
Electrochemical synthesis of 7-membered carbocycles through cascade 5-exo-trig/7-endo-trig radical cyclization†
Hao
Long
a,
Jinshuai
Song
b and
Hai-Chao
Xu
 *a
*a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, Key Laboratory of Chemical Biology of Fujian Province, iChEM and College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China. E-mail: haichao.xu@xmu.edu.cn
bFujian Institute of Research on Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, P. R. China
First published on 26th September 2018
Abstract
7-endo-trig cyclizations, especially for unhindered terminal alkenes, remain underdeveloped. We report herein an electrochemical synthesis of functionalized 7-membered carbocycles through a 5-exo-trig/7-endo-trig radical cyclization cascade. The first cyclization step of the cascade process forms a 5-membered ring with trans-disposition of the radical center and the remaining alkene. This trans configuration forces the 6-heptenyl radical to undergo regioselective 7-endo cyclization.
Introduction
Radical cyclization reactions have become an indispensable tool for the synthesis of cyclic structures.1 Among these reactions, those that form 7-membered carbocycles remain rare. The 6-heptenyl radicals usually undergo preferentially 6-exo-trig cyclization over the 7-endo-trig alternative (Scheme 1a) and the 7-octenyl radicals frequently form a mixture of 7- and 8-membered rings because of the competing 7-exo- and 8-endo-trig processes.2 Several strategies have been developed to reverse the general preference of the 6-heptenyl radicals to favor the 7-endo mode of cyclization by introducing substituents at positions 1, 5, or 6 to increase the steric hindrance for the 6-exo-trig process (Scheme 1b and c).3 Alternatively, annealing a trans-fused 5-membered ring has been found to be effective for reversing the 6-exo/7-endo selectivity even for a monosubstituted alkene (Scheme 1d).4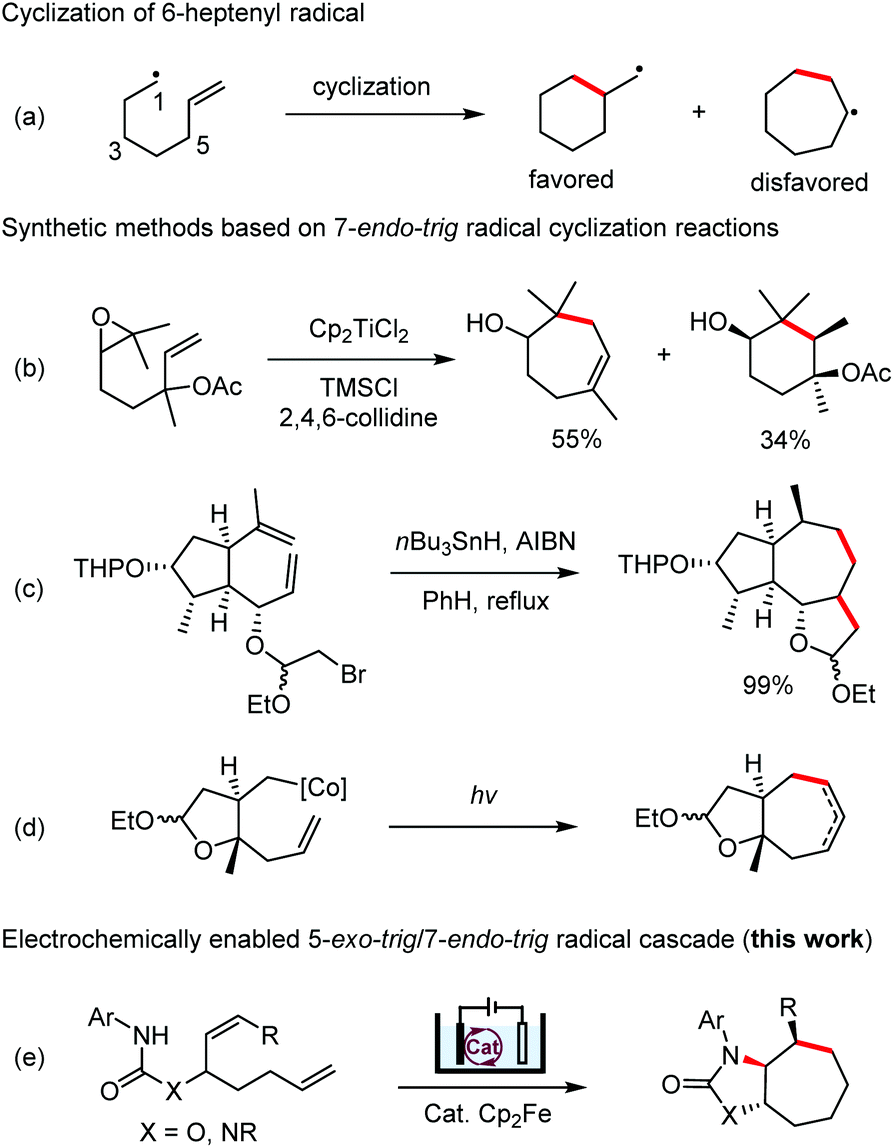 | ||
| Scheme 1 7-endo-trig radical cyclization reactions. | ||
The promiscuous radical species can participate in various transformations including dimerization, H-atom abstraction, fragmentation, addition to π systems, etc.5 To channel these reactive species toward cyclization reactions, it is important to control the rate of radical formation. Organic electrochemistry, which is an enabling and innately sustainable tool for organic synthesis, has been attracting increasing interest from synthetic chemists.6 The electron transfer on the electrode can be controlled easily through adjusting the electric current or electrode potential. Hence, the rate of radical formation under electrochemical conditions can be fine-tuned conveniently. In this area, we have reported electrochemical methods for the generation of nitrogen-centered radicals (NCRs) from N–H precursors, enabling the development of several cascade radical cyclization reactions.7 Building on these studies, we report herein an electrochemically enabled 5-exo-trig/7-endo-trig cascade radical cyclization for the stereoselective preparation of functionalized 7-membered carbocycles.
Results and discussion
Our studies had begun by optimizing the electrolysis conditions for the cyclization of carbamate 1 that contained a disubstituted cis-alkene and a monosubstituted alkene. The cis-alkene was important to ensure a stereoselective 5-exo-trig cyclization for the carbamate.7a Our previously developed reaction conditions7a for alkene hydroamidation were also effective for this cascade cyclization. Hence the electrolysis was conducted in a three-necked round bottomed flask using a reticulated vitreous carbon (RVC) anode, a Pt plate cathode, and a constant current of 5 mA. The reaction employed ferrocene (Cp2Fe)8 as the catalyst, Na2CO3 as the basic additive, and 1,4-cyclohexadiene (1,4-CHD) as the reducing reagent (Table 1). Under these conditions, the biscyclized 7-membered ring product 2 was formed stereoselectively in 60% yield along with 9% of monocyclized product 3. Attempts to increase the yield of 2 by reducing the amount of 1,4-CHD (entries 2–4), changing the solvent system (entries 5–9), or varying the amount of basic additive (entries 10 and 11) failed.|
|
|||
|---|---|---|---|
| Entry | Deviation from the standard conditions | Yieldb [%] | |
| 2 | 3 | ||
| a Undivided cell, RVC anode (j ≅ 0.06 mA cm−2), Pt cathode, 1a (0.2 mmol), nBu4NBF4 (0.1 M), MeOH (1 mL), THF (5 mL), reflux, argon, 2 h, 1.9 F mol−1. b Determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as the internal standard. c Isolated yield. d Reaction for 2.5 h. PMP = p-methoxyphenyl. | |||
| 1 | None | 60c | 9 |
| 2 | 1,4-CHD (3 equiv.) | 45 | 13 |
| 3 | 1,4-CHD (1 equiv.) | 50 | 8 |
| 4d | No 1,4-CHD | 37c | 18c |
| 5 | ClCH2CH2Cl/MeOH (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
60 | 6 |
| 6 |
tBuOMe/MeOH (5:1) |
35 | 18 |
| 7 | 1,4-Dioxane/MeOH (5:1) |
47 | 16 |
| 8d | PhCl/MeOH (5:1) |
40 | 16 |
| 9 | MeCN/THF (5:1) |
54 | 7 |
| 10 | 0.5 equiv. of Na2CO3 | 57 | 11 |
| 11 | No Na2CO3 | 44 | 19 |
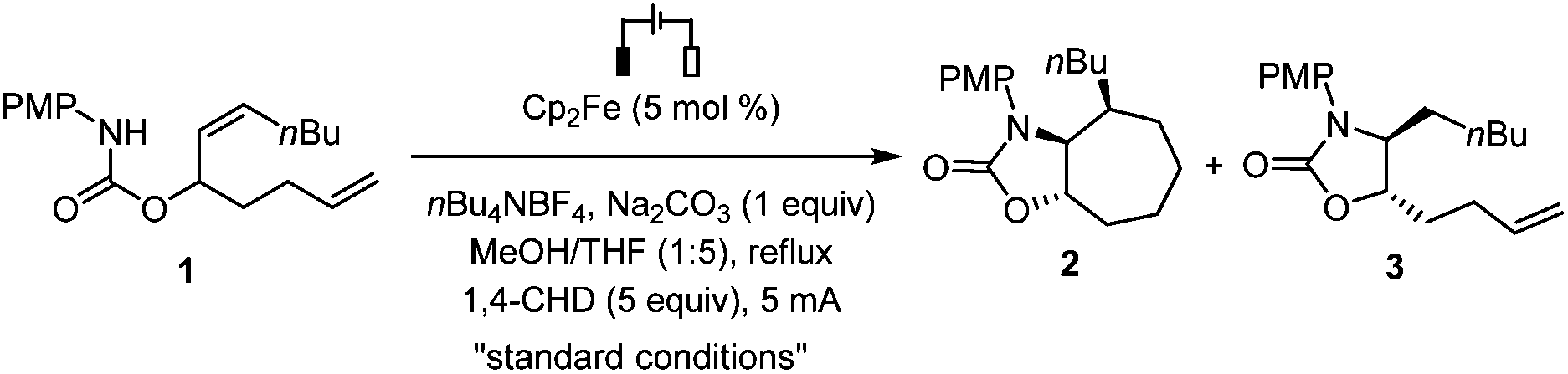
We then investigated the reaction scope (Scheme 2). The internal alkene could be substituted with primary (4) or secondary (5) alkyl groups, but not a bulky tBu group (6) probably because of the increased difficulty for the formation of the 7-membered ring.9 The cyclization reaction was compatible with N-phenyl groups bearing substituents with diverse electronic properties at the para position, including H (7), Me (8), halogens (F, Cl, Br; 9–11), CF3 (12) and CN (13). The structure of 12 was further confirmed by single crystal X-ray analysis. A meta-substituted N-phenyl group was also tolerated (14). Besides carbamates, urea type substrates bearing at the linking nitrogen atom a Me, Ph, or PMP group were also cyclized stereoselectively under the standard conditions to give cycloheptane-fused 2-imidazolones (15–17). The 7-membered ring formation was sensitive to the steric hindrance of the alkene distal to the reacting nitrogen atom as evidenced by the formation of monocyclized compound 18 as the only identifiable product in 62% yield from a substrate bearing two internal alkenes.
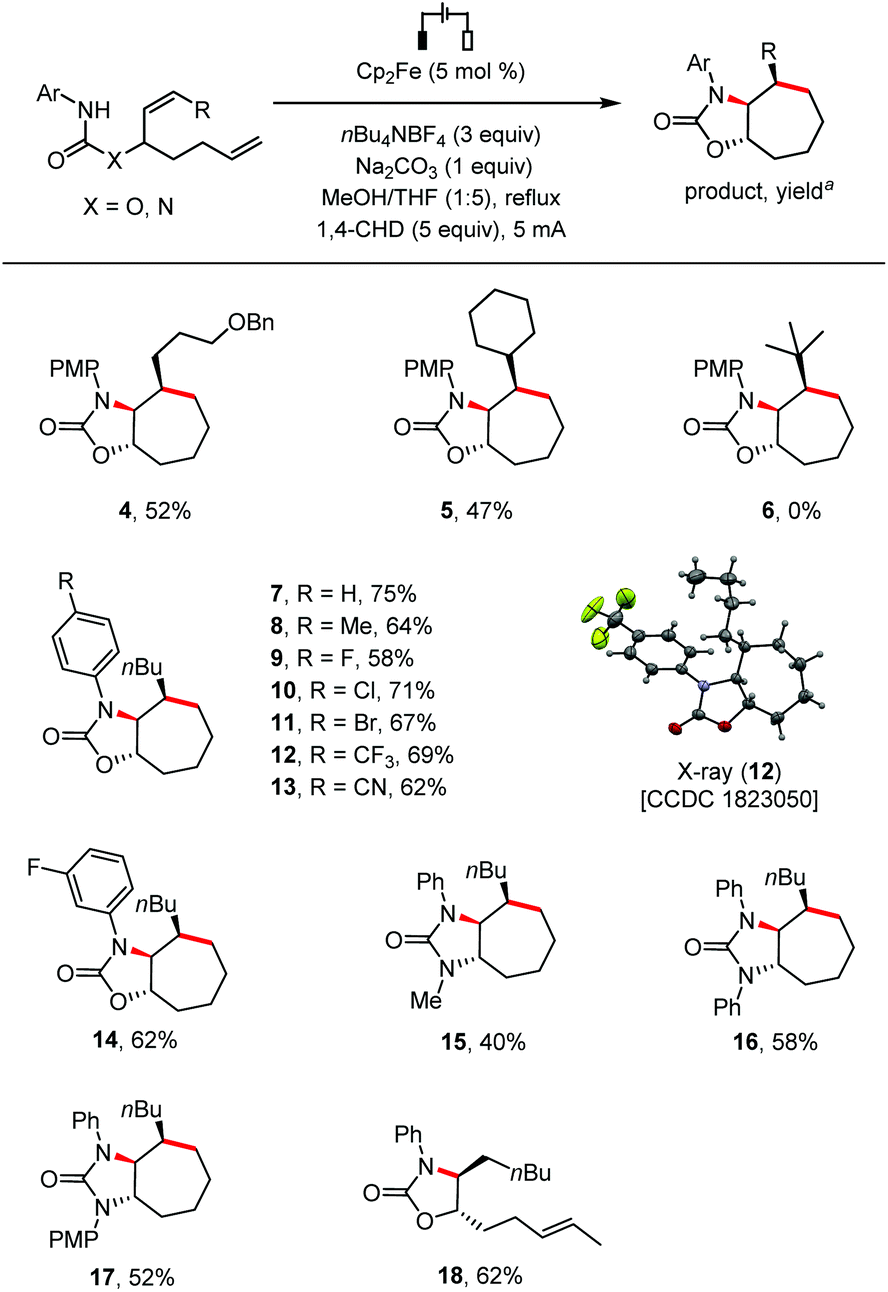 | ||
| Scheme 2 Scope of substrates. Reaction conditions: Undivided cell, substrate (0.2 mmol), THF (5 mL), MeOH (1 mL), argon, 2.0–3.0 h. aYield of the isolated product. | ||
A possible mechanism for the electrosynthesis is proposed based on the results of this work and those of previously reported (Scheme 3).7a When charge is passed through the cell, Cp2Fe loses one electron to the anode to afford Cp2Fe+. Meanwhile, the solvent MeOH is reduced at the cathode to afford methoxide (MeO−) and H2. The base generated at the cathode deprotonates substrate 1 to give its conjugate base A. The anodically generated Cp2Fe+ oxidizes A through single-electron transfer (SET) to regenerate Cp2Fe and form NCR B.10–12 The latter radical undergoes stereoselective 5-exo-trig cyclization to give carbon-centered radical species C.7a This radical abstracts a H-atom from 1,4-CHD or the solvent molecules to give monocyclized compound 3, or undergoes 7-endo-trig cyclization with the remaining terminal alkene to give the bicyclic radical intermediate D. The reduction of radical Dvia H-atom transfer affords the final 7-membered ring product 2. The 6-heptenyl radical bearing a monosubstituted alkene usually cyclizes preferentially in a 6-exo fashion instead of 7-endo.2a The trans disposition of the C-centered radical and the alkene in intermediate C is critical in channelling the cyclization to the 7-endo pathway. The alternative 6-exo-trig cyclization is probably inhibited by the high ring strain because of the trans-fusion of the forming bicyclic ring system.
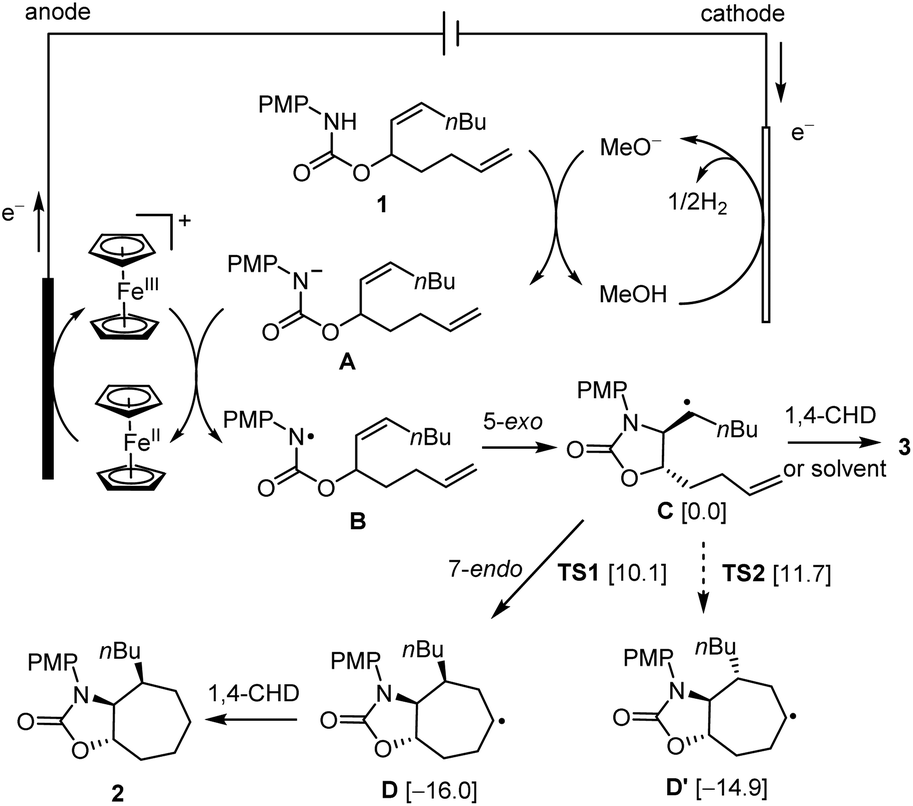 | ||
| Scheme 3 Proposed mechanism. The numbers in the brackets are DFT (UB3LYP/6-31G*) calculated Gibbs free energies (kcal mol−1) in the gas phase. Energies of D, D′, TS1 and TS2 are relative to C. | ||
Density functional theory (DFT) calculations were carried out to shed light on the origin of the stereochemistry of the 7-membered ring formation. The results suggested that the 7-endo-trig cyclization to form intermediate D with 3,4-cis stereochemistry was kinetically and thermodynamically favored over the alternative 3,4-trans stereochemistry as shown for D′ (Scheme 3). Analysis of the computed transition states TS1 and TS2 for the two cyclization pathways revealed that the steric repulsion between the PMP group and the nBu group in TS2 is probably responsible for the unfavorable formation of D′ (Fig. 1). To reduce the steric repulsion, the PMP group in TS2 rotates away from the nBu group leading to reduced conjugation of the PMP group with the lone pair of the nitrogen atom as evidenced by a much larger dihedral angle of ∠C2–N1–C12–C17 for TS2 (61.3°) compared with TS1 (35.0°).
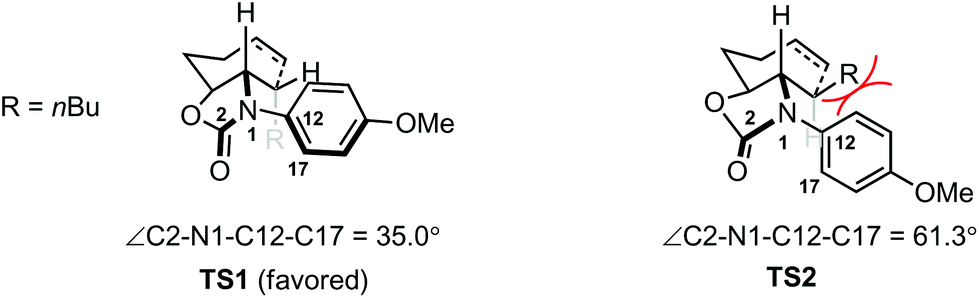 | ||
| Fig. 1 Computed transition states for the cyclization of radical C. | ||
Conclusions
In summary, we have developed a catalytic radical cascade cyclization reaction featuring a 7-endo-trig cyclization of an unhindered monosubstituted alkene. These reactions provide efficient and stereoselective access to functionalized 7-membered carbocyclic compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21672178) and the Ministry of Science and Technology (2016YFA0204100).Notes and references
- (a) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed; (b) C. P. Jasperse, D. P. Curran and T. L. Fevig, Chem. Rev., 1991, 91, 1237–1286 CrossRef CAS; (c) J. Justicia, L. Alvarez de Cienfuegos, A. G. Campana, D. Miguel, V. Jakoby, A. Gansauer and J. M. Cuerva, Chem. Soc. Rev., 2011, 40, 3525–3537 RSC.
- (a) A. L. J. Beckwith and C. H. Schiesser, Tetrahedron, 1985, 41, 3925–3941 CrossRef CAS; (b) E. Lee, C. H. Yoon and T. H. Lee, J. Am. Chem. Soc., 1992, 114, 10981–10983 CrossRef CAS; (c) X. Fang, K. Liu and C. Li, J. Am. Chem. Soc., 2010, 132, 2274–2283 CrossRef CAS PubMed.
- (a) J. Justicia, J. L. Oller-Lopez, A. G. Campana, J. E. Oltra, J. M. Cuerva, E. Bunuel and D. J. Cardenas, J. Am. Chem. Soc., 2005, 127, 14911–14921 CrossRef CAS PubMed; (b) E. Lee, J. W. Lim, C. H. Yoon, Y. S. Sung, Y. K. Kim, M. Yun and S. Kim, J. Am. Chem. Soc., 1997, 119, 8391–8392 CrossRef CAS.
- A. Ali, D. C. Harrowven and G. Pattenden, Tetrahedron Lett., 1992, 33, 2851–2854 CrossRef CAS.
- H. Togo, in Advanced Free Radical Reactions for Organic Synthesis, ed. H. Togo, Elsevier Science, Amsterdam, 2004, pp. 1–37; DOI:10.1016/B978-008044374-4/50002-5.
- Selected recent reviews on organic electrosynthesis: (a) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC; (b) J. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265–2299 CrossRef CAS PubMed; (c) J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed; (d) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS PubMed; (e) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706–6765 CrossRef CAS PubMed; (f) S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 CrossRef CAS; (g) E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 CrossRef CAS PubMed; (h) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (i) M. Yan, Y. Kawamata and S. Baran Phil, Angew. Chem., Int. Ed., 2017, 57, 4149–4155 CrossRef PubMed; (j) Q. L. Yang, P. Fang and T. S. Mei, Chin. J. Chem., 2018, 36, 338–352 CrossRef CAS; (k) C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179–7189 CrossRef CAS; (l) N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS; (m) K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed; (n) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and R. Waldvogel Siegfried, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed; (o) S. Mohle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed; (p) Y. Okada and K. Chiba, Chem. Rev., 2018, 118, 4592–4630 CrossRef CAS PubMed; (q) J.-i. Yoshida, A. Shimizu and R. Hayashi, Chem. Rev., 2018, 118, 4702–4730 CrossRef CAS PubMed.
- (a) L. Zhu, P. Xiong, Z. Y. Mao, Y. H. Wang, X. Yan, X. Lu and H.-C. Xu, Angew. Chem., Int. Ed., 2016, 55, 2226–2229 CrossRef CAS PubMed; (b) Z. W. Hou, Z. Y. Mao, H. B. Zhao, Y. Y. Melcamu, X. Lu, J. Song and H. C. Xu, Angew. Chem., Int. Ed., 2016, 55, 9168–9172 CrossRef CAS PubMed; (c) P. Xiong, H.-H. Xu, J. Song and H.-C. Xu, J. Am. Chem. Soc., 2018, 140, 2460–2464 CrossRef CAS PubMed; (d) Z.-W. Hou, Z.-Y. Mao, J. Song and H.-C. Xu, ACS Catal., 2017, 7, 5810–5813 CrossRef CAS; (e) Z.-W. Hou, H. Yan, J.-S. Song and H.-C. Xu, Chin. J. Chem., 2018, 36, 909–915 CrossRef CAS; (f) H.-B. Zhao, Z.-W. Hou, Z.-J. Liu, Z.-F. Zhou, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 587–590 CrossRef CAS PubMed; (g) Z.-W. Hou, Z.-Y. Mao, Y. Y. Melcamu, X. Lu and H.-C. Xu, Angew. Chem., Int. Ed., 2018, 57, 1636–1639 CrossRef CAS PubMed.
- For examples of Cp2Fe-mediated electroorganic synthesis, see ref. 7a–e and: (a) L. Li and S. Luo, Org. Lett., 2018, 20, 1324–1327 CrossRef CAS PubMed; (b) A. J. J. Lennox, J. E. Nutting and S. S. Stahl, Chem. Sci., 2018, 9, 356–361 RSC; (c) M. Y. Lin, K. Xu, Y. Y. Jiang, Y. G. Liu, B. G. Sun and C. C. Zeng, Adv. Synth. Catal., 2018, 360, 1665–1672 CrossRef CAS; (d) Z.-J. Wu and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 4734–4738 CrossRef CAS PubMed; (e) Z.-J. Wu, S.-R. Li, H. Long and H.-C. Xu, Chem. Commun., 2018, 54, 4601–4604 RSC.
- A 2-oxazolidinone derivative, which was similar to compound 18 and derived from intramolecular hydroamidation, was detected.
- Examples of Cp2Fe+-promoted oxidative radical reactions: (a) U. Jahn and P. Hartmann, Chem. Commun., 1998, 209–210 RSC; (b) F. Kafka, M. Holan, D. Hidasova, R. Pohl, I. Cisarova, B. Klepetarova and U. Jahn, Angew. Chem., Int. Ed., 2014, 53, 9944–9948 CrossRef CAS PubMed; (c) U. Jahn, M. Muller and S. Aussieker, J. Am. Chem. Soc., 2000, 122, 5212–5213 CrossRef CAS; (d) M. P. Sibi and M. Hasegawa, J. Am. Chem. Soc., 2007, 129, 4124–41245 CrossRef CAS PubMed.
- Examples of electrochemical generation of NCRs, see ref. 7 and: (a) S. Zhang, L. Li, M. Xue, R. Zhang, K. Xu and C. Zeng, Org. Lett., 2018, 20, 3443–3446 CrossRef CAS PubMed; (b) M.-Y. Lin, K. Xu, Y.-Y. Jiang, Y.-G. Liu, B.-G. Sun and C.-C. Zeng, Adv. Synth. Catal., 2018, 360, 1665–1672 CrossRef CAS; (c) T. Gieshoff, A. Kehl, D. Schollmeyer, K. D. Moeller and S. R. Waldvogel, J. Am. Chem. Soc., 2017, 139, 12317–12324 CrossRef CAS PubMed; (d) H.-C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2010, 132, 2839–2844 CrossRef CAS PubMed.
- (a) J. Xuan and A. Studer, Chem. Soc. Rev., 2017, 46, 4329–4346 RSC; (b) J. R. Chen, X. Q. Hu, L. Q. Lu and W. J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1823050. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qo00803e |
| This journal is © the Partner Organisations 2018 |