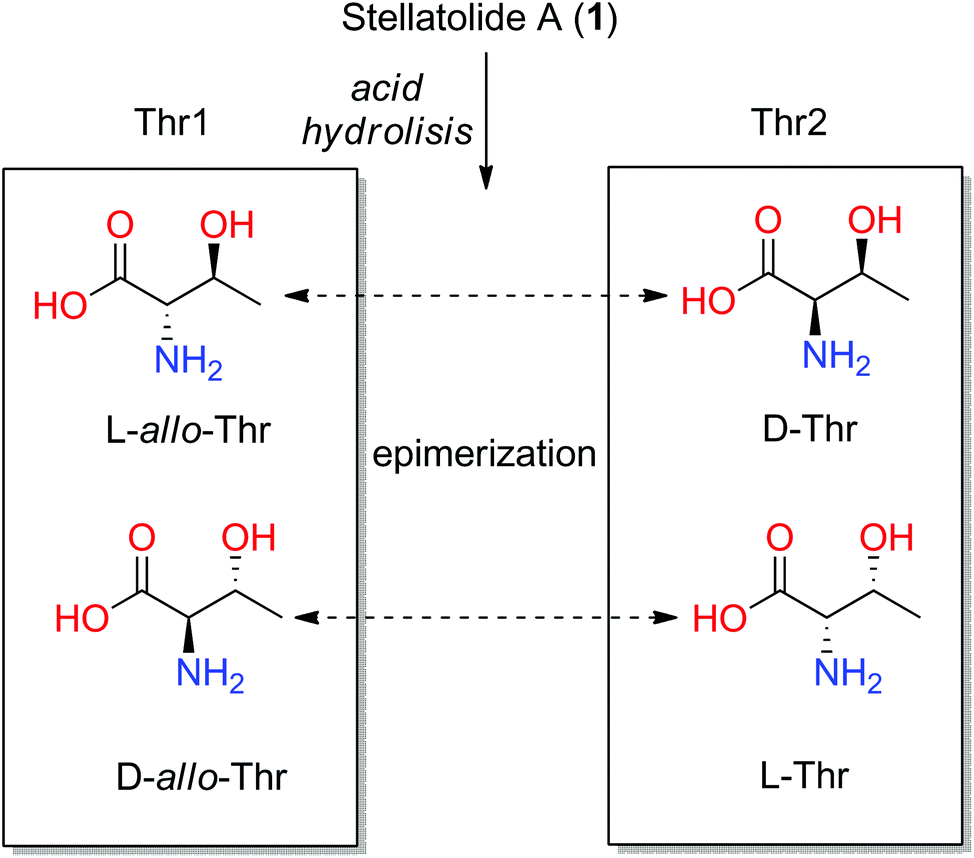 | ||
| Fig. 5 Four configurations of threonines and the possible interchange among them by epimerization after acid hydrolysis of stellatolide A (1). | ||
In order to know if this problem could be avoided by the application of our approach, we elucidated the relative configuration of each threonine in stellatolide A (1) by JBCA. The pure in-phase PIP-HSQMBC NMR experiment was used to measure the heteronuclear coupling constants.30
The values found for Thr1 indicated a gauche disposition in an anti 2-amino-3-hydroxy configuration, and so, the relative configuration of this threonine was assigned as 2S*3S*. Nevertheless, the values obtained for Thr2 displayed a gauche disposition in a syn configuration corresponding to a 2R*3S* relative configuration which is not compatible with the allo configuration. Thus, performing a previous JBCA of the threonines present in 1 would have prevented incorrectly concluding that they were the same diastereoisomer, the D-allo-Thr, deduced by Marfey's analysis of 1 (see Fig. 4) and the time consuming synthetic efforts.
Gunungamide A (2) did not show cytotoxic activity in a panel of four human cancer cell lines: A-549 (lung), HT-29 (colon), MDA-MB-231(breast) and PSN-1 (colon) at a concentration of 15 μM.
Experimental
Materials and methods
Optical rotations were determined using a Jasco P-1020 polarimeter. UV spectra were performed using an Agilent 8453 UV-vis spectrometer. IR spectra were obtained with a PerkinElmer Spectrum 100 FT-IR spectrometer with ATR sampling. NMR spectra were recorded on a Varian “Unity 500” spectrometer at 500/125 MHz (1H/13C). Chemical shifts were reported in ppm using residual CD3OH (δ 3.30 ppm for 1H and 49.0 ppm for 13C) and DMSO-d6 (δ 2.50 ppm for 1H and 39.5 ppm for 13C) as an internal reference. HRESITOFMS was performed on an Agilent 6230 TOF LC/MS chromatograph spectrometer. (+)-ESIMS were recorded using an Agilent 1100 Series LC/MSD spectrometer. HRESITOFMS was performed on an Agilent 6230 TOF LC/MS chromatograph spectrometer.Biological material
The sponge Discodermia sp. (242 g) was collected by hand using rebreather diving near Palau Gunung, Indonesia (129°51.952′ E/04°31.584′ S) at a depth of 100 m and frozen immediately after collection. A voucher specimen (ORMA131384) is deposited at PharmaMar.Extraction and isolation
The sponge Discodermia sp. (242 g) was triturated and exhaustively extracted with MeOH:DCM (1:1, 3 × 600 mL). The combined extracts were concentrated to yield a crude mass of 6.5 g. The crude product was subjected to VLC on Lichroprep RP-18 with a stepped gradient from H2O to MeOH to CH2Cl2. The fraction eluted with MeOH gave a mass of 402 mg which was subjected to semipreparative HPLC Ascentis C18 5 μm, 4.6 × 150 mm, (H2O + 0.04% TFA/AcN + 0.04% TFA gradient from 20% to 100% over 20 min, flow 3 mL min−1, UV detection) to obtain Gunungamide A (2, 14.0 mg, 0.0058% wet wt).
Gunungamide A (2)
Reddish oil; [α]D −33.0° (c 0.1, MeOH); IR νmax 3378, 2966, 2311, 1675 and 1140; UV (MeOH) λmax 261 nm. 1H NMR (500 MHz) and 13C NMR (125 MHz) see Table 1; (+)-HREI-TOFMS m/z 672.1518 [M + H]+ (calcd for C25H3135ClN7O11S m/z 672.1485).Absolute configuration
Ozonolysis (O3, CH2Cl2, 6 h) was performed using approximately 0.5 mg of 2. The product was evaporated to dryness and hydrolyzed (6 N HCl, 110 °C for 15 h). To the hydrolysate, a solution of 700 μg of L-FDAA (N-(3-fluoro-4,6-dinitrophenyl)-L-alanine-amide) in acetone (160 μL), H2O (100 μL) and NaHCO3 1 N (50 μL) was added. The vial was heated at 40 °C for 1 h, and the contents neutralized with 2 N HCl (20 μL) after cooling to room temperature. The resulting solution was dried in vacuum and reconstituted in H2O (800 μL) before being filtered. The resultant aqueous solution was analyzed by HPLC-MS (Symmetry 10 × 150 mm, 5 μm, flow 2.3 mL min−1, H2O/AcN + 0.1%TFA from 20 to 50% in 30 min). Threonines were analyzed by HRESITOF (ACQUITY UPLC BEH C18 1.7 μm, flow 0.65 mL min−1, H2O/AcN + 0.1% TFA from 10 to 25% in 30 min).Computational analysis
Calculations were carried out using the Gaussian09 W, v. A-02 software package. Structures and energies of the six staggered conformers of the (2S,3R)- and (2S,3S)- methyl O-acetyl-N-((benzyloxy)carbonyl)-L-threoninate were optimized at the DFT levels using the functional B3LYP and the 6-31G(d) basis set. 180° (anti), 60° (gauche-(+)), and −60° (gauche-(−)) between protons H2 and H3 were fixed for each conformer. The coupling constants were calculated using a GIAO approach by DFT methods employing the combination B3LYP//6-311G(d).Cytotoxicity assay
The cytotoxic activity of the isolated compound was evaluated against a panel of four human tumor cell lines33 (A549, HT-29, MDA-MB-231 and PSN-1). This compound did not show activity at 15 μM in any of the four human tumor cell lines tested (A-549, HT-29, MDA-MB-231 and PSN-1).Conclusions
A new cyclic depsipeptide bearing an unusual chloropyrrol moiety, gunungamide (2), was isolated from a marine sponge belonging to the Discodermia genus. Hormaomycin,31 from Streptomyces griseoflavus, and the cyclocinamides,32 isolated from a marine sponge of the genus Psammocinia, provide other examples of depsipeptides containing a similar chloropyrrol but bearing a different pattern of substitution. The combination of the JBCA method and Marfey's analysis constitutes a powerful tool to determine the absolute configuration of peptides bearing threonine residues or α-amino acids with two or more adjacent chiral centers. An initial JBCA of such residues allows possible stereoisomers to be discarded and helps detect unwanted epimerization during the acid hydrolysis required for Marfey's analysis. This was demonstrated by the application of this approach to the structural elucidation of the cyclodepsipeptides 1 and 2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the help of our PharmaMar colleagues C. de Eguilior, for collecting the marine samples, S. Bueno for determining the sponge taxonomy, B. de Castro for the realization of the biological assays and S. Munt for revision of the manuscript. The present research was financed by Grants from Ministerio de Economía y Competitividad of Spain (AGL2015-63740-C2-2-R and RTC-2016-4611-1 co-funded by the FEDER Programme from the European Union). J. Rodríguez and C. Jiménez acknowledge Xunta de Galicia and CESGA for the computational facilities.Notes and references
- E. E. Kwan and S. G. Huang, Eur. J. Org. Chem., 2008, 2671 CrossRef CAS
.
-
(a) J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munroe and M. R. Prinsep, Nat. Prod. Rep., 2017, 34, 235 RSC
- R. Bhushan and H. Brückner, Amino Acids, 2004, 27, 231 CrossRef CAS
- R. Bhushan and H. Brückner, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 3148 CrossRef CAS
- J. A. V. Lopez, S. S. Al-Lihaibi, W. M. Alarif, A. Abdel-Lateff, Y. Nogata, K. Washio, M. Morikawa and T. Okino, J. Nat. Prod., 2016, 79, 1213 CrossRef CAS
- L. Coello, F. Reyes, M. J. Martín, C. Cuevas and R. Fernández, J. Nat. Prod., 2014, 77, 298 CrossRef CAS
- J. Gao, C. Caballero-George, B. Wang, K. V. Rao, A. G. Shilabin and M. T. Hamann, J. Nat. Prod., 2009, 72, 2172 CrossRef CAS
- C. Urda, M. Pérez, J. Rodríguez, C. Jiménez, C. Cuevas and R. Fernández, Tetrahedron Lett., 2016, 57, 3239 CrossRef CAS
- A. Raveh, S. Moshe, Z. Evron, E. Flescher and S. Carmeli, Tetrahedron, 2010, 66, 2705 CrossRef CAS
- F. Marner and R. E. Moore, J. Org. Chem., 1977, 42, 2815–2819 CrossRef CAS
- M. J. Martín, R. Rodríguez-Acebes, Y. García-Ramos, V. Martinez, C. Murcia, I. Digón, I. Marco, M. Pelay-Gimeno, R. Fernández, F. Reyes, A. M. Francesch, S. Munt, J. Tulla-Puche, F. Albericio and C. Cuevas, J. Am. Chem. Soc., 2014, 136, 6754 CrossRef
- A. Cüalimsiz, I. Morales and A. L. M. Ramos, J. Org. Chem., 2006, 71, 6351 CrossRef
- A. Plaza and C. A. Bewley, J. Org. Chem., 2006, 71, 6898 CrossRef CAS
-
(a) G. Tarazona, G. Benedit, R. Fernández, M. Pérez, J. Rodríguez, C. Jiménez and C. Cuevas, J. Nat. Prod., 2018, 81, 343 CrossRef CAS
- N. U. Sata, S. Matsunaga, N. Fusetani and R. W. van Soest, J. Nat. Prod., 1999, 62, 969 CrossRef CAS
- M. Kimura, T. Wakimoto, Y. Egami, K. C. Tan, Y. Ise and I. Abe, J. Nat. Prod., 2012, 75, 290 CrossRef CAS
- D. P. Clark, J. Carroll, S. Naylor and P. Crews, J. Org. Chem., 1998, 63, 8757 CrossRef CAS
- H. Tajima, T. Wakimoto, K. Takada, Y. Ise and I. Abe, J. Nat. Prod., 2014, 77, 154 CrossRef CAS
- Y. Kato, N. Fusetani, S. Matsunaga, K. Hashimoto, S. Fujita and T. Furuya, J. Am. Chem. Soc., 1986, 108, 2780 CrossRef CAS
- M. Suganuma, H. Fujiki, H. Furuya-Suguri, S. Yoshizawa, S. Yasumoto, Y. Kato, N. Fusetani and T. Sugimura, Cancer Res., 1990, 50, 3521 CAS
- P. G. Cruz, A. H. Daranas, J. J. Fernández and M. Norte, Org. Lett., 2007, 9, 3045 CrossRef CAS
- S. P. Gunasekera, M. Gunasekera, R. E. Longley and G. K. Schulte, J. Org. Chem., 1990, 55, 4912 CrossRef CAS
- T. N. Nils, J. Ø. Duus and O. W. Sørensen, J. Am. Chem. Soc., 2005, 127, 6154 CrossRef
-
(a) R. T. Williamson, A. V. Buevich, G. E. Martin and T. Parella, J. Org. Chem., 2014, 79, 3887 CrossRef CAS
- K. Matsubara, Magn. Reson. Chem., 2001, 39, 633 CrossRef CAS
- G. Nuzzo, M. L. Ciavatta, R. Kiss, V. Mathieu, H. Leclercqz, E. Manzo, G. Villani, E. Mollo, F. Lefranc, L. D'Souza, M. Gavagnin and G. Cimino, Mar. Drugs, 2012, 10, 1799 CrossRef CAS
- B. R. Clark, R. J. Capon, E. Lacey, S. Tennant and J. H. Gill, Org. Lett., 2006, 8, 701 CrossRef CAS
- G. Bifulco, C. Bassarello, R. Riccio and L. Gomez-Paloma, Org. Lett., 2004, 6, 1025 CrossRef CAS
- J. Hong, Chem. – Eur. J., 2014, 20, 10204 CrossRef CAS
- L. Castañar, J. Saurí, R. T. Williamson, A. Virgili and T. Parella, Angew. Chem., Int. Ed., 2014, 53, 8379 CrossRef
- I. Höfer, M. Crüsemann, M. Radzom, B. Geers, D. Flachshaar, X. Cai, A. Zeeck and J. Piel, Chem. Biol., 2011, 18, 381 CrossRef
- W. D. Clark, T. Corbett, F. Valeriote and P. Crews, J. Am. Chem. Soc., 1997, 119, 9285 CrossRef CAS
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qo00961a |
| This journal is © the Partner Organisations 2019 |