DOI:
10.1039/C8QO00849C
(Research Article)
Org. Chem. Front., 2018,
5, 2934-2939
Enantioselective isoquinuclidine synthesis via sequential Diels–Alder/visible-light photoredox C–C bond cleavage: a formal synthesis of the indole alkaloid catharanthine†
Received
9th August 2018
, Accepted 4th September 2018
First published on 5th September 2018
Abstract
An enantioselective route to substituted chiral isoquinuclidines, present in alkaloids such as catharanthine, deserpidine, ibogamine and ibogaine, has been developed using a “ketene equivalent” approach. Organocatalyzed Diels–Alder reaction of an N-protected dihydropyridine with acrolein using a valine derived 1,2-aminoalcohol catalyst occurs with high er and dr. Subsequent Ru(Bipy)3Cl2·(H2O)6 catalyzed photoredox cleavage generates an N-protected isoquinuclidinone, providing rapid access to the core structures of a variety of important indole alkaloids. Optimized conditions for the enamine mediated photocleavage employed the use of silica gel, acetic acid (1.5 equiv.) and piperidine (3.0 equiv.), a blue LED light source, and pure oxygen rather than air. The two-step sequence utilizes acrolein as a ketene dienophile equivalent providing a new approach to asymmetric “ketene” Diels–Alder reactions. Further elaboration of the resultant isoquinuclidines permitted an enantioselective route to intermediates previously employed in total syntheses of catharanthine and deserpidine.
Introduction
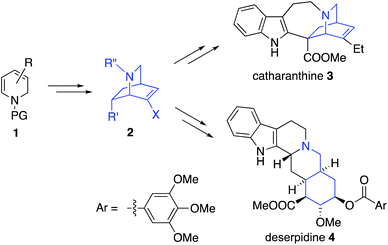
There has been a remarkable explosion in the development of Diels–Alder based methods for chiral isoquinuclidine formation,1 especially since Fukuyama's synthesis of Oseltamivir (which used an organocatalytic Diels–Alder reaction).2–5 As part of a general interest in utilizing Diels–Alder reactions for heterocyclic synthesis we became interested in taking advantage of enantioselective dihydropyridine (DHP) 1 Diels–Alder reactions for isoquinuclidine 2 formation (Fig. 1).6 Enantioselective routes toward substituted isoquinuclidine 2 enables access to both isoquinuclidine and other alkaloid families, including the venerable iboga alkaloids such as catharanthine 3, the Rauvolfia derived-alkaloids such as deserpidine 4, and the alkaloid manzamine A.7 The Madagascan periwinkle derived isoquinuclidine alkaloid catharanthine 3 was chosen as an initial target due to its biological activity and its structural complexity, comprising a pentacyclic skeleton, three stereocentres, and a quaternary carbon. Biomedical interest has been primarily focused on 3 serving as a chemical precursor to clinical anti-cancer natural products, vinblastine and vincristine. Notably, the unnatural analogues, vinorelbine and vinflunine, are also used for the clinical treatment of cancer.8 Moreover, Watanabe has recently shown that 3 can affect cold induced pain signals in mammals as a potent TRPM8 antagonist.9 The low natural abundance (approximately 0.0003% of leaf by mass) has driven significant synthetic interest in the development of new routes to 310 which has long been considered a quintessential alkaloid target since Büchi's seminal synthetic work.10a Most recent synthetic attention has focused upon enantioselective total syntheses of 3,11 and Stephenson's photochemical transformations of 3.12 Also relevant are synthetic studies13–15 toward the structurally related isoquinuclidine alkaloids ibogamine and ibogaine which have interesting CNS biological activity, reportedly lessening withdrawal symptoms in opioid addiction.
 |
| | Fig. 1 Isoquinuclidine route to alkaloids. | |
Results and discussion
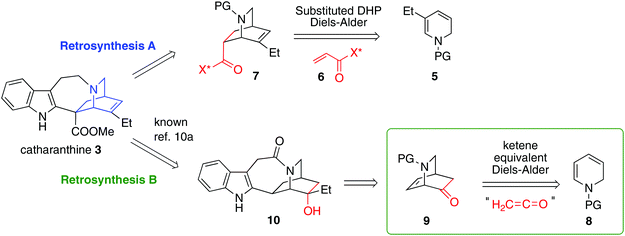
We initially planned two retrosynthetic analyses based on different modes of chiral Diels–Alder isoquinuclidine formation (Fig. 2). Both routes were explored in parallel. Retrosynthetic analysis (path A) was based on a Matsuyama-type Diels–Alder union of a dihydropyridine 5 and Oppolzer's sultam acrylamine 6.3b This strategy would enable an extremely concise synthesis of catharanthine, but, problematically, a highly diastereoselective union of a suitably substituted dihydropyridine with an acrylate to give 7, is unknown. An alternative ketene equivalent based hetero Diels–Alder retrosynthetic strategy using 8 to give 9 (retrosynthesis path B) would instead target intermediate 10 a compound on the Doris–Büchi route to 3.10a,11a Pentacycle 10 was also attractive due to its possible application toward other isoquinuclidines, such as ibogamine, ibogaine and the anti-angiogenic voacangine.16
 |
| | Fig. 2 Dihydropyridine based Diels–Alder retrosynthetic analyses to isoquinuclidines. | |
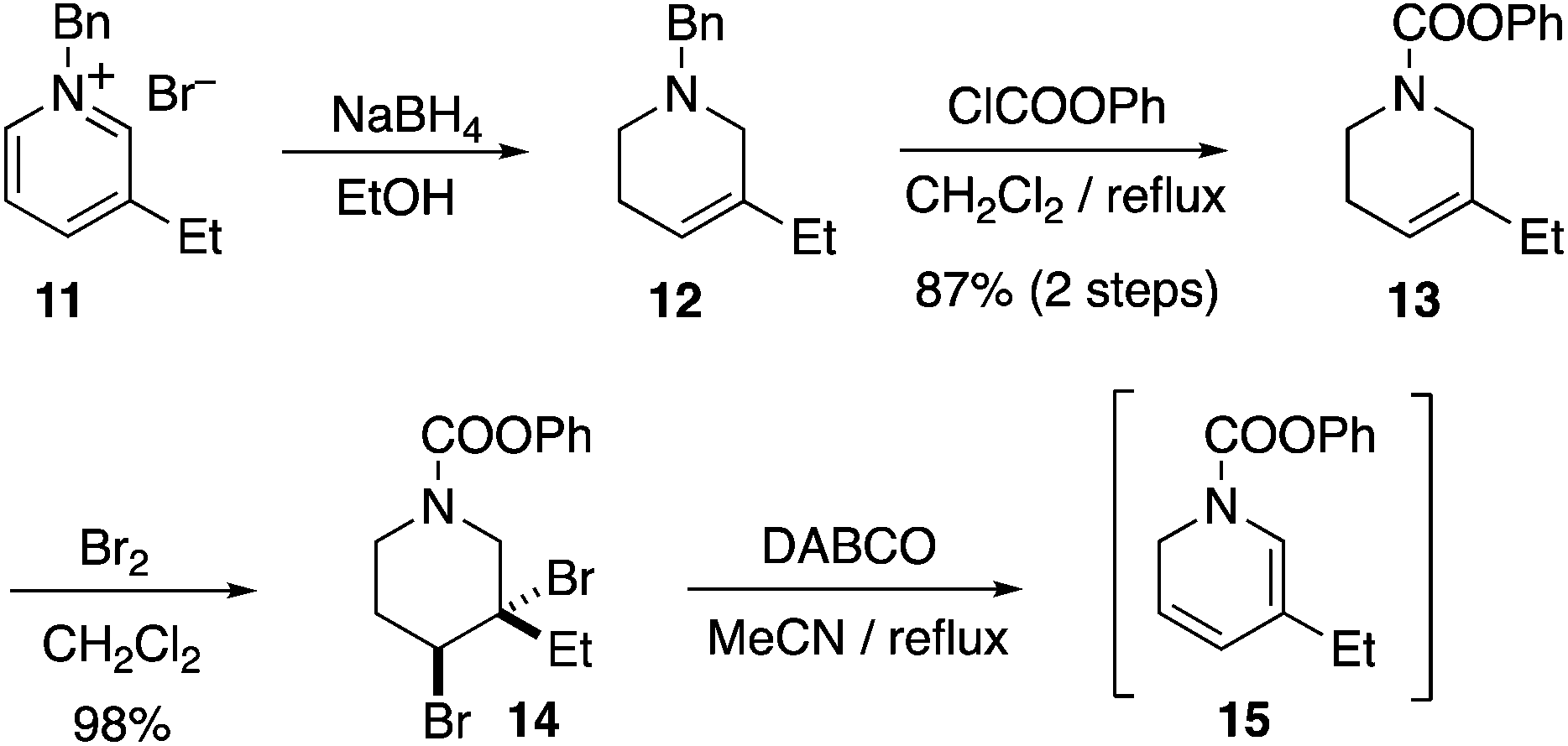
Several of the previous syntheses of ibogamine, ibogaine, and catharanthine have employed an intermolecular DHP Diels–Alder route analogous to retrosynthetic path A, including racemic syntheses by Büchi,10a,13a,15a Raucher,10h Sundberg,10l and Fukuyama,10m while intramolecular examples have been achieved by Kuehne10i and Oguri.11b However, since the pioneering studies of Büchi10a an enantioselective intermolecular DHP Diels–Alder based total synthesis of 3 has remained elusive. Using the chemistry developed by Raucher and Fukuyama,10h,m we subjected the known compound 11 to reduction with NaBH4 to yield known compound 12, which on carbamate introduction afforded 13 in 87% yield (2 steps) (Scheme 1). The alkene 13 was dibrominated to garner 14 in 98% yield. The dibromide 14 was then subjected to double elimination with DABCO, furnishing the desired dihydropyridine 15. The Cbz carbamate protected analogue of 15 is known to be unstable, and is best used crude.10m Therefore, we also used crude 15 immediately after synthesis for the next step, vide infra.
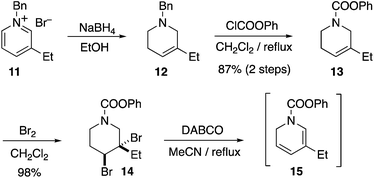 |
| | Scheme 1 Synthesis of dihydropyridine 15. | |
Unfortunately, attempts to react 15 and known dienophile 16 were unsuccessful using a variety of Lewis acid catalysts (Scheme 2). For example, the use of HfCl4 or ZrCl4 resulted in a slow decomposition of dihydropyridine 15 to form a mixture of unidentified aromatic products (crude 1H NMR spectroscopy), while various lanthanide(III) triflates led to no conversion of either the diene or the dienophile. The Matsuyama work shows the success of an unsubstituted dihydropyridine as a diene.3 We reasoned that the slightly increased steric bulk imparted by the ethyl group in 15 thwarted its cycloaddition with 16 (relative to the unsubstituted dihydropyridine, which is known to react successfully). The result is less surprising given that even in a racemic mode, the Diels–Alder reaction of substituted dihydropyridines is a difficult transformation.17
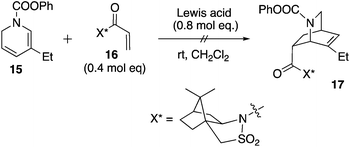 |
| | Scheme 2 Attempted Diels–Alder reaction of 15 and 16. | |
The key issue of constructing the isoquinuclidine core 9 enantioselectively hinged on the development of an asymmetric ketene dienophile equivalent.18 The challenge was considerable, as previous efforts to selectively react a dihydropyridine and a chiral ketene equivalent were fraught with issues, and particularly low diastereoselectivities.19 Contemporaneous with the studies outlined above, we explored retrosynthetic pathway B, with the intention of utilizing acrolein as a ketene equivalent in a reaction with diene 18 (Scheme 3). Thus, a Nakano-type Diels–Alder reaction using catalyst 19 afforded 20 in 71% yield on decagram scale.3c20 was obtained in 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 er (HPLC) and 30:1 dr (1H NMR), although crude 1H NMR spectroscopy indicates that the reaction mixture contained ≥98:2 dr.20 The yield of the Diels–Alder step is comparable with analogous enantioselective dihydropyridine Diels–Alder reactions used in total synthesis.2,21 The key challenge occurred at this stage. Attempted conversion of the aldehyde 20 into 21 through a multistep process involving α-acetoxylate/ester hydrolysis/oxidative cleavage was problematic.22 Fortunately, the key conversion of aldehyde 20 into ketone 21 was achieved using a Xia Ru(Bipy)3Cl2·(H2O)6 photoredox catalyzed cleavage with molecular oxygen on gram scale in 83% yield.23 The originally reported Xia conditions employed the catalyst (5 mol%) in 0.1 M in CH3CN, with piperidine (3 equiv.), and irradiation with a 15 W fluorescent light bulb under air at 25 °C on a 0.1 mmol scale. This protocol (using a 23 W fluorescent light bulb) gave poor reaction conversion and was not suitable for scale-up. Thus, substantial reaction optimization of these conditions was required in order to achieve reaction of the isoquinuclidine 20. Three factors were found to be important to achieve effective reaction conversion and high yield of 21 on a gram scale, compared to the original Xia conditions. (i) The use of a pure O2 atmosphere (1 atm) provides a greater oxygen concentration compared to the use of air. (ii) The use of 1.5 equiv. of acetic acid and silica as an acidic drying agent presumably results in improved enamine formation (the intermediate that undergoes oxidation). (iii) The use of a blue LED light source, rather than a fluorescent bulb (23 W), using a vessel wrapped in aluminum foil, provides improved photochemical efficiency. Overall, this two-step route compares favorably to Ishihara's five-step synthesis of 21 from 18, reported during the course of our studies.11d This is the first example of a Xia photoredox oxidative cleavage being used in a target oriented synthesis. Given that MacMillan and others have utilized organocatalytic Diels–Alder reactions of acrolein and α,β-unsaturated enals with a broad range of dienes,24 this sequence has potential applications beyond isoquinuclidine synthesis for which a ketene equivalent Diels–Alder tactic might be envisaged.
:3 er (HPLC) and 30:1 dr (1H NMR), although crude 1H NMR spectroscopy indicates that the reaction mixture contained ≥98:2 dr.20 The yield of the Diels–Alder step is comparable with analogous enantioselective dihydropyridine Diels–Alder reactions used in total synthesis.2,21 The key challenge occurred at this stage. Attempted conversion of the aldehyde 20 into 21 through a multistep process involving α-acetoxylate/ester hydrolysis/oxidative cleavage was problematic.22 Fortunately, the key conversion of aldehyde 20 into ketone 21 was achieved using a Xia Ru(Bipy)3Cl2·(H2O)6 photoredox catalyzed cleavage with molecular oxygen on gram scale in 83% yield.23 The originally reported Xia conditions employed the catalyst (5 mol%) in 0.1 M in CH3CN, with piperidine (3 equiv.), and irradiation with a 15 W fluorescent light bulb under air at 25 °C on a 0.1 mmol scale. This protocol (using a 23 W fluorescent light bulb) gave poor reaction conversion and was not suitable for scale-up. Thus, substantial reaction optimization of these conditions was required in order to achieve reaction of the isoquinuclidine 20. Three factors were found to be important to achieve effective reaction conversion and high yield of 21 on a gram scale, compared to the original Xia conditions. (i) The use of a pure O2 atmosphere (1 atm) provides a greater oxygen concentration compared to the use of air. (ii) The use of 1.5 equiv. of acetic acid and silica as an acidic drying agent presumably results in improved enamine formation (the intermediate that undergoes oxidation). (iii) The use of a blue LED light source, rather than a fluorescent bulb (23 W), using a vessel wrapped in aluminum foil, provides improved photochemical efficiency. Overall, this two-step route compares favorably to Ishihara's five-step synthesis of 21 from 18, reported during the course of our studies.11d This is the first example of a Xia photoredox oxidative cleavage being used in a target oriented synthesis. Given that MacMillan and others have utilized organocatalytic Diels–Alder reactions of acrolein and α,β-unsaturated enals with a broad range of dienes,24 this sequence has potential applications beyond isoquinuclidine synthesis for which a ketene equivalent Diels–Alder tactic might be envisaged.
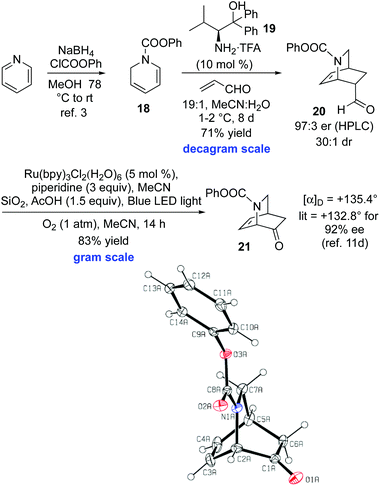 |
| | Scheme 3 Enantioselective Diels–Alder/catalytic photoredox sequence and X-Ray crystal structure of 21. | |
Conversion of 21 into 24 was achieved via ketalization, carbamate deprotection of 22 to 23, and subsequent amide formation using EDCI coupling with 3-indole acetic acid (Scheme 4). We had already synthesized racemic 24 using an approach based upon the synthesis of an ethyl carbamate variant of 21 which was reported by MaGee.25,26 Deprotection of 24 to 25 followed by application of the Trost cyclization conditions with stoichiometric (MeCN)2PdCl2 and AgBF4 afforded 26 in 25% yield.14a In our hands the latter reaction gave poorer yields than those obtained by Doris.11a On the other hand, direct cyclization of 24 using the Trost protocol afforded the pentacycle 27, in improved yield. Unfortunately, attempts to achieve deprotection of the dioxolane to form the ketone 26 were unsuccessful (see ESI† for details).
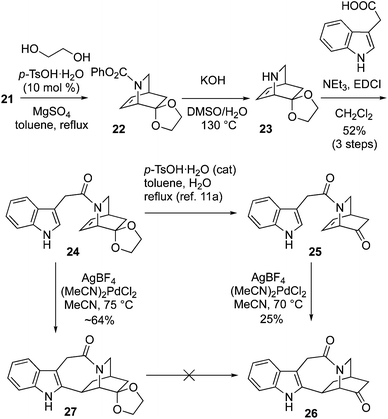 |
| | Scheme 4 Synthesis of pentacycle 27A. | |
Therefore, the ketone 21 was protected with 1,3-propanediol and p-TsOH (10 mol%), to give 1,3-dioxane ketal 28 in 96% yield (Scheme 5). Phenyl carbamate deprotection of 28 was smoothly achieved with refluxing hydrazine and KOH in a MeOH/H2O mixture. The crude amine 29 was coupled to 3-indole acetic acid with EDC, garnering amide 30 in 63% yield (2 steps). Application of the Trost cyclization conditions to 30 using stoichiometric (MeCN)2PdCl2 and AgBF4 afforded 31 in 59% yield.14a Attempted use of the Sames indole bromination/reductive Heck sequence on 30 failed, with none of compound 31 detected.13o
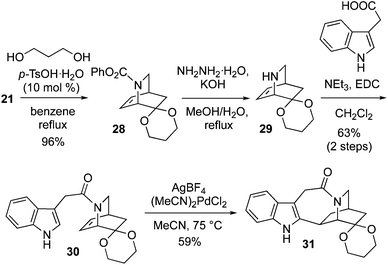 |
| | Scheme 5 Synthesis of pentacyclic skeleton 31. | |
The deprotection of 31 occurred slowly over 1 week with p-TsOH in acetone/H2O, furnishing 26 in 85% yield (Scheme 6). Reaction of 26 with ethynyl magnesium bromide afforded 33 with a >19:1 dr (crude 1H NMR).27 Hydrogenation of 32 to yield 10 in 80% yield over 2 steps constitutes an enantioselective formal synthesis of catharanthine. Notably, our access to 26 in just 8 steps from pyridine compares well with the elegant work of Doris to obtain 26 in 16 steps from N-CBz-serine.11a The comparison underscores the advancement of synthetic reach enabled by both enantioselective dihydropyridine Diels–Alder methods and photoredox catalysis.28 The synthesis of the intermediates 10 and 26, used by Büchi and Doris, constitute a formal synthesis of catharanthine.
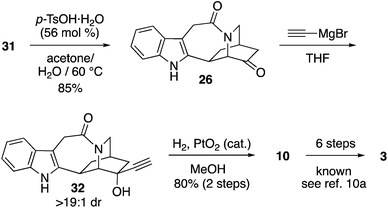 |
| | Scheme 6 Completion of formal synthesis of catharanthine. | |
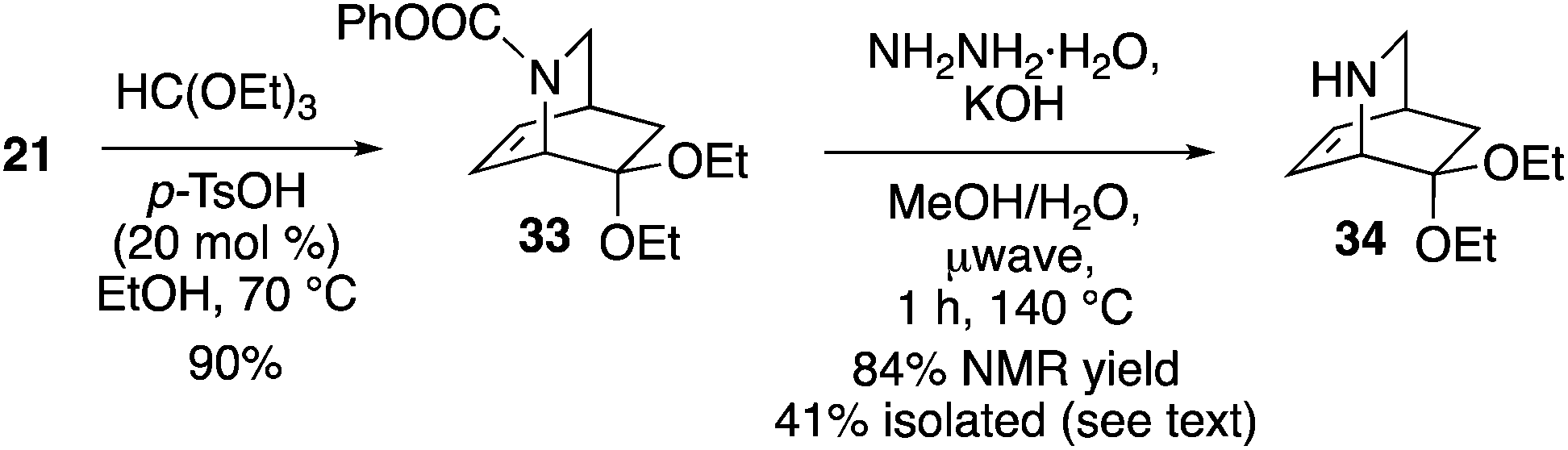
Access to enantioenriched isoquinuclidine 21 also provides a route to the Rauvolfia derived-alkaloids. Deserpidine 4 (and the more famed reserpine) display notable biological activity on hypertension,29 and impressive pentacyclic fused carbon skeletons. Mariano employed a racemic mixture of 34 as a key intermediate to synthesize 4.30 Thus, we reacted 21 with triethoxyorthoformate yielding 33 in 90% yield (Scheme 7). Removal of the carbamate protecting group from 33 occurred with KOH/hydrazine under microwave conditions to give 34 in a moderate 41% yield. However, an NMR yield (1H NMR, 3-chloropyridine as a standard) of the crude material was found to be 84%, indicating that the amine is best used directly. The sequence shown thus constitutes an enantioselective formal synthesis of deserpidine.
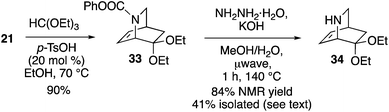 |
| | Scheme 7 Synthesis of deserpidine intermediate 34. | |
Conclusions
An enantioselective formal synthesis of catharanthine from pyridine has been developed. Key steps include the application of organocatalytic Diels–Alder reaction and photoredox catalyzed oxidative cleavage. Considerable optimization of the Xia photoredox oxidative cleavage conditions was required to achieve the enamine mediated oxidative deformylation on the isoquinuclidine core to achieve complete reaction conversion and on gram scale. Combined with the organocatalytic Diels–Alder reaction this establishes the use of acrolein as a Diels–Alder ketene equivalent, a tactical gambit that has potential synthetic ramifications beyond the isoquinuclidine alkaloids. This principle was further demonstrated by making an enantioenriched key synthetic intermediate for Mariano's synthesis of deserpidine. The work reinforces the applicability of Ru(Bipy)3Cl2 photoredox catalysis in the framework of total synthesis. Further studies on generalizing this strategy to isoquinuclidine and other alkaloid syntheses will be reported in due course.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful for financial support through a Natural Science and Engineering Research Council (NSERC) of Canada Discovery Grant. We thank Dr Matthew Forbes for MS analysis, Dr Alan Lough for X-ray structure determination of 21, and Dr Jeffrey St Denis and Prof. Andrei K. Yudin for the loan of a blue LED light strip.
Notes and references
- For examples of chiral isoquinuclidine formation using Diels–Alder reactions before 2007, see:
(a) C. Marazano, Y. Yannic, M. Mehmandoust and B. C. Das, Tetrahedron Lett., 1990, 31, 1995–1998 CrossRef;
(b) C. Kouklovsky, A. Pouihes and Y. Langlois, J. Am. Chem. Soc., 1990, 112, 6672–6679 CrossRef;
(c) D. C. Dos Santos, R. P. De Freitas Gil, L. Gil and C. Marazano, Tetrahedron Lett., 2001, 42, 6109–6111 CrossRef;
(d) N. Takenaka, Y. Huang and V. H. Rawal, Tetrahedron, 2002, 58, 8299–8305 CrossRef;
(e) H. Nakano, N. Tsugawa and R. Fujita, Tetrahedron Lett., 2005, 46, 5677–5681 CrossRef;
(f) H. Nakano, N. Tsugawa, K. Takahashi, Y. Okuyama and R. Fujita, Tetrahedron, 2006, 62, 10879–10887 CrossRef.
- N. Satoh, T. Akiba, S. Yokoshima and T. Fukuyama, Angew. Chem., Int. Ed., 2007, 46, 5734–5736 CrossRef PubMed.
- For examples of chiral isoquinuclidine formation using Diels–Alder reactions after 2007, see:
(a) M. Hirama, Y. Kato, C. Seki, H. Matsuyama, N. Oshikiri and M. Iyoda, Chem. Lett., 2008, 37, 924–925 CrossRef;
(b) M. Hirama, Y. Kato, C. Seki, H. Nakano, M. Takeshita, N. Oshikiri, M. Iyoda and H. Matsuyama, Tetrahedron, 2010, 66, 7618–7624 CrossRef;
(c) H. Nakano, K. Osone, M. Takeshita, E. Kwon, C. Seki, H. Matsuyama, N. Takano and Y. Kohari, Chem. Commun., 2010, 46, 4827–4829 RSC;
(d) C. Seki, M. Hirama, N. D. M. Romauli Hutabarat, J. Takada, C. Suttibut, C. Takahashi, T. Takaguchi, Y. Kohari, H. Nakano, K. Uwai, N. Takano, M. Yasui, Y. Okuyama, M. Takeshita and H. Matsuyama, Tetrahedron, 2012, 68, 1774–1781 CrossRef;
(e) Y. Kohari, Y. Okuyama, E. Kwon, T. Furuyama, N. Kobayashi, T. Otuki, J. Kumagai, C. Seki, K. Uwai, G. Dai, T. Iwasa and H. Nakano, J. Org. Chem., 2014, 79, 9500–9511 CrossRef PubMed;
(f) See also ref. 11b–d.
- For a review of chiral isoquinuclidine formation using Diels–Alder reactions, see: E. M. P. Silva, D. H. A. Rocha and A. M. S. Silva, Synthesis, 2018, 50, 1773–1782 CrossRef.
- For a review of chiral isoquinuclidine formation, see: M. Faisal, D. Shahzad, A. Saeed, B. Lal, S. Saeed, F. A. Larik, P. A. Channar, P. A. Mahesar and J. Mahar, Tetrahedron: Asymmetry, 2018, 28, 1445–1461 CrossRef.
- For examples, see:
(a) D. A. Powell and R. A. Batey, Org. Lett., 2002, 4, 2913–2916 CrossRef PubMed;
(b) D. A. Powell and R. A. Batey, Tetrahedron Lett., 2003, 44, 7569–7573 CrossRef;
(c) C. A. Miller and R. A. Batey, Org. Lett., 2004, 6, 699–702 CrossRef PubMed;
(d) H. Twin and R. A. Batey, Org. Lett., 2004, 6, 4913–4916 CrossRef PubMed;
(e) H. Twin, W. W. Wen, D. A. Powell, A. J. Lough and R. A. Batey, Tetrahedron Lett., 2007, 48, 1841–1844 CrossRef;
(f) C. D. Smith and R. A. Batey, Tetrahedron, 2008, 64, 652–663 CrossRef;
(g) C. D. Smith, J. I. Gavrilyuk, A. J. Lough and R. A. Batey, J. Org. Chem., 2010, 75, 702–715 CrossRef PubMed;
(h) R. R. R. Taylor and R. A. Batey, J. Org. Chem., 2013, 78, 1404–1420 CrossRef PubMed.
- For conversion of an isoquinuclidine to a potential manzamine A core, see: D. I. MaGee and M. L. Lee, Tetrahedron Lett., 2001, 42, 7177–7180 CrossRef.
-
(a) A. Duflos, A. Kruczynski and J.-M. Barret, Curr. Med. Chem.: Anti-Cancer Agents, 2002, 2, 55–70 CrossRef PubMed;
(b) M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef PubMed.
- Y. Terada, M. Kitajima, F. Taguchi, H. Takayama, S. Horie and T. Watanabe, J. Nat. Prod., 2014, 77, 1831–1838 CrossRef PubMed.
- For racemic synthesis of catharanthine, see for example:
(a) G. Büchi, P. Kulsa, K. Ogasawara and R. L. Rosati, J. Am. Chem. Soc., 1970, 92, 999–1005 CrossRef;
(b) J. P. Kutney and F. Bylsma, Helv. Chim. Acta, 1975, 58, 1672–1689 CrossRef PubMed;
(c) B. M. Trost, S. A. Godleski and J. L. Belletire, J. Org. Chem., 1979, 44, 2052–2054 CrossRef;
(d) R. Z. Andriamialisoa, N. Langlois and Y. Langlois, Heterocycles, 1980, 14, 1457–1460 CrossRef;
(e) T. Imanishi, H. Shin, N. Yagi and M. Hanaoka, Tetrahedron Lett., 1980, 21, 3285–3288 CrossRef;
(f) C. Marazano, M. LeGoff, J. Fourrey and B. C. Das, J. Chem. Soc., Chem. Commun., 1981, 389–391 RSC;
(g) T. Imanishi, N. Yagi, H. Shin and M. Hanaoka, Chem. Pharm. Bull., 1982, 30, 4052–4059 CrossRef;
(h) S. Raucher and B. L. Bray, J. Org. Chem., 1985, 50, 3236–3237 CrossRef;
(i) M. E. Kuehne, W. G. Bornmann, W. G. Earley and I. Marko, J. Org. Chem., 1986, 51, 2913–2927 CrossRef;
(j) S. Raucher, B. L. Bray and R. F. Lawrence, J. Am. Chem. Soc., 1987, 109, 442–446 CrossRef;
(k) C. Szántay, H. Bölcskei and E. Gács-Baitz, Tetrahedron, 1990, 46, 1711–1732 CrossRef;
(l) R. J. Sundberg, J. Hong, S. Q. Smith and M. Sabat, Tetrahedron, 1998, 54, 6259–6292 CrossRef;
(m) M. T. Reding and T. Fukuyama, Org. Lett., 1999, 1, 973–976 CrossRef.
- For enantioselective syntheses of catharanthine, see:
(a) L. Moisan, P. Thuéry, M. Nicolas, E. Doris and B. Rousseau, Angew. Chem., Int. Ed., 2006, 45, 5334–5336 CrossRef PubMed;
(b) H. Mizoguchi, H. Oikawa and H. Oguri, Nat. Chem., 2014, 6, 57–64 CrossRef PubMed;
(c) K. Ishihara, H. Yamada and M. Akakura, Chem. Commun., 2014, 50, 6357–6363 RSC;
(d) M. Hatano, Y. Goto, A. Izumiseki, M. Akakura and K. Ishihara, J. Am. Chem. Soc., 2015, 137, 13472–13475 CrossRef PubMed;
(e) Y. Zhang, Y. Xue, G. Li, H. Yuan and T. Luo, Chem. Sci., 2016, 7, 5530–5536 RSC.
- J. W. Beatty and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 10270–10273 CrossRef PubMed.
- For racemic syntheses of ibogamine, see for example:
(a) G. Büchi, D. L. Coffen, K. Kocsis, P. E. Sonnet and F. E. Ziegler, J. Am. Chem. Soc., 1965, 87, 2073–2075 CrossRef;
(b) S. I. Sallay, J. Am. Chem. Soc., 1967, 89, 6762–6763 CrossRef PubMed;
(c) W. Nagata, H. Hirai, T. Okumura and K. Kawata, J. Am. Chem. Soc., 1968, 90, 1650–1651 CrossRef PubMed;
(d) S. Hirai, K. Kawata and W. Nagata, Chem. Commun., 1968, 1016–1017 RSC;
(e) P. Rosenmund, W. H. Haase, J. Bauer and R. Frische, Chem. Ber., 1975, 108, 1871–1895 CrossRef;
(f) T. Imanishi, N. Yagi, H. Shin and M. Hanaoka, Tetrahedron Lett., 1981, 22, 4001–4004 CrossRef;
(g) J. W. Huffman, G. Shanmugasundaram, R. Sawdaye, P. C. Raveendranath and R. C. Desai, J. Org. Chem., 1985, 50, 1460–1464 CrossRef;
(h) M. E. Kuehne and P. J. Reider, J. Org. Chem., 1985, 50, 1464–1467 CrossRef;
(i) T. Imanishi, N. Yagi and M. Hanaoka, Chem. Pharm. Bull., 1985, 33, 4202–4211 CrossRef;
(j) G. R. Krow, D. A. Shaw, B. Lynch, W. Lester, S. W. Szczepanski, K. Raghavachari and A. E. Derome, J. Org. Chem., 1988, 53, 2258–2262 CrossRef;
(k) C. Herdeis and C. Hartke-Karger, Liebigs Ann. Chem., 1991, 99–104 CrossRef;
(l) K. J. Henry, P. A. Grieco Jr. and W. J. DuBay, Tetrahedron Lett., 1996, 37, 8289–8292 CrossRef;
(m) G. K. Jana and S. Sinha, Tetrahedron Lett., 2012, 53, 1671–1675 CrossRef;
(n) G. K. Jana and S. Sinha, Tetrahedron, 2012, 68, 7155–7165 CrossRef;
(o) A. C. Kruegel, S. Rakshit, X. Li and D. Sames, J. Org. Chem., 2015, 80, 2062–2071 CrossRef PubMed;
(p) G. Zhao, X. Xie, H. Sun, Z. Yuan, Z. Zhong, S. Tang and X. She, Org. Lett., 2016, 18, 2447–2450 CrossRef PubMed.
- For enantioselective syntheses of ibogamine, see:
(a) B. M. Trost, S. A. Godleski and J. P. Genêt, J. Am. Chem. Soc., 1978, 100, 3930–3931 CrossRef;
(b) J. D. White and Y. Choi, Org. Lett., 2000, 2, 2373–2376 CrossRef PubMed;
(c) D. M. Hodgson and J.-M. Galano, Org. Lett., 2005, 7, 2221–2224 CrossRef PubMed;
(d) See also ref. 11e.
- For racemic total syntheses of ibogaine, see:
(a) G. Büchi, D. L. Coffen, K. Kocsis, P. E. Sonnet and F. E. Ziegler, J. Am. Chem. Soc., 1966, 88, 3099–3109 CrossRef;
(b) See also ref. 13n and 13p.
- Y. Kim, H. J. Jung and H. J. Kwon, Biochem. Biophys. Res. Commun., 2012, 417, 330–334 CrossRef PubMed.
- For a notable racemic exception, see: R. M. Martin, R. G. Bergman and J. A. Ellman, Org. Lett., 2013, 15, 444–447 CrossRef PubMed.
- Examples of ketene equivalents in Diels–Alder reactions include α-substituted acrylonitriles and acrylates, vinylsulfoxides, nitroalkenes, alkenylboranes, etc. For a review, see: V. K. Aggarwal, A. Ali and M. P. Coogan, Tetrahedron, 1999, 55, 293–312 CrossRef.
- V. K. Aggarwal, Z. Gültekin, R. S. Grainger, H. Adams and P. L. Spargo, J. Chem. Soc., Perkin Trans. 1, 1998, 2771–2781 RSC.
- Aldehyde 20 can be reduced with sodium borohydride to the corresponding primary alcohol 35 which was obtained in 94% ee (see ESI†).
-
(a) G. Barbe and A. B. Charette, J. Am. Chem. Soc., 2008, 130, 13873–13875 CrossRef PubMed;
(b) G. Barbe, D. Fiset and A. B. Charette, J. Org. Chem., 2011, 76, 5354–5362 CrossRef PubMed.
- Although the α-benzoyloxy group was introduced to 20 to give 36 in 71% yield (dr = 1.25:1) (see ESI†), attempts at its selective deprotection were unsuccessful. See: C. S. Beshara, A. Hall, R. L. Jenkins, K. L. Jones, T. C. Jones, N. M. Killeen, P. H. Taylor, S. P. Thomas and N. C. O. Tomkinson, Org. Lett., 2005, 7, 5729–5732 CrossRef PubMed.
- H. Sun, C. Yang, F. Gao, Z. Li and W. Xia, Org. Lett., 2013, 15, 624–627 CrossRef PubMed.
- See for example: K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef.
- MaGee used the α-acetylvinyl 2,4-dinitrophenylbenzoate as a dienophile: D. I. MaGee and M. L. Lee, Synlett, 1997, 786–788 CrossRef.
- Racemic 24 was initially synthesized via an approach mimicking the MaGee route for the synthesis of the ethylcarbamate analog of 21 (rather than the methyl carbamate, see ref. 25) and its subsequent conversion to racemic 24. In our hands attempts to separate the enantiomers of 24 using chiral HPLC as described by Doris (see ref. 11a) were unsuccessful.
- The use of an ethyl Grignard reagent was reported to be unsuccessful for addition to substrates related to 2, with reduction to the secondary alcohol occurring through β-hydride transfer instead. As a result the addition of a vinyl Grignard was employed (see ref. 10a and 11a). Luo an co-workers used an organocerium reagent for the incorporation of the ethyl group in the formation of the amine analog of 10 (i.e., lacking the lactam functionality) in 72% yield (see ref. 11e). The two step protocol reported here in using an ethynyl Grignard reagent is somewhat more efficient than these approaches occurring with a combined yield of 80%.
- For recent reviews on photoredox catalysis, see:
(a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC;
(b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef PubMed;
(c) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef PubMed;
(d) M. D. Karkas, J. A. Porco, Jr. and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed;
(e) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef.
- T. Winsor, Dis. Chest, 1959, 35, 415–421 CrossRef PubMed.
- E. W. Baxter, D. Labaree, H. L. Ammon and P. S. Mariano, J. Am. Chem. Soc., 1990, 112, 7682–7692 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1861258. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qo00849c |
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*