
Base-promoted [3 + 3] cyclization of cyclopropenones and cyclopropenethiones with amides for the synthesis of 6H-1,3-oxazin-6-ones and 6H-1,3-thiazin-6-ones†
Ben
Niu
a,
Bo
Jiang
b,
Liu-Zhu
Yu
a and
Min
Shi
 *abc
*abc
aKey Laboratory for Advanced Materials and Institute of Fine Chemicals, School of Chemistry & Molecular Engineering, East China University of Science and Technology, Meilong Road No. 130, Shanghai, 200237, China
bState Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: mshi@mail.sioc.ac.cn
cState Key Laboratory and Institute of Elemento-organic Chemistry, Nankai University, Tianjin 300071, P. R. China
First published on 28th February 2018
Abstract
A facile synthetic method to access 6H-1,3-oxazin-6-ones and 6H-1,3-thiazin-6-ones has been disclosed via a base-promoted [3 + 3] cyclization reaction of cyclopropenones and cyclopropenethiones with amides. These reactions exhibited excellent yields and good functional group tolerance under metal free and mild conditions.
6H-1,3-Oxazin-6-one frameworks have been recognized as important core structures that widely exist in medicinal agents, pharmaceuticals and biologically active molecules,1 such as salinazinone A, salinazinone B and AX-9657, as shown in Scheme 1. Consequently, synthetic methods of 6H-1,3-oxazin-6-ones have garnered much attention over the past few years and various useful methods have been developed for the synthesis of 6H-1,3-oxazin-6-ones.2 For example, in 2013, Guan and co-workers achieved palladium-catalyzed oxidative carbonylation of enamides with equivalent Cu(OAc)2 as an oxidant for the construction of 6H-1,3-oxazin-6-ones (Scheme 2, eqn (1)).2a In 2015, Liu's group described a gold-catalyzed cycloaddition reaction of tert-butyl propiolates with nitriles (Scheme 2, eqn (2)).2b More recently, Liu and co-workers have successfully realized the formal carbonylation reaction of enamides with Togni's reagent as the CO surrogate for the generation of 6H-1,3-oxazin-6-ones (Scheme 2, eqn (3)).2c Obviously, these reaction procedures are often accompanied by transition metal catalysts and the use of complex starting materials is unavoidable. Sometimes, undesired side products were also formed in these reactions. Therefore, the exploration of a simple, more general and convenient synthetic protocol for the synthesis of 6H-1,3-oxazin-6-ones is still highly desirable.
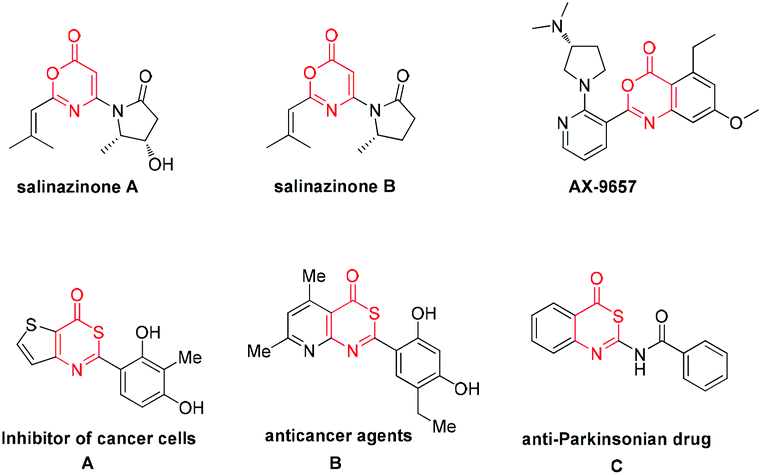 | ||
| Scheme 1 Biologically active molecules containing 6H-1,3-oxazin-6-ones and 6H-1,3-thiazin-6-one. | ||
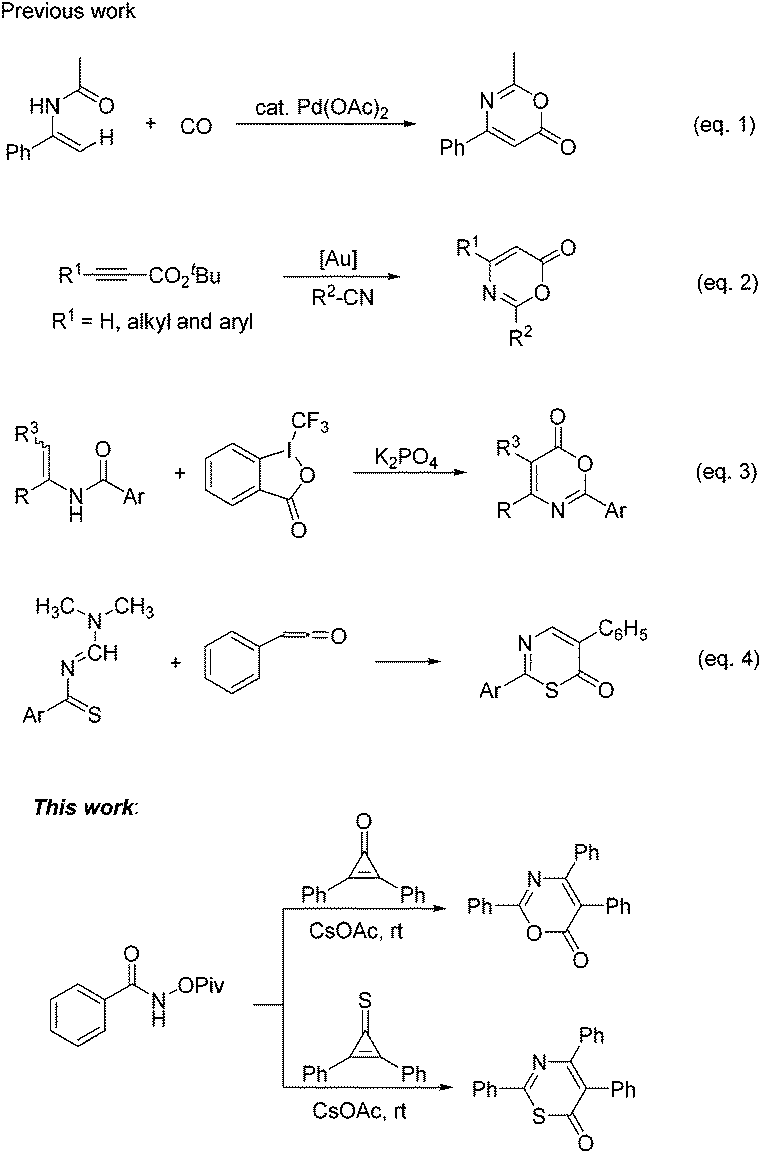 | ||
| Scheme 2 Previous work for the synthesis of 6H-1,3-oxazin-6-ones and 6H-1,3-thiazin-6-one. | ||
On the other hand, their sulfur-containing analogues, 6H-1,3-thiazin-6-ones, are also an important class of molecules that are widely used in various fields, including the pharmaceutical industry, agrochemistry and materials sciences.3 For example, molecule A has the ability to suppress DNA synthesis in cancer cells,3a especially in C6 cells (Scheme 1). Compound B also has anticancer activity in vitro (Scheme 1).3a Molecule C, structurally neither related to xanthines nor to adenine, has been identified as an anti-Parkinsonian drug (Scheme 1).3b However, until now, a lack of methodological investigation on the construction of 6H-1,3-thiazin-6-ones was a gap in the organic synthetic chemistry domain. Thus far, easily available methods for synthesizing the 6H-1,3-thiazin-6-one unit were very limited and have a lot of drawbacks,4 such as substrate dependence, poor yields, harsh reaction conditions and so on. For example, in 1975, Quiniou and co-workers synthesized 6H-1,3-thiazin-6-one through an intermolecular cyclization reaction of N-methylenebenzothioamide with ketene (Scheme 2, eqn (4)).4a Obviously, complex and labile starting materials were used. Undoubtedly, the development of new methods to access 6H-1,3-thiazin-6-ones in a simple and efficient way is also very meaningful in the agrochemical field and in medicinal chemistry.
In recent years, the chemistry of strained small rings, particularly three-membered rings,5 has been extensively investigated as a class of activated coupling partners.6 Among them, cyclopropenones, a kind of representative highly reactive molecules, have also been broadly used in organic synthesis,7 because of their unique chemical properties that can react readily with both nucleophilic and electrophilic reagents. Thus, we envisioned whether cyclopropenones could react with amides to afford the desired 6H-1,3-oxazin-6-one or 6H-1,3-thiazin-6-one scaffold (Scheme 2, this work).
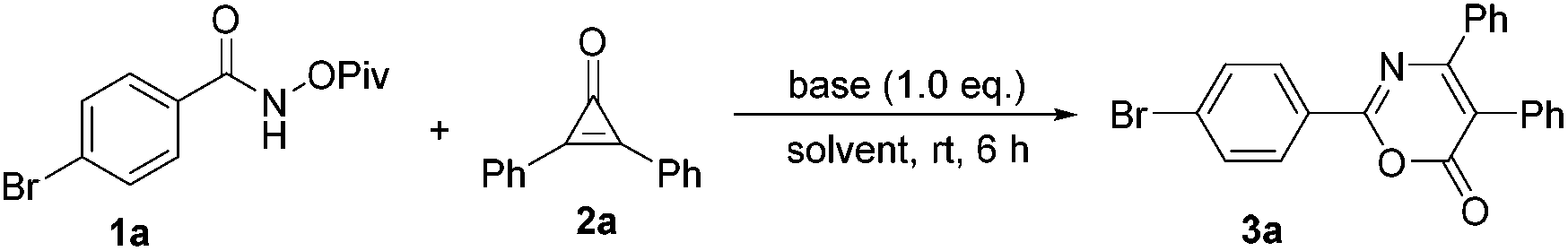
We initially investigated the reaction outcome of 4-bromo-N-(pivaloyloxy)benzamide 1a with diphenylcyclopropenone 2a. As shown in Table 1, we first tested a set of representative bases, such as Cs2CO3, NaOH, DBU and K2CO3, using methanol as a solvent, but none of the desired transformations was observed (Table 1, entries 1–4). To our delight, when the reaction mixture was treated with PhCOONa, the desired cyclization product 3a was obtained in 70% yield (Table 1, entry 5). The use of Et3N to replace PhCOONa gave 3a in 85% yield under otherwise identical conditions (Table 1, entry 6). Gratifyingly, the yield was further improved to 90% when CsOAc was used as a base (Table 1, entry 7). The examination of the solvent effect revealed that no better result could be obtained (Table 1, entries 8–11). However, when the reaction was carried out in 1,2-dichloroethane (DCE), we found that the reaction proceeded more cleanly and smoothly. The use of 0.2 equiv. CsOAc afforded the desired product 3a in 42% yield (Table 1, entry 12). Therefore, the reaction should be carried out in DCE and 1.0 equiv. CsOAc should be used as a base.
|
|
|||
|---|---|---|---|
| Entrya | Base | Solvent | Yieldb/% |
| a The reactions were carried out using 1a (0.2 mmol), 2a (0.2 mmol), base (1.0 equiv.), and solvent (2.0 mL) in a Schlenk tube. b Isolated yields. c CsOAc (0.2 equiv.). | |||
| 1 | Cs2CO3 | MeOH | nr |
| 2 | NaOH | MeOH | nr |
| 3 | DBU | MeOH | nr |
| 4 | K2CO3 | MeOH | nr |
| 5 | PhCOONa | MeOH | 70 |
| 6 | NEt3 | MeOH | 85 |
| 7 | CsOAc | MeOH | 90 |
| 8 | CsOAc | CH2Cl2 | 60 |
| 9 | CsOAc | Toluene | 72 |
| 10 | CsOAc | DCE | 90 |
| 11 | CsOAc | THF | 85 |
| 12c | CsOAc | DCE | 42 |
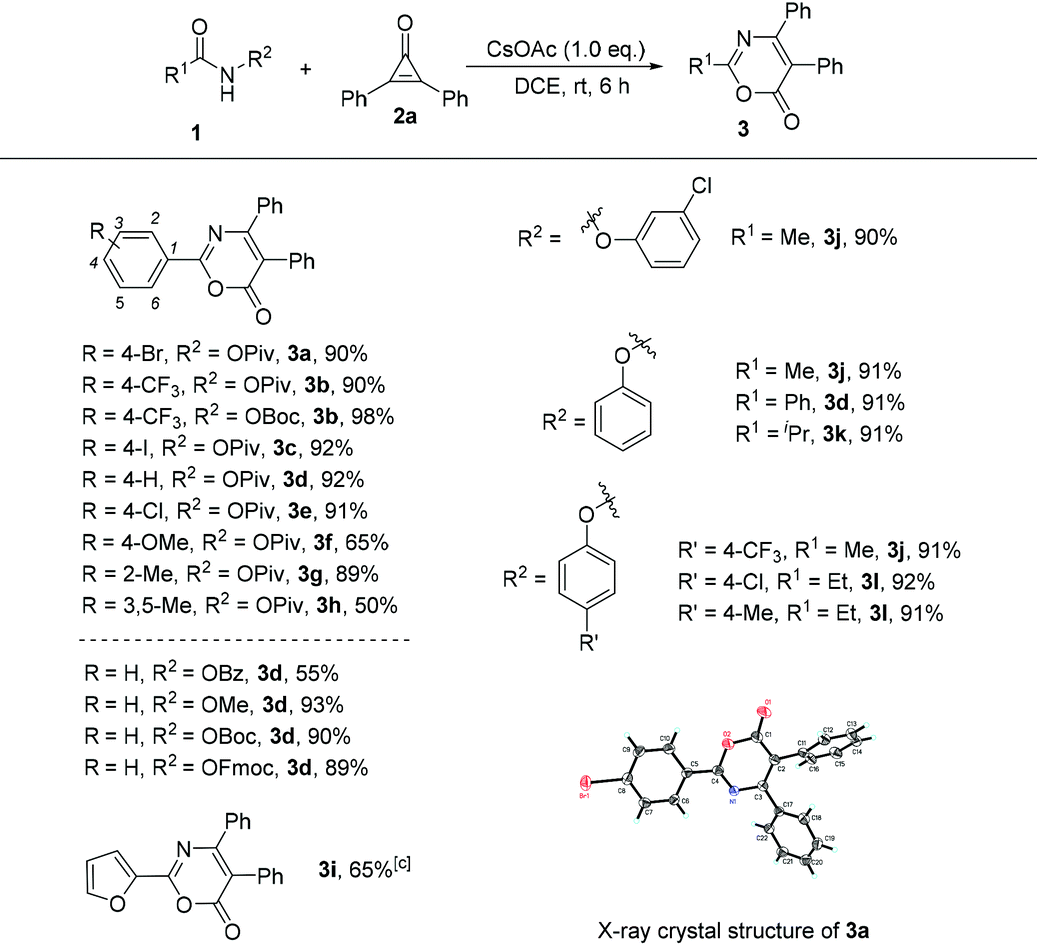
Under the optimized conditions (Table 1, entry 10), we next focused our attention on the investigation of the scope of amides in the reaction with cyclopropenone 2a. As shown in Table 2, when R1 is an aromatic ring and the leaving group is the OPiv anion, we first examined the electronic effect at the para-position of the aromatic ring. As for substrates 1b–1f, regardless of whether an electron-donating or electron-withdrawing substituent was introduced, the reaction proceeded smoothly, giving the desired products 3b–3f in good to excellent yields ranging from 65 to 98%. In the case of the ortho-substituted substrate (2-methyl) or disubstituted substrate (3,5-dimethyl), the reaction also performed very well, providing the corresponding products 3g and 3h in 65% and 89% yields, respectively. Afterwards, we screened a set of leaving groups such as OBz, OMe, OBoc and OFmoc when R1 is a phenyl group. As can be seen from Table 2, all of them afforded the desired product 3d in good yields. The heteroaryl-substituted amide 1i was also compatible, affording the corresponding product 3i in 65% yield. The structure of 3a was determined by X-ray diffraction and its ORTEP drawing is shown in Table 2.8
To make this cyclization reaction even more integrated, next, R1 was switched from an aryl group to an alkyl group and R2 was changed to a phenoxyl group. We found that these substrates were also well tolerated when R1 is a methyl, an ethyl or an isopropyl group, furnishing the target products 3d, 3j, 3k and 3l in excellent yields varying from 91–92% regardless of the electronic nature of the phenoxyl leaving group.
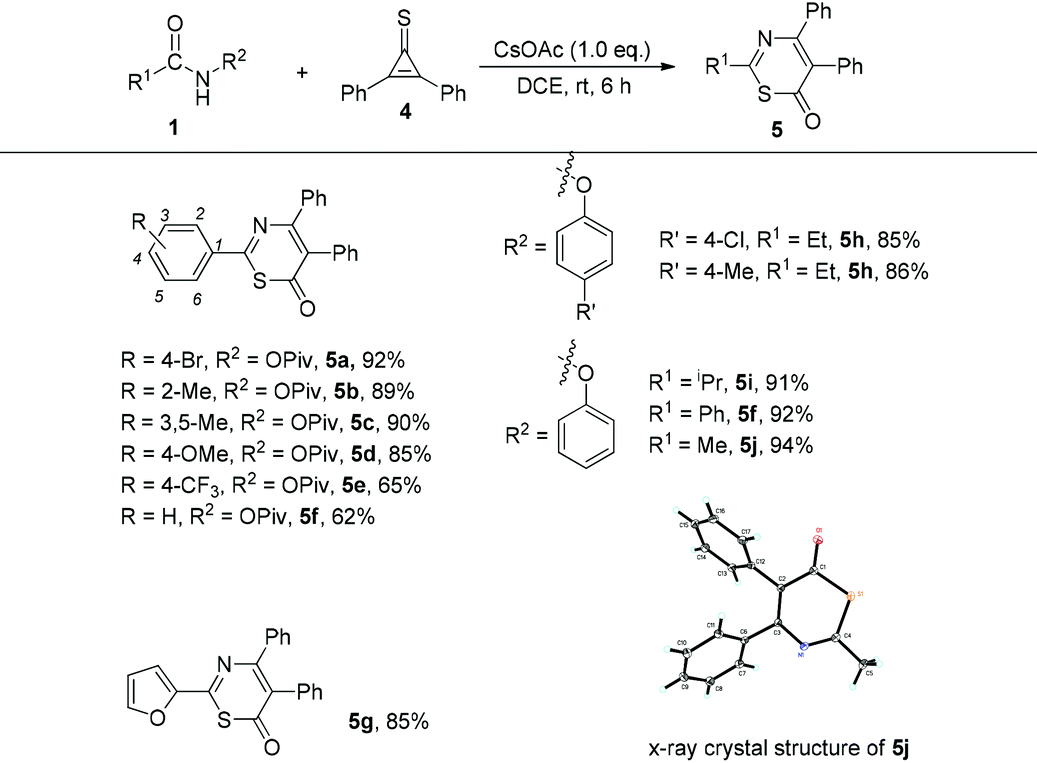
Encouraged by the above results, we next tried to replace the cyclopropenone with cyclopropenethione 4. Initially, we commenced the investigation in the reaction of N-phenoxyacetamide 1j (R1 = Me, R2 = OPh) with cyclopropenethione 4 under the optimal conditions. We found that 6H-1,3-thiazin-6-one 5j was obtained in 94% yield and its structure was unambiguously determined by X-ray diffraction9 (Table 3). After that, we further explored the substrate scope of this cycloaddition reaction and the results are shown in Table 3. Firstly, when cyclopropenethione 4 was treated with various benzamides, the desired products 5a–5f were obtained in 62–92% yields regardless of whether an electron-donating or electron-withdrawing substituent was introduced at different positions of the aromatic ring. The use of N-phenoxyacetamides 1h–1j as substrates also gave the corresponding products 5h–5j in good yields ranging from 85 to 92%. Heteroaryl-substituted amide was tolerated in this case, giving the desired product 5g in 85% yield under the standard conditions.
| a Reaction conditions: 1 (0.2 mmol), 4 (0.2 mmol), CsOAc (1.0 equiv.), DCE (2.0 mL). NR = no reaction. |
|---|
|
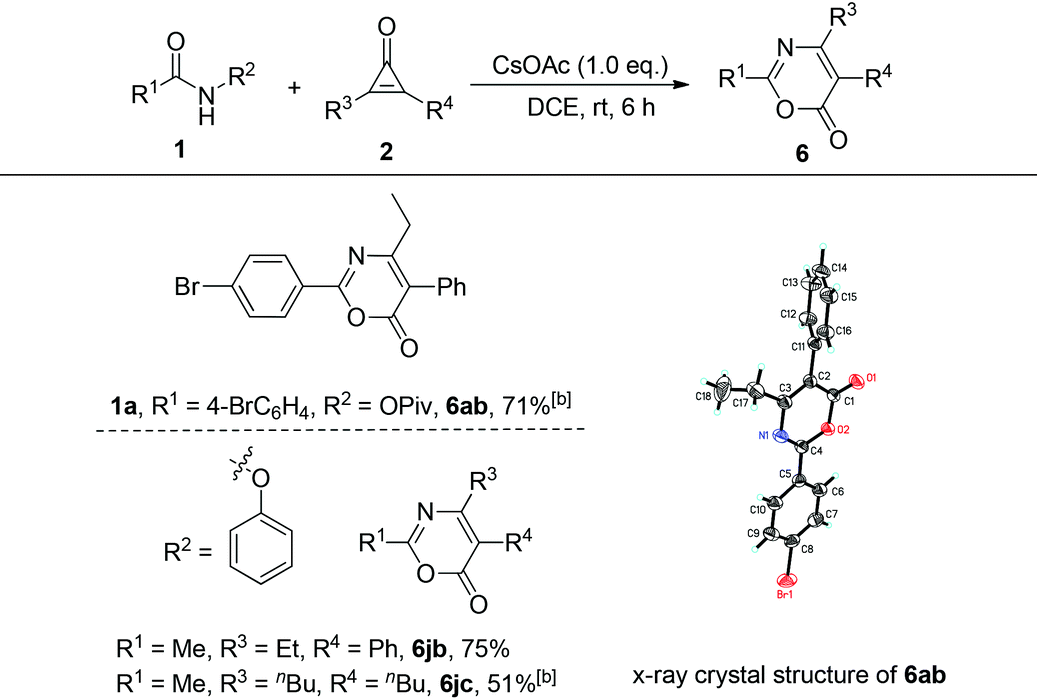
The substrate scope of cyclopropenones was also explored, using two representative amides, 4-bromo-N-(pivaloyloxy)benzamide 1a and N-phenoxyacetamide 1j (R1 = Me, R2 = OPh), for this cyclization reaction. Upon treatment of 4-bromo-N-(pivaloyloxy)benzamide 1a with 2-ethyl-3-phenylcyclopropenone 2b at 60 °C under the standard conditions provided the desired product 6ab in 71% yield. Its crystal structure has been determined by X-ray diffraction and the ORTEP drawing is shown in Table 4.10 On the other hand, the reaction of 1j with 2b proceeded smoothly at room temperature, affording the target product 6jb in 75% yield. However, the reaction of 1j with 2,3-dibutylcyclopropenone 2c should be carried out at 60 °C, giving the desired product 6jc in 51% yield, suggesting that the phenoxyl group might be a better leaving group in this transformation. When R3 and R4 are different aryl groups, a cycloadduct mixture is formed under the standard conditions. All these results indicated a wide substrate scope in this base-promoted cyclization reaction.
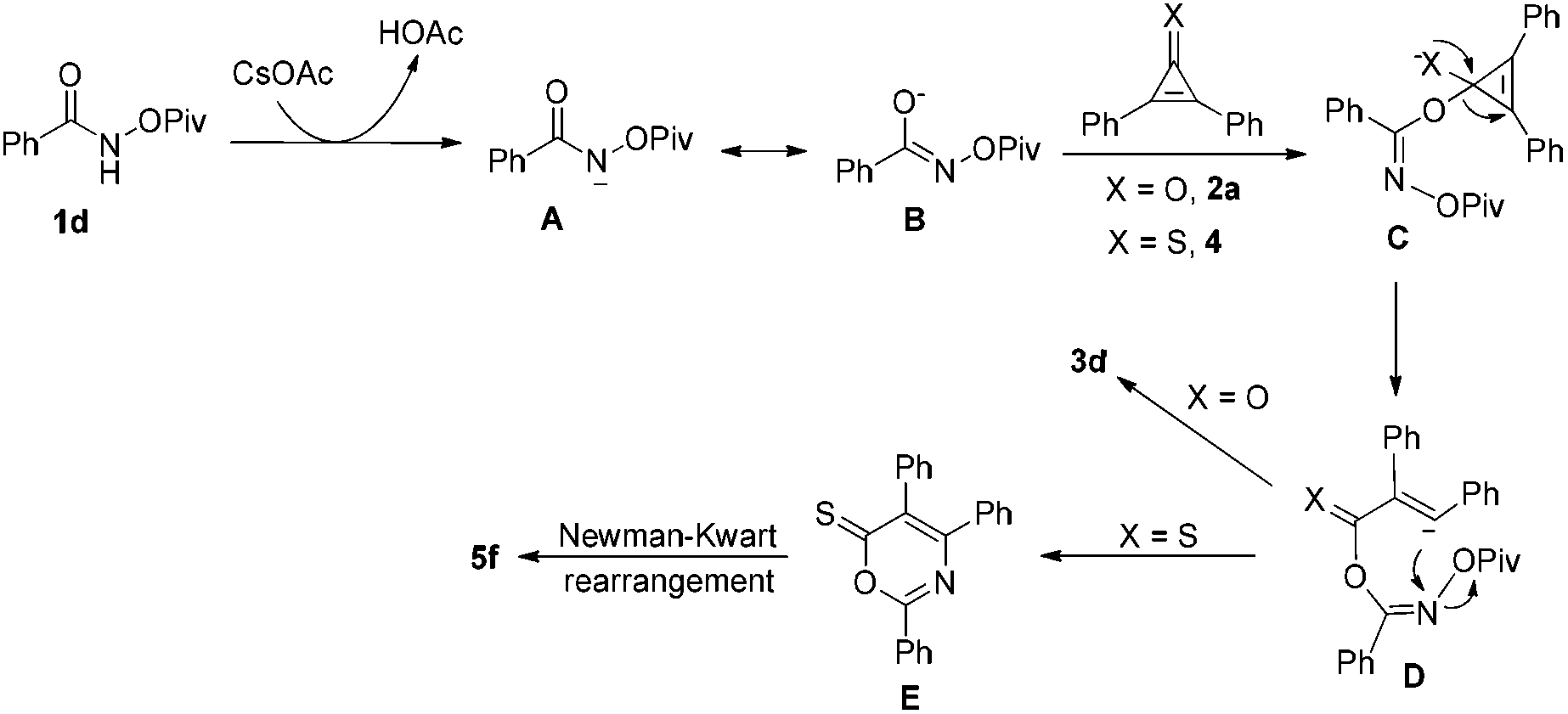
Based on the above results, a plausible mechanism has been outlined in Scheme 3. As for the synthesis of 6H-1,3-oxazin-6-ones (X = O), it is commonsense that the intermediate A is formed upon treatment of 1d with CsOAc, which undergoes an isomerization to give the intermediate B. The reaction of the intermediate B with 2a provides the intermediate C, which undergoes a ring-opening process to yield the intermediate D. Then, the desired product 3d is formed through an intramolecular nucleophilic attack reaction along with the release of the OPiv anion. On the other hand, as for the synthesis of 6H-1,3-thiazin-6-one (X = S), we believe that the intermediate E undergoes a Newman–Kwart rearrangement process11 to produce a thermodynamically more stable product 5f.
 | ||
| Scheme 3 A plausible reaction mechanism. | ||
In summary, we have developed a novel and efficient synthetic protocol to easily access 6H-1,3-oxazin-6-ones and 6H-1,3-thiazin-6-ones via a base-promoted [3 + 3] cyclization reaction of cyclopropenones and cyclopropenethiones with amides. The reaction exhibits a wide substrate scope using easily available starting materials, excellent yields and good functional group tolerance under metal free and mild conditions. The potential utilization and extension of the scope of this new synthetic methodology are currently under investigation in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Basic Research Program of China ((973)-2015CB856603), the Strategic Priority Research Program of the Chinese Academy of Sciences, Grant no. XDB20000000 and sioczz201808, the National Natural Science Foundation of China (20472096, 21372241, 21572052, 20672127, 21421091, 21372250, 21121062, 21302203, 20732008, 21772037 and 21772226), and the Fundamental Research Funds for the Central Universities (222201717003).Notes and references
- (a) M. C. Kim, J. H. Lee, B. Shin, L. Subedi, J. W. Cha, J.-S. Park, D.-C. Oh, S. Y. Kim and H. C. Kwon, Org. Lett., 2015, 17, 5024 CrossRef CAS PubMed; (b) P. Fu, S. La and J. B. MacMillan, J. Nat. Prod., 2016, 79, 455 CrossRef CAS PubMed; (c) R. L. Jarvest, M. J. Parratt, C. M. Debouck, J. G. Gorniak, L. J. Jennings, H. T. Serafinowska and J. E. Strickler, Bioorg. Med. Chem. Lett., 1996, 6, 2463 CrossRef CAS; (d) M. Gütschow and U. Neumann, Bioorg. Med. Chem., 1997, 5, 1935 CrossRef; (e) U. Neumann, N. M. Schechter and M. Gütschow, Bioorg. Med. Chem., 2001, 9, 947 CrossRef CAS PubMed; (f) P. Kopelman, A. Bryson, R. Hickling, A. Rissanen, S. Rossner, S. Toubro and P. Valensi, Int. J. Obes., 2007, 31, 494 CrossRef CAS PubMed; (g) Y. Yamada, T. Kato, H. Ogino, S. Ashina and K. Kato, Horm. Metab. Res., 2008, 40, 539 CrossRef CAS PubMed; (h) R. Padwal, Curr. Opin. Invest. Drugs, 2008, 9, 414 CAS.
- (a) M. Chen, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Angew. Chem., Int. Ed., 2013, 52, 14196 CrossRef CAS PubMed; (b) S. N. Karad, W.-K. Chung and R.-S. Liu, Chem. Sci., 2015, 6, 5964 RSC; (c) P. Song, P. Yu, J.-S. Lin, Y. Q. Li, N.-Y. Yang and X.-Y. Liu, Org. Lett., 2017, 19, 1330 CrossRef CAS PubMed; (d) C. Zhang, S. Li, F. Bureš, R. Lee, X. Ye and Z. Jiang, ACS Catal., 2016, 6, 6853 CrossRef CAS; (e) X. F. Wu, J. Schranck, H. Neumann and M. Beller, Chem. – Eur. J., 2011, 17, 12246 CrossRef CAS PubMed; (f) Q. Liu, P. Chen and G. Liu, ACS Catal., 2013, 3, 178 CrossRef CAS; (g) W. Li and X. F. Wu, J. Org. Chem., 2014, 79, 10410 CrossRef CAS PubMed; (h) S. Munusamy, S. Venkatesan and K. I. Sathiyanarayanan, Tetrahedron Lett., 2015, 56, 203 CrossRef CAS; (i) J. Yu, D. Zhang-Negrerie and Y. Du, Eur. J. Org. Chem., 2016, 562 CrossRef; (j) A. Verma and S. Kumar, Org. Lett., 2016, 18, 4388 CrossRef CAS PubMed; (k) D. L. Boger and R. J. Wysocki, J. Org. Chem., 1989, 54, 714 CrossRef CAS; (l) K. Liu, M. Z. Zou and A. W. Lei, J. Org. Chem., 2016, 81, 7088 CrossRef CAS PubMed.
- (a) J. Matysiak, M. Juszczak, M. M. Karpińska, E. Langner, K. Walczak, M. K. Lemieszek, A. Skrzypek, A. Niewiadomy and W. Rzeski, Mol. Diversity, 2015, 19, 725 CrossRef CAS PubMed; (b) A. Stößel, M. Schlenk, S. Hinz, P. Küppers, J. Heer, M. Gütschow and C. E. Müller, J. Med. Chem., 2013, 56, 4580 CrossRef PubMed; (c) C. Landreau, D. Deniaud, F. Reliquet, A. Reliquet and J. C. Meslin, Heterocycles, 2000, 53, 2667 CrossRef CAS; (d) A. Niewiadomy, J. Matysiak and M. M. Karpińska, Arch. Pharm. Chem. Life Sci., 2011, 11, 224 CrossRef PubMed; (e) C. B. Vicentine, S. Guccione, L. Giurato, R. Ciaccio, D. Mares and G. Forlani, J. Agric. Food Chem., 2005, 53, 3848 CrossRef PubMed.
- (a) J. C. Meslin and H. Quiniou, Tetrahedron, 1975, 31, 3055 CrossRef CAS; (b) C. Landreau, D. Deniaud, F. Reliquet, A. Reliquet and J. C. Meslin, Heterocycles, 2000, 53, 2667 CrossRef CAS; (c) H. Sheibania, M. H. Mosslemin, S. Behzadi, M. R. Islami, H. Foroughi and K. Saidi, ARKIVOC, 2005, 88–96 Search PubMed; (d) H.-G. Häcker, F. Grundmann, F. Lohr, P. A. Ottersbach, J. Zhou, G. Schnakenburg and M. Gütschow, Molecules, 2009, 14, 378 CrossRef PubMed; (e) S. Leistner, M. GüSchow and G. Wagner, Synthesis, 1986, 466 Search PubMed; (f) J. W. Lown and K. Matsumo, Can. J. Chem., 1972, 50, 584 CrossRef CAS.
- (a) I. Nakamura, B.-H. Oh, S. Saito and Y. Yamamoto, Angew. Chem., Int. Ed., 2001, 40, 1298 CrossRef CAS; (b) J. P. Markham, S. T. Staben and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 9708 CrossRef CAS PubMed; (c) M. Murakami, N. Ishida and T. Miura, Chem. Commun., 2006, 643 RSC; (d) G. Bhargava, B. Trillo, M. Araya, F. López, L. Castedo and J. L. Mascareñas, Chem. Commun., 2010, 46, 270 RSC; (e) B. Yao, Y. Li, Z. Liang and Y. Zhang, Org. Lett., 2011, 13, 640 CrossRef CAS PubMed; (f) S. Li, Y. Luo and J. Wu, Org. Lett., 2011, 13, 3190 CrossRef CAS PubMed; (g) L.-Z. Yu, K. Chen, Z.-Z. Zhu and M. Shi, Chem. Commun., 2017, 53, 5935 RSC; (h) C. L. Ladd and A. B. Charette, Org. Lett., 2016, 18, 6046 CrossRef CAS PubMed.
- (a) K. T. Potts and J. S. Baum, Chem. Rev., 1974, 74, 189 CrossRef CAS; (b) K. Komatsu and T. Kitagawa, Chem. Rev., 2003, 103, 1371 CrossRef CAS PubMed; (c) D. K. Nielsen and A. G. Doyle, Angew. Chem., Int. Ed., 2011, 50, 6056 CrossRef CAS PubMed; (d) Y.-L. Yang, Z. Zhang, X.-N. Zhang, D. Wang, Y. Wei and M. Shi, Chem. Commun., 2014, 50, 115 RSC; (e) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2014, 114, 7317 CrossRef CAS PubMed; (f) Y. Liang, L. Jiao, Y. Wang, Y. Chen, L. Ma, J. Xu, S. Zhang and Z.-X. Yu, Org. Lett., 2006, 8, 5877 CrossRef CAS PubMed.
- (a) P. A. Wender, T. J. Paxton and T. J. Williams, J. Am. Chem. Soc., 2006, 128, 14814 CrossRef CAS PubMed; (b) P. A. O'Gorman, T. Chen, H. E. Cross, S. Naeem, A. Pitard, M. I. Qamar and K. Hemming, Tetrahedron Lett., 2008, 49, 6316 CrossRef; (c) Y.-L. Yang, Z. Zhang, X.-N. Zhang, D. Wang, Y. Wei and M. Shi, Chem. Commun., 2014, 50, 115 RSC.
- The crystal data of 3a have been deposited at CCDC with the number 1535814.†.
- The crystal data of 5j have been deposited at CCDC with the number 1045751.†.
- The crystal data of 6ab have been deposited at CCDC with the number 1576769.†.
- (a) G. C. Lloyd-Jones, J. D. Moseley and J. S. Renny, Synthesis, 2008, 661 CAS; (b) H. Kwart and E. R. Evans, J. Org. Chem., 1966, 31, 410 CrossRef CAS; (c) M. S. Newman and H. A. Karnes, J. Org. Chem., 1966, 31, 3980 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of new compounds. CCDC 1535814, 1045751 and 1576769. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qo00091c |
| This journal is © the Partner Organisations 2018 |