
Expanding the scope of peropyrenes and teropyrenes through a facile InCl3-catalyzed multifold alkyne benzannulation†
Wenlong
Yang
 ,
Rezvan R.
Kazemi
,
Nelum
Karunathilake
,
Vincent J.
Catalano
,
Mario A.
Alpuche-Aviles
and
Wesley A.
Chalifoux
*
,
Rezvan R.
Kazemi
,
Nelum
Karunathilake
,
Vincent J.
Catalano
,
Mario A.
Alpuche-Aviles
and
Wesley A.
Chalifoux
*
Department of Chemistry, University of Nevada—Reno, Reno, Nevada 89557, USA. E-mail: wchalifoux@unr.edu
First published on 10th May 2018
Abstract
Herein, we report a facile synthesis of bay-region-functionalized peropyrenes and teropyrenes through an InCl3-catalyzed double or quadruple benzannulation reaction of alkynes. This method also allows for the formation of persistently twisted, and thus chiral, peropyrenes. In contrast to using Brønsted acids to facilitate the alkyne benzannulation reaction, these InCl3-catalyzed reactions are capable of cyclizing electron-rich, electron-poor, and even alkyl-substituted alkynes; this drastically improves the scope of products that can be accessed. X-ray crystallographic analysis showed that the substituents in the bay-region play an important role, not only in the crystal packing, but also for their chirality in the solid-state. The excellent solubility and broad scope of both the peropyrenes and teropyrenes obtained by this method, enable us to fully study their interesting optical and electrochemical properties for the first time.
Introduction
A relatively simple and efficient method to produce polycyclic aromatic hydrocarbons (PAHs) containing a wide scope of functional groups is important in electronic and optoelectronic device applications.1 There are some bottom-up synthetic methods which have been widely used for the controlled synthesis of large π-extended PAHs, such as aryl–aryl oxidation reactions (Scholl reaction),2 and transition metal-catalyzed3 or light-induced4 C–C arylation reactions. Moreover, the preparation of carbon-rich materials from precursors containing C–C triple bonds is gaining more attention.5 This is because there is a large thermodynamic driving force for the formation of new C–C bonds that lead to conjugated materials on account of the high energy of the alkyne moieties in the precursors. Alkyne benzannulation reactions6 have been shown to proceed using a variety of π-Lewis acids,6c,7 Brønsted acids,8 electrophiles,9 as well as radical reagents.10 Transition-metal-catalyzed alkyne benzannulation reactions have been applied to the synthesis of pyrene derivatives.11 Plenty of π-extended pyrene derivatives have been investigated because pyrene is commercially available and relatively easy to chemically modify.12 Peropyrene and teropyrene, which can be viewed as 1D π-extended analogues of pyrene, are attractive targets for synthesis owing to their interesting properties. For example, peropyrene13 and its derivatives14 have been reported as potential singlet fission materials, and it has been proposed that their singlet fission properties could be tuned by altering the crystal packing through chemical substitution. However, there are limited methods for the preparation and functionalization of peropyrene.13,14 Thus, having an easy way to introduce different substituents onto peropyrenes to tune their crystal packing behavior, and other physical properties, would be an important contribution to the field. Teropyrenes absorb and emit light at much longer wavelengths than peropyrene,15 which make them enticing candidates for optoelectronic devices. Recently, we reported the synthesis of bay-region-functionalized peropyrenes and teropyrenes through triflic acid (TfOH) catalyzed alkyne benzannulation chemistry (Fig. 1a).15,16 This method was also successfully applied to the preparation of soluble narrow graphene nanoribbons.17 However, efforts from our lab showed that only electron-rich ethynylaryl moieties, such as p-alkyloxyphenylethynyl and ethynylthiophene, were effective cyclization candidates. Neither electron-neutral, electron-deficient ethynylaryls, nor alkyl-substituted alkyne moieties gave cyclization products when using Brønsted acids. Therefore, the creation of a new method to synthesize both peropyrenes and teropyrenes from a broad scope of alkyne-substituted precursors is highly desirable. Compared to the limited scope of Brønsted acid catalyzed alkyne benzannulation (Fig. 1a), in this paper, we disclose an InCl3-catalyzed multifold alkyne benzannulation methodology to construct peropyrenes, teropyrenes, as well as chiral peropyrenes, that is tolerant to a wide scope of alkyne substituents; this includes electron-rich, electron-neutral, and electron-deficient ethynylaryl groups and even ethynyl groups terminated with alkyl substituents (Fig. 1b). The substituent effects were studied by UV-Vis, fluorescence, and cyclic voltammetry. | ||
| Fig. 1 Synthesis of peropyrenes and teropyrenes through (a) triflic acid-catalyzed and (b) InCl3-catalyzed benzannulation reactions of alkynes. | ||
Results and discussion
Synthesis
Precursor 1a, containing electron-rich ethynylaryl substituents, was used as a representative substrate to optimize the benzannulation reaction conditions. Seven different Lewis acid catalysts with the same loadings (30 mol%) were screened in toluene at 100 °C for 12 hours and the results are summarized in Table 1 (entries 1–7). Alkyne benzannulations using Au(I),11b,18 Au(III),7d Pt(II),7e Ru(II),7e and In(III),7g catalysts have been reported to make functionalized PAHs and heteroaromatics. We observed that none of the Au(I) catalysts gave cyclized product 2a (entries 1–3, Table 1). AuCl3 as a catalyst resulted in trace amounts of mono-cyclized intermediate15 along with a significant amount of starting material (entry 4). Compared to Au(I) and Au(III) catalysts, In(III) catalysts exhibited higher catalytic ability for this transformation. It is important to point out that after one of the two alkynes reacts to produce the mono-cyclized intermediate, this generates a rigid planar system and makes the second alkyne cyclization more difficult.15,17 Thus, our reported double cyclization, via a planar intermediate, is different (and more difficult to do) than previously reported InCl3-catalyzed alkyne benzannulations.7d,g Full conversion of starting material 1a to peropyrene 2a was observed in the presence of both In(OTf)3 and InCl3, where peropyrene 2a was isolated in 73% and 88% yields, respectively (entries 5 and 6). We found that InCl3 resulted in a cleaner reaction than In(OTf)3. We also investigated Sc(OTf)3 for alkyne benzannulation, which, to the best of our knowledge, had never been used before for alkyne benzannulations. Sc(OTf)3 also worked to give 2a as the major product along with a trace amount of mono-cyclized intermediate. Because this was an inseparable mixture and since Sc(III) was less effective than In(III), the yield was not determined (entry 7). With InCl3 proving to be an excellent, and cost-effective catalyst for this benzannulation reaction, we decided not to screen any other precious metal catalysts. During our control study (30 mol%, 100 °C, and 12 h reaction time; entry 6), we noticed that the reaction seemed to be complete before 12 h. Upon repeating this reaction and monitoring it closely, we found that full cyclization was indeed accomplished within 2 hours to provide peropyrene 2a in 89% yield (entry 8). Decreasing the catalytic loading to 20 mol% required a prolonged reaction time but still gave full conversion of starting material to provide 2a in good yield (entry 9). With InCl3 being a relatively cheap catalyst and 30 mol% catalyst loading (15 mol% per alkyne) giving a four-fold increase in reaction rate, we decided to use this catalyst loading for the remainder of the study.|
|
|||
|---|---|---|---|
| Entry | Catalysta | Time (h) | 2a (%) |
| a 30 mol% catalyst loading was used. b Isolated yield. c Inseparable mixture of 2a and mono-cyclized intermediate. d 20 mol% InCl3 was used. ND = not determined. | |||
| 1 | AuCl(PPh3) | 12 | 0 |
| 2 | AuClP(p-CF3-C6H4)3 | 12 | 0 |
| 3 | dppm(AuBr)2 | 12 | 0 |
| 4 | AuCl3 | 12 | 0 |
| 5 | In(OTf)3 | 12 | 73 |
| 6 | InCl3 | 12 | 88 |
| 7 | Sc(OTf)3 | 12 | NDc |
| 8 | InCl3 | 2 | 89 |
| 9d | InCl3 | 8 | 83 |

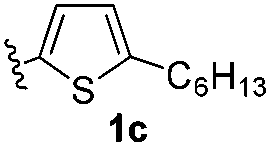
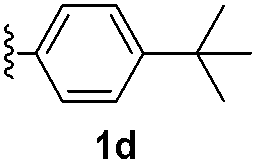
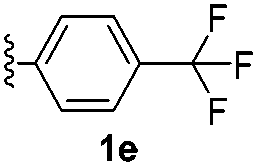
With optimized reaction conditions in hand, we began exploring the scope of this reaction. As expected, electron-rich 1b and 1c successfully produced peropyrenes 2b and 2c in 85% and 86% yields, respectively (entries 1 and 2, Table 2). Interestingly, the yield for 2c is higher than that obtained for the benzannulation reaction using TfOH.16b It is noteworthy that we were able to successfully synthesize the 4-tert-butylphenyl-substituted peropyrene 2d in 91% yield (entry 3), a reaction that failed with TfOH. However, after treating the electron-deficient precursor 1e with 30 mol% InCl3 in toluene at 100 °C for 36 hours, we observed the formation of peropyrene 2e along with a significant amount of mono-cyclized intermediate. Full conversion could not be achieved even after 72 hours at 100 °C using 50 mol% catalyst loading. Finally, by switching to xylene as solvent and running the reaction for 24 hours at 150 °C, we were able to obtain peropyrene 2e in 72% yield (entry 4). Alkyl-substituted alkyne derivative 1f was investigated, and upon treatment with 30 mol% InCl3 in toluene at 100 °C for 16 hours, alkyl-substituted peropyrene 2f was isolated in 87% yield (entry 5). With this new methodology we now have easy access to a broad scope of functionalized peropyrenes – soft materials that are attractive for various electronic and optoelectronic applications.



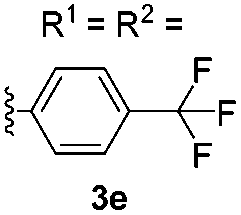
After the successful preparation of bay-functionalized peropyrenes, we began looking at a four-fold annulation reaction to prepare teropyrene derivatives. After treating precursor 3a under the optimized reaction conditions for 10 hours, we found that the reaction was incomplete with only some desired product formed. As expected, increasing the catalyst loading to 50 mol% significantly enhanced the reaction rate with full conversion to product being observed after 8 hours at 100 °C. However, desired teropyrene 4a was only isolated in 20% yield along with a significant amount of deep purple side product(s) (not characterized) (entry 1, Table 3). Lowering the reaction temperature to 80 °C substantially suppressed the formation of side products with teropyrene 4a being isolated in a much improved 85% yield (entry 1, Table 3). Under the same reaction conditions, thiophene-functionalized teropyrene 4b was isolated in 79% yield (entry 2). Interestingly, carrying out the synthesis of teropyrenes 4c and 4d at 100 °C resulted in 90% and 75% yields, respectively, with no apparent side product formation (entries 3 and 4). Unfortunately, we failed to fully cyclize 3e – containing four p-trifluoromethylphenyl substituents – to afford the teropyrene product 4e. Although partial cyclization was observed, the formation of 4e was not observed in any appreciable amount, even after prolonged reaction time in xylene at 150 °C with 50 mol% catalyst loading (entry 5). This is likely because, as the alkynes begin to cyclize in succession, the polyaromatic backbone becomes more electron-deficient (i.e. less nucleophilic), making the ensuing cyclizations more challenging. Since we were successful in cyclizing two electron-deficient alkynes to make peropyrene 2e (Table 2), we suspected that we might be able to make a teropyrene that contains at least two electron-poor aryl substituents. To test our hypothesis, precursor 3f – containing two p-hexyloxyphenylethynyl and two p-trifluoromethylphenylethynyl groups – was synthesized. Indeed, teropyrene 4f, a “push–pull” system, was isolated in 88% yield (entry 6), which proved that the electron density of the backbone can affect the reaction. Since electron-rich alkynes cyclize faster (see Tables 2 and 3), we suspect that one or both of these electron-rich alkyne groups cyclize first to produce a peropyrene intermediate that is relatively electron-rich, which makes cyclization of the remaining electron-poor alkynes more facile (see Scheme S9†).






Recently, we reported the first synthesis of chiral peropyrenes through a Brønsted acid-catalyzed four-fold benzannulation reaction of alkynes. This reaction allowed the formation of two new doubly-substituted (thus, helical) bay-regions within the same reaction.16a As shown above, teropyrenes could be effectively obtained through an InCl3 catalyzed four-fold benzannulation reaction of alkynes. Therefore, we treated precursor 5a with 50 mol% of InCl3, and although we observed formation of 6a, partly cyclized intermediates16a could still be observed, even after stirring for 48 hours at 100 °C. Increasing to 120 °C with a 100 mol% of InCl3 (25 mol% catalyst loading per alkyne) provided chiral peropyrene 6a in 63% yield (entry 1, Table 4). The attempted preparation of chiral peropyrene 6b – having four thiophene groups in the bay-positions – failed, with significant decomposition being observed with prolonged reaction time (entry 2). Chiral peropyrene 6c, with four bulky p-tert-butylphenyl groups within the two bay-regions, was obtained in 52% yield (entry 3). Similar to the preparation of teropyrene 4e, we failed to fully cyclize the electron-deficient tetrayne precursor 5d to the corresponding chiral peropyrene 6d, even at elevated temperature (entry 4). An alkyl-substituted alkyne derivative was also investigated, and chiral peropyrene 6e was successfully isolated, albeit in a modest 8% yield, with the rest being intractable material (entry 5). Very bulky groups were employed to see if we could further twist the peropyrene backbone. We were able to successfully obtain the extremely sterically congested chiral peropyrene 6f in 59% yield with its structure unambiguously confirmed by X-ray crystallography (vide infra). From the NMR analysis of 6f, we observed that the rotation of aryl substituents in the bay-positions was significantly restricted, with the tert-butyl groups also showing up as broad signals in the 1H NMR spectrum, even at elevated temperature (see ESI†). This addition suggests that this InCl3-catalyzed alkyne benzannulation methodology broadens the scope of products, increases the yields, and also results in cleaner reactions (relative to using Brønsted acids), making it much easier to purify all of these interesting and highly desirable compounds.






Crystal structures
Single crystals suitable for X-ray crystallographic analysis were obtained for peropyrene 2a, teropyrenes 4c and 4d, and chiral peropyrene 6f. Compared to the crystal structures of 2b15 and 2c,16b the peropyrene backbone of 2a is slightly less twisted with a bay-region dihedral angle of 13.5° and end-to-end twist angle of 12° (Fig. 2a). Twisted compound 2a is chiral in the solid-state and packs in a slip-stacked arrangement with its symmetry-generated enantiomer (Fig. 2b) and a minimum interplanar spacing of 3.3 Å (Fig. 2c). Unlike 4a15 and 4d, teropyrene 4c is chiral in the solid-state (Fig. 2d) and crystalizes with 1.5 molecules (with opposite handedness; red and blue structures in Fig. 2e) in the asymmetric unit. The second half of the partial molecule is generated by a two-fold symmetry operation. The bay-region of the more twisted molecule of 4c (blue, Fig. 2e) within the asymmetric unit has dihedral angles up to 21°, possibly due to the high steric repulsion of the tert-butylphenyl groups (Fig. 2d). Furthermore, the two structures of 4c in the unit cell have different end-to-end twist angles of 35° and 27° (Fig. 2e), resulting in a layered packing motif (Fig. 2f). The structure of teropyrene 4d shows C2v symmetry that is similar to the solid-state structure of 4a,15 in which the central naphthyl unit of the PAH is twisted 23° out of planarity with respect to the coplanar ends (Fig. 2g). This twisting is a result of steric repulsion between the alkyl groups and the hydrogen atom in the bay-region of 4d, resulting in dihedral angles of 23° and 25° (Fig. 2g). More interestingly, 4d adopts a slip-stacking arrangement (Fig. 2h) with a close interplanar spacing of 3.5 Å (Fig. 2i), making this material a promising candidate in organic electronic and optical devices. We were able to obtain single crystals of 6f that were suitable for X-ray crystallographic analysis and to probe whether the twist angle could be increased by increasing the sterics (Fig. 2j). Compound 6f is indeed more twisted 6a16a (by about 3° end-to-end) due to the high repulsion between the 3,5-di-tert-butylphenyl substituents, with dihedral angles within the bay-regions of up to 32° and the end-to-end twist angle of 30° (Fig. 2j–k). The π-stacking distance between two aryl substituents within the same bay-region is 4 Å, which is about 1 Å farther apart than that observed in 6a.16a Peropyrene 6f showed a slip-stacking structure (Fig. 2l), while 6a adopted a face-to-face molecular packing.16a | ||
| Fig. 2 X-ray structures (ellipsoids at 50% probability, hydrogens removed for clarity) and packing structures (hydrogens and side chains removed for clarity) of (a–c) 2a, (d) 4c, which contained 1.5 molecules within the asymmetric cell but only the most twisted molecule is shown, (e, f) the two independent molecules of 4c with different end-to-end twist angles, (g–i) 4d, and (j–l) 6f. | ||
Optical properties
Our relatively mild method of producing peropyrenes and teropyrenes allows for incorporation of a variety of functionality, leading to these compounds exhibiting excellent solubility in a number of common organic solvents. For the first time, we are now able to study substituent effects on optical and electronic properties of these interesting molecules. The comparison of their UV-vis absorption in toluene solution is shown in Fig. 3. The absorption spectra and the λmax of aryl-substituted peropyrenes 2b–f are quite similar, which means that electronic communication between the aryl side chains in the bay-region and the peropyrene backbone is negligible (Fig. 3a). This insignificant substituent effect becomes even more evident when comparing the λmax of the aryl-substituted peropyrenes 2b–e to the alkyl-substituted peropyrene 2f. The same phenomena were also observed in the absorption spectra of teropyrenes 4 (Fig. 3b); this being particularly evident when comparing all other teropyrene spectra to the alkyl-substituted derivative 4d and also to 4f, which is effectively a push–pull system. These results are in accordance with X-ray analysis in which the aryl substituents in the bay-region are twisted out of conjugation by 57–68° relative to the PAH backbone, leading to very little orbital overlap. More interestingly, we found that the highly twisted chiral peropyrenes 6 exhibited a ∼40 to 50 nm red-shift compared with peropyrenes 2. This red-shift is also observed for the all alkyl-substituted chiral peropyrene 6e, albeit to a lesser degree. This suggests that the red-shift is due, at least in part, to twisting of the PAH backbone. In contrast with the results obtained for compounds 2, we found that the absorption spectra of chiral peropyrenes 6 did shift slightly when varying the substituents on the aryl group (Fig. 3c). This is because the aryl group in the bay-region of 6 has better π-overlap with the PAH backbone and is twisted out of conjugation by only 33–37° (less than observed for compounds 2, vide supra). This increase in conjugation is evident by a red-shift (∼20 nm) in UV-vis absorbance in going from the tetraalkyl derivative 6e to the tetraaryl derivative 6a. The good solubility of peropyrenes 2 and teropyrenes 4 allows us to study their thin-film absorption properties. Broad absorption bands were observed along with red-shifts in absorbance (10–15 nm) (Fig. S7†). The broadening and red-shift observed in the absorbance spectra is a strong indication of intermolecular π–π interactions between these molecules in the solid-state. However, the tert-butylphenyl-substituted peropyrene 2d and teropyrene 4c had negligible red-shifts in the solid-state (Fig. S7†). This can be attributed the bulky substituents causing large packing distances between molecules, leading to weak π–π interactions (as seen in the X-ray packing). The fluorescence spectra were also recorded for all compounds (Fig. S2, S4, and S6†). Peropyrenes emit green light and teropyrenes red light, as observed in analogous systems.15 There were very small Stokes shifts observed for all compounds, which is expected on account of their high rigidity.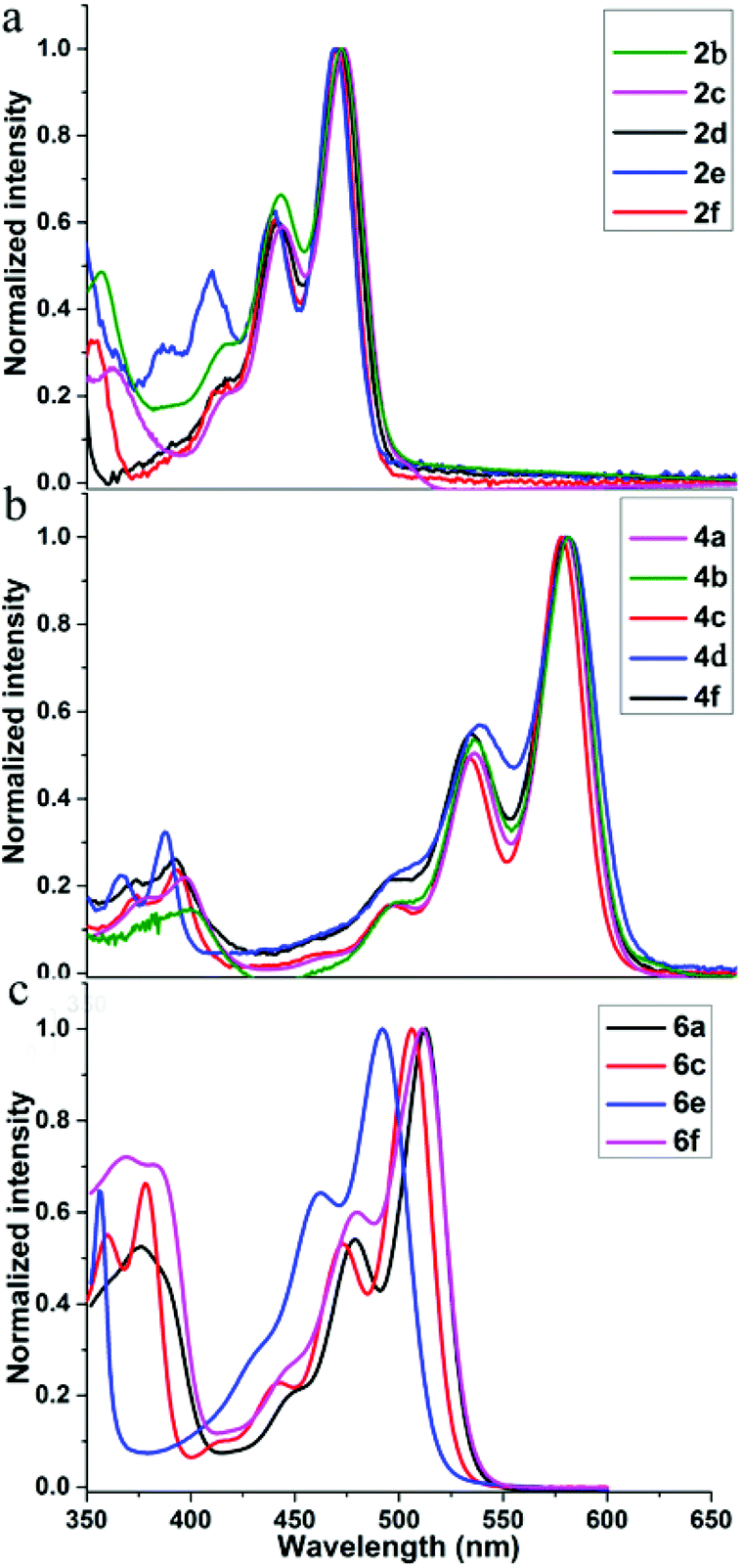 | ||
| Fig. 3 Normalized UV-vis absorption spectra of (a) peropyrenes 2, (b) teropyrenes 4, and (c) chiral peropyrenes 6, in toluene at room temperature. | ||
Electrochemistry
To the best of our knowledge, the comparison of the electrochemistry of peropyrenes, chiral peropyrenes, and teropyrenes, has not been reported. The electrochemical behavior of peropyrene, chiral peropyrene, and teropyrene compounds were studied using cyclic voltammetry (CV), and the results are summarized in Table 5 with CV results of some representative compounds shown in Fig. 4. All the compounds exhibit two reversible peaks for reduction and oxidation reactions, respectively. Herein, we focus on the first reduction and oxidation waves and discuss substituent effects on the bandgap. A complete discussion of the electrochemistry of these compounds will be presented elsewhere. As can be seen in Fig. 4, all compounds show chemically reversible waves for a one-electron reduction and a one-electron oxidation process. The assignment of a one-electron process is justified by the excellent agreement between the digital simulations and the experimental data (Fig. S8 and S9†). As expected, the reduction process is harder for the peropyrene containing electron-rich aryl substituents (2b) and easier for electron-poor aryl substituents (2e) (Fig. 4a). Compared to alkyl-substituted peropyrene 2f, all aryl-substituted peropyrenes (2b–e) exhibited lower HOMO and LUMO energies. However, their bandgaps remain consistent (∼2.5 eV). Compared to peropyrene 2b, chiral peropyrene 6a showed increased HOMO and decreased LUMO energies (Fig. 4b), which caused a slight decrease of the bandgap (0.1 eV). Similar changes were also observed between 2d and 6c. Interestingly, the bandgap of 6e is also 0.1 eV lower than 2f. In this case, the alkyl substituents are expected to have a negligible effect on the bandgap; therefore, this result suggests that increasing the twist of the peropyrene core causes the bandgap to decrease. However, we found that the HOMO of the very sterically hindered compound 6f dramatically decreased while its LUMO remained consistent with other chiral peropyrene derivatives, resulting in a larger bandgap than expected. There is a slight difference (0.21 eV) between the electronic and optical bandgap for 6f, which suggests there is a slightly higher barrier of electron transfer for this substrate, relative to the other pero- and teropyrene derivatives. We are currently investigating these differences and the effect of twisting in these molecules. Teropyrenes 4 exhibited a smaller bandgap (∼2.1 eV) than peropyrenes 2 and 6, which is clearly shown in Fig. 4c. Compared to 4a, both reduction and oxidation peaks of 4f are shifted towards more positive potentials due to the two electron-withdrawing CF3 groups (Fig. 4c). From this study we can conclude that altering the substituents in the bay-region can be a method to modulate the HOMO/LUMO levels but has little effect on the bandgap. However, bandgap engineering can be achieved by modulating the conjugation length of the PAH backbone. It is also noteworthy to point out that all of the optical and electrochemical bandgap energies show excellent agreement, except for compound 6f (as discussed above).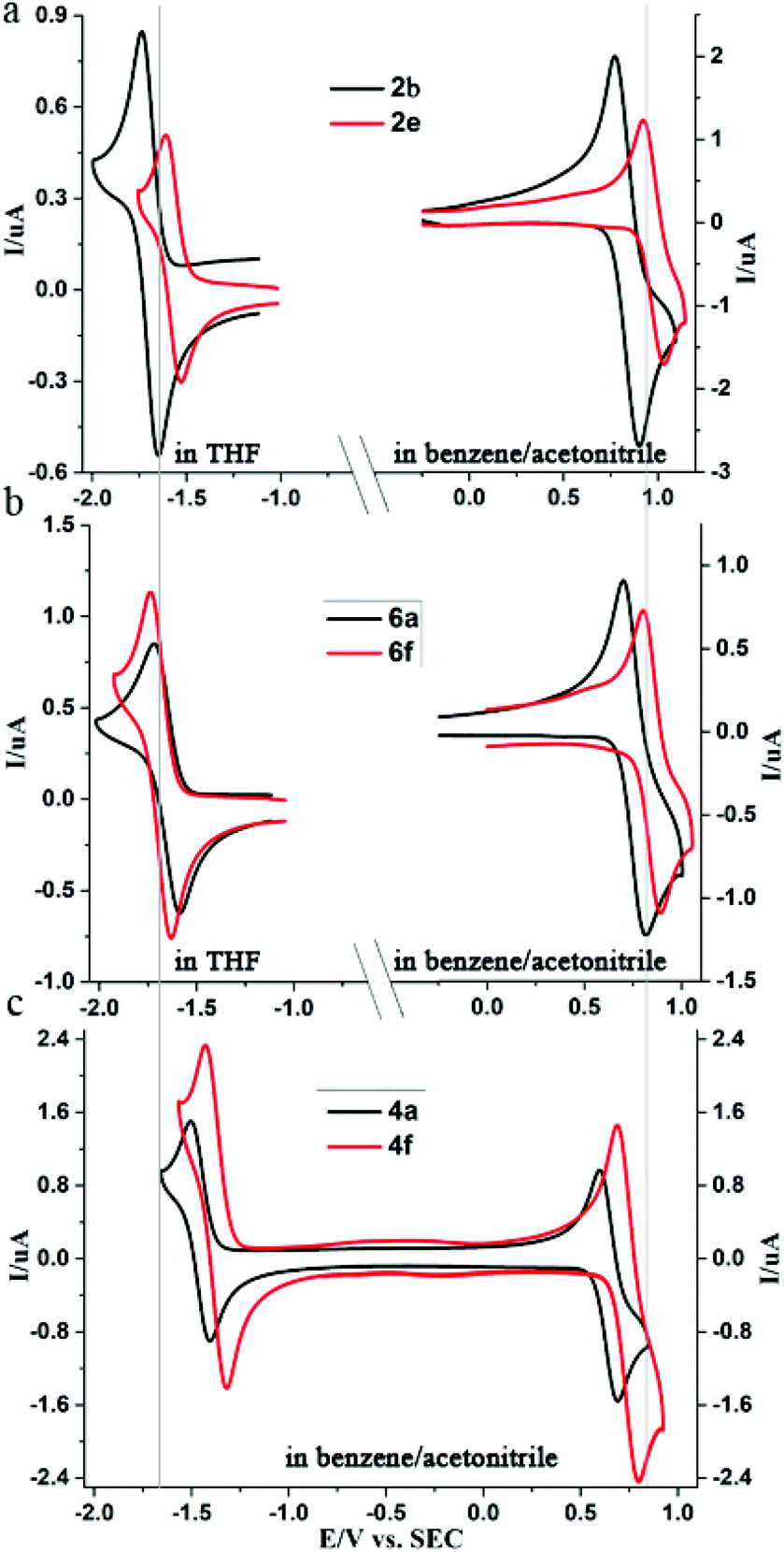 | ||
Fig. 4 Cyclic voltammograms of (a) 2b and 2e in THF (0.4 mM) for reduction and in benzene/acetonitrile (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture (0.8 mM) for oxidation, (b) 6a and 6f in THF (0.4 mM) for reduction and in benzene/acetonitrile (5:1) mixture (0.8 mM) for oxidation, (c) 4a and 4f in benzene/acetonitrile (5:1) mixture (0.8 mM). 0.1 M TBAP was used as the supporting electrolyte. Gray line (left and right lines are E0 of the first reduction, at 1.686, and oxidation, at 0.824, of 2b, respectively) added as a guide to the eye to show relative changes in reduction and oxidation energies. :1) mixture (0.8 mM) for oxidation, (b) 6a and 6f in THF (0.4 mM) for reduction and in benzene/acetonitrile (5:1) mixture (0.8 mM) for oxidation, (c) 4a and 4f in benzene/acetonitrile (5:1) mixture (0.8 mM). 0.1 M TBAP was used as the supporting electrolyte. Gray line (left and right lines are E0 of the first reduction, at 1.686, and oxidation, at 0.824, of 2b, respectively) added as a guide to the eye to show relative changes in reduction and oxidation energies. | ||
| Compounda |
b/V |
b/V |
E
HOMOc/eV |
E
LUMOc/eV |
E
g(EC)d/eV |
λ
abse/nm |
E
g(opt)f/eV |
|---|---|---|---|---|---|---|---|
| a The electrochemistry of 2a was not studied since 2a and 2b are similar. b Redox potential obtained by digital simulation versus SCE. c Calculated according to EHOMO = −(EOx + 4.4 eV) and ELUMO = −(ERed + 4.4 eV). d Calculated from redox potentials. e Absorption wavelengths of the first absorption onset. f Estimated from the UV-vis spectra. | |||||||
| 2b | 0.82 | −1.68 | −5.22 | −2.71 | 2.51 | 467 | 2.50 |
| 2c | 0.86 | −1.63 | −5.26 | −2.77 | 2.49 | 469 | 2.50 |
| 2d | 0.82 | −1.66 | −5.22 | −2.74 | 2.52 | 465 | 2.53 |
| 2e | 0.96 | −1.56 | −5.36 | −2.84 | 2.52 | 465 | 2.54 |
| 2f | 0.77 | −1.70 | −5.17 | −2.70 | 2.47 | 468 | 2.52 |
| 4a | 0.63 | −1.45 | −5.03 | −2.95 | 2.08 | 572 | 2.07 |
| 4b | 0.69 | −1.37 | −5.09 | −3.03 | 2.06 | 573 | 2.06 |
| 4c | 0.64 | −1.45 | −5.04 | −2.95 | 2.09 | 569 | 2.08 |
| 4d | 0.58 | −1.43 | −4.98 | −2.97 | 2.01 | 572 | 2.05 |
| 4f | 0.73 | −1.38 | −5.13 | −3.02 | 2.11 | 571 | 2.06 |
| 6a | 0.76 | −1.66 | −5.16 | −2.74 | 2.42 | 506 | 2.34 |
| 6c | 0.80 | −1.62 | −5.20 | −2.78 | 2.42 | 513 | 2.34 |
| 6e | 0.72 | −1.66 | −5.12 | −2.74 | 2.38 | 492 | 2.37 |
| 6f | 0.87 | −1.67 | −5.27 | −2.73 | 2.54 | 511 | 2.33 |


Conclusions
In summary, we have demonstrated that the synthesis of bay-region-functionalized peropyrenes and teropyrenes can be achieved using multifold alkyne benzannulations catalyzed by InCl3. We established that various aryl- and alkyl-substituted alkyne moieties can be cyclized, enabling access to a wide scope of these useful compounds. Incorporation of a broad scope of substituents into these conjugated materials not only allows for tuning of their optical and electronic properties but also imparts excellent solubility, making these conjugated materials easily processable for a breadth of applications. A decrease in the HOMO–LUMO gap (2.50 to 2.10 eV) from peropyrene to teropyrene was observed in both the optical and electrochemical studies. Within a certain class of PAH (i.e., peropyrene, chiral peropyrene, or teropyrene), small changes in HOMO and LUMO energy levels were observed when varying the bay-region substituents, with the bandgaps remaining essentially constant. There did seem to be an effect on the bandgap energies as a function of the PAH being twisted out of planarity, with bandgaps decreasing as a function of twist angle. X-ray crystallographic analysis showed that the substituents in the bay-regions can effectively tune their crystal packing motifs to modulate the intermolecular π–π interactions. We believe that this new methodology will lead to the development of new functional PAHs, and that these multifaceted studies will attract scientists in other fields to explore their use in electronic and optoelectronic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
WAC acknowledges the National Science Foundation (NSF) for supporting this work through a CAREER Award (CHE-1555218). MAA thanks the NSF for support through a CAREER Award (CHE-1255387).Notes and references
- (a) T. Wöhrle, I. Wurzbach, J. Kirres, A. Kostidou, N. Kapernaum, J. Litterscheidt, J. C. Haenle, P. Staffeld, A. Baro, F. Giesselmann and S. Laschat, Chem. Rev., 2016, 116, 1139 CrossRef PubMed; (b) M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267 CrossRef PubMed; (c) Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463 CrossRef PubMed; (d) W. Pisula, X. Feng and K. Müllen, Adv. Mater., 2010, 22, 3634 CrossRef PubMed; (e) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718 CrossRef PubMed.
- (a) K. Y. Cheung, X. Xu and Q. Miao, J. Am. Chem. Soc., 2015, 137, 3910 CrossRef PubMed; (b) T. Fujikawa, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 7763 CrossRef PubMed; (c) J. Liu, B. W. Li, Y. Z. Tan, A. Giannakopoulos, C. Sanchez-Sanchez, D. Beljonne, P. Ruffieux, R. Fasel, X. Feng and K. Müllen, J. Am. Chem. Soc., 2015, 137, 6097 CrossRef PubMed; (d) A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616 RSC; (e) T. Fujikawa, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2016, 138, 3587 CrossRef PubMed; (f) Y. Li, Z. Jia, S. Xiao, H. Liu and Y. Li, Nat. Commun., 2016, 7, 11637 CrossRef PubMed; (g) Y. Sakamoto and T. Suzuki, J. Am. Chem. Soc., 2013, 135, 14074 CrossRef PubMed; (h) K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739 CrossRef PubMed; (i) A. Pradhan, P. Dechambenoit, H. Bock and F. Durola, Angew. Chem., Int. Ed., 2011, 50, 12582 CrossRef PubMed; (j) H.-A. Lin, Y. Sato, Y. Segawa, T. Nishihara, N. Sugimoto, L. T. Scott, T. Higashiyama and K. Itami, Angew. Chem., Int. Ed., 2018, 57, 2874 CrossRef PubMed; (k) J. Luo, X. Xu, R. Mao and Q. Miao, J. Am. Chem. Soc., 2012, 134, 13796 CrossRef PubMed.
- (a) Y. Koga, T. Kaneda, Y. Saito, K. Murakami and K. Itami, Science, 2018, 359, 435 CrossRef PubMed; (b) W. Matsuoka, H. Ito and K. Itami, Angew. Chem., Int. Ed., 2017, 56, 12224 CrossRef PubMed.
- (a) M. Daigle, A. Picard-Lafond, E. Soligo and J.-F. Morin, Angew. Chem., Int. Ed., 2016, 55, 2042 CrossRef PubMed; (b) R. K. Mohamed, S. Mondal, J. V. Guerrera, T. M. Eaton, T. E. Albrecht-Schmitt, M. Shatruk and I. V. Alabugin, Angew. Chem., Int. Ed., 2016, 55, 12054 CrossRef PubMed.
- (a) E. T. Chernick and R. R. Tykwinski, J. Phys. Org. Chem., 2013, 26, 742 CrossRef; (b) F. Diederich and M. Kivala, Adv. Mater., 2010, 22, 803 CrossRef PubMed; (c) R. S. Jordan, Y. Wang, R. D. McCurdy, M. T. Yeung, K. L. Marsh, S. I. Khan, R. B. Kaner and Y. Rubin, Chem., 2016, 1, 78 CrossRef; (d) I. V. Alabugin and E. Gonzalez-Rodriguez, Acc. Chem. Res., 2018, 51, 1206 CrossRef PubMed.
- (a) W. Yang and W. A. Chalifoux, Synlett, 2017, 28, 625 CrossRef; (b) S. J. Hein, D. Lehnherr, H. Arslan, F. J. Uribe-Romo and W. R. Dichtel, Acc. Chem. Res., 2017, 50, 2776 CrossRef PubMed; (c) H. Arslan, J. D. Saathoff, D. N. Bunck, P. Clancy and W. R. Dichtel, Angew. Chem., Int. Ed., 2012, 51, 12051 CrossRef PubMed.
- (a) S. Yamaguchi and T. M. Swager, J. Am. Chem. Soc., 2001, 123, 12087 CrossRef PubMed; (b) A. Fürstner and V. Mamane, J. Org. Chem., 2002, 67, 6264 CrossRef; (c) A. Fürstner and V. Mamane, Chem. Commun., 2003, 2112 RSC; (d) V. Mamane, P. Hannen and A. Furstner, Chem. – Eur. J., 2004, 10, 4556 CrossRef PubMed; (e) T. A. Chen, T. J. Lee, M. Y. Lin, S. M. Sohel, E. W. Diau, S. F. Lush and R. S. Liu, Chem. – Eur. J., 2010, 16, 1826 CrossRef PubMed; (f) J. Carreras, G. Gopakumar, L. Gu, A. Gimeno, P. Linowski, J. Petuškova, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2013, 135, 18815 CrossRef PubMed; (g) R. Stężycki, M. Grzybowski, G. Clermont, M. Blanchard-Desce and D. T. Gryko, Chem. – Eur. J., 2016, 22, 5198 CrossRef PubMed.
- M. B. Goldfinger, K. B. Crawford and T. M. Swager, J. Am. Chem. Soc., 1997, 119, 4578 CrossRef.
- T. Yao, M. A. Campo and R. C. Larock, J. Org. Chem., 2005, 70, 3511 CrossRef PubMed.
- K. Pati, C. Michas, D. Allenger, I. Piskun, P. S. Coutros, G. D. P. Gomes and I. V. Alabugin, J. Org. Chem., 2015, 80, 11706 CrossRef PubMed.
- (a) M. M. Machuy, C. Würtele and P. R. Schreiner, Synthesis, 2012, 44, 1405 CrossRef; (b) T. Matsuda, T. Moriya, T. Goya and M. Murakami, Chem. Lett., 2011, 40, 40 CrossRef.
- (a) J. M. Casas-Solvas, J. D. Howgego and A. P. Davis, Org. Biomol. Chem., 2014, 12, 212 RSC; (b) T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260 CrossRef PubMed.
- V. M. Nichols, M. T. Rodriguez, G. B. Piland, F. Tham, V. N. Nesterov, W. J. Youngblood and C. J. Bardeen, J. Phys. Chem. C, 2013, 117, 16802 Search PubMed.
- (a) K. Uchida, T. Kubo, D. Yamanaka, A. Furube, H. Matsuzaki, R. Nishii, Y. Sakagami, A. Abulikemu and K. Kamada, Can. J. Chem., 2017, 95, 432 CrossRef; (b) M. Martinez-Abadia, G. Antonicelli, A. Saeki and A. Mateo-Alonso, Angew. Chem., Int. Ed. DOI:10.1002/anie.201804453.
- W. Yang, J. H. S. K. Monteiro, A. de Bettencourt-Dias, V. J. Catalano and W. A. Chalifoux, Angew. Chem., Int. Ed., 2016, 55, 10427 CrossRef PubMed.
- (a) W. Yang, G. Longhi, S. Abbate, A. Lucotti, M. Tommasini, C. Villani, V. J. Catalano, A. O. Lykhin, S. A. Varganov and W. A. Chalifoux, J. Am. Chem. Soc., 2017, 139, 13102 CrossRef PubMed; (b) W. Yang, J. H. S. K. Monteiro, A. de Bettencourt-Dias and W. A. Chalifoux, Can. J. Chem., 2016, 95, 341 CrossRef.
- W. Yang, A. Lucotti, M. Tommasini and W. A. Chalifoux, J. Am. Chem. Soc., 2016, 138, 9137 CrossRef PubMed.
- A. Davis, D. Walker and J. Howgego, Synthesis, 2010, 3686 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra, X-ray crystallographic data, supplementary absorption and fluorescence spectra, and electrochemistry. CCDC 1826979–1826982. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qo00389k |
| This journal is © the Partner Organisations 2018 |